Organization Of Genetic Material Chromatin
When chromatin is isolated from interphase nuclei, the individual chromosomes are not recognizable. Instead one observes an irregular aggregate of nucleoprotein. Chemical analysis of isolated chromatin shows that it consists primarily of DNA and proteins and lesser amounts of RNA. These proteins are of two major classes:
Eukaryotic Genome
- Basic proteins (positively charged at neutral pH) called histones; and
- A heterogenous, largely acidic (negatively charged at neutral pH) group of proteins collectively referred to as non-histone chromosomal proteins.
Histones: Histones play a major structural role in chromatin. They are present in the chromatin of all higher eukaryotes in amounts equivalent to the amounts of DNA (weight to weight).
- Indeed histones are present in such enormous quantities in the cell (about 60 million molecules of each l type per human cell) that their total mass in chromatin is about equal to that of DNA.
- The histones of all the higher plants and animals consist of five different major proteins. These five major histones, called H1, H2a, H2b, H3, and H4 are present in almost all cell types.
- The five histones are present in molar ratios of approximately 1H1:2H2a:2H2b:2H3:2H4. They are specifically complexed with DNA to produce
The Constituents Of Calf Thymus Chromatin:
“Understanding eukaryotic genome organization through FAQs: Q&A explained”
- Weight relative to 100 units of DNA. the basic structural subunits of chromatin, small (approximately 110 A° in diameter by 60 A° high) ellipsoidal “beads” called nucleosomes by P. Oudet et al. (1975).
- The histones have been highly conserved during evolution, four of the five types of histones being very similar in all higher eukaryotes. Indeed, the amino acid sequence of histone 4 from a pea and a cow differ at only at 2 of the 102 positions.
- This strong evolutionary conservation suggests that a change in any position is deleterious to the cell. This suggestion has been tested directly in yeast cells, in which it is possible to mutate a given histone gene in vitro and introduce it into the yeast genome in place of the normal gene.
- As might be expected, most changes in histone sequences are lethal; the few that are not lethal cause changes in the normal pattern of gene expression, as well as, other abnormalities.
- The histones are basic because they contain 20 – 30 percent arginine and lysine, two positively charged amino acids.
- The exposed – NH3+ group of arginine and lysine allow histones to act as polycations. This is important in their interaction with DNA, which is polyanionic, because of the negatively charged phosphate groups.

Composition Of Histones:
“How is the eukaryotic genome structured and regulated? FAQ answered”
- The remarkable constancy of histones H2A, H2B, H3, and H4 in all cell types of an organism and even between widely divergent species is consistent with the idea that they are important in chromatin structure (“DNA packaging” and are only specifically involved in the regulation of gene.
- In contrast, the nonhistone protein fraction of chromatin consists of a large number of very heterogeneous proteins. The composition of the nonhistone chromosomal protein fraction varies widely among different cell types of the same organism.
- Thus, the nonhistone chromosomal proteins are likely candidates for roles in the regulation of expression of specific genes or sets of genes.
DNA Arrangement in Eukaryotic Chromosome: The Unineme Model: How is the 1 – 20 cm (104 to 2 × 105μm) of DNA, which is present in an average eukaryotic chromosome, arranged in the highly condensed mitotic and meiotic structures that are seen with the light microscope?
- Are there many DNA molecules that run parallel throughout the chromosome (the “multigene” or “multistrand” model), or is there just one DNA double helix extending from one end of the chromosome to the other end(the “unnamed” or “single – strand” Model)?
- Are there many DNA molecules joined end-to-end or arranged in some other fashion in the chromosome, or does one giant, continuous molecule of DNA extend from one end to the other in a highly coiled and folded form? The evidence supporting the unnamed model of chromosome structure is now overwhelming.
- Thus, according to the current view, each eukaryotic chromosome appears to contain a single, giant molecule of DNA that extends from one end through the centromere to the other end of the chromosome.
“Importance of studying eukaryotic genome organization for biology students: Questions explained”
Evidence In Support of The Unineme Model: Evidence that the eukaryotic chromosome is uncinematic comes from several sources as follows:
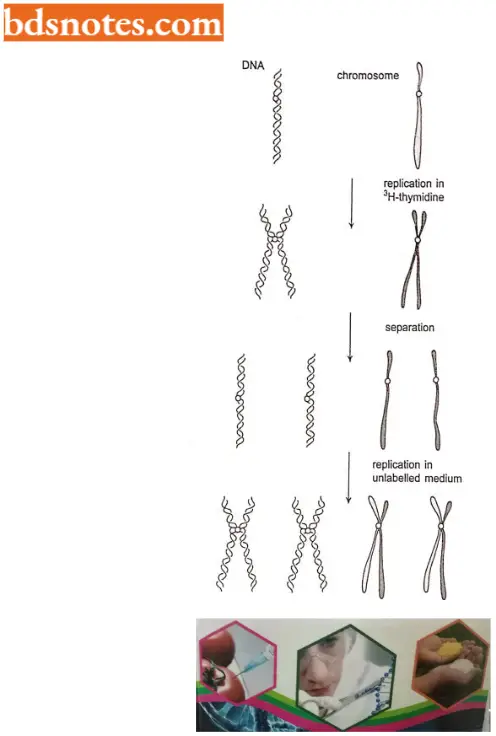
- Radioactive experiments. The best data are provided by radioactive labeling studies, first done by J. Taylor and his colleagues in 1957.
- If a eukaryote is allowed to undergo one DNA replication in the presence of tritiated (3H- 1) thymidine, each of the daughter chromatids would be expected to contain a double helix with one unlabeled DNA template strand and one labeled with each chromatid containing one double helix.
- A second round of DNA replication, in the absence of 3H-thymidine, should produce chromosomes in which one chromatid would have unlabelled DNA and one would have labeled DNA. Thus, one chromatid of every pair is labeled and one is not.
- Lampbrush chromosomes in vertebrate oocytes. Another strong piece of evidence supporting the unnamed model of chromosome structure has come from the studies on the lampbrush chromosomes. Such chromosomes are present during prophase I of oogenesis in many vertebrates, particularly amphibians.
- Lambrush chromosomes are up to 800 pm long; they thus provide very favorable material for cytological studies.
- The homologous chromosomes are paired, and each is duplicated to produce two chromatids at the lampbrush stage.
- Each lampbrush chromosome contains a central axial region, where the two chromatids are highly condensed and numerous pairs of lateral loops.
- The loops are transcriptionally active regions of single chromatids. The integrity of both the central axis and the lateral loops is dependent on DNA. Treatment of both of these with DNase enzyme fragments them.
- In contrast, treatment with RNA or protease enzyme removes surrounding matrix material but does not destroy the continuity of either the axis or the loops.
- Electron microscopy of RNase – and protease-treated lampbrush chromosomes reveals a central filament of just over 20 A° in diameter in the lateral loops.
- Since each loop is a segment of one chromatid, and since the diameter of a DNA double helix is 20A°, these lampbrush chromosomes must be unnamed structures.
- The above conclusion is also supported by studies on the kinetics of the nuclear digestion of lampbrush chromosomes.
- That is, the kinetics observed are those expected if a single double helix of DNA is the central filament in the loops.
- The axial region then contains two DNA molecules, one from each of the two tightly paired chromatids.
- Viscoelsotometric evidence. In 1974, biophysicists such as R.Kavenoff, L.Klotf, and B. Zimin used a technique called viscoelastometry (a procedure for analyzing viscosity parameters of molecules in solution) to estimate the sizes of DNA molecules from eukaryotic chromosomes.
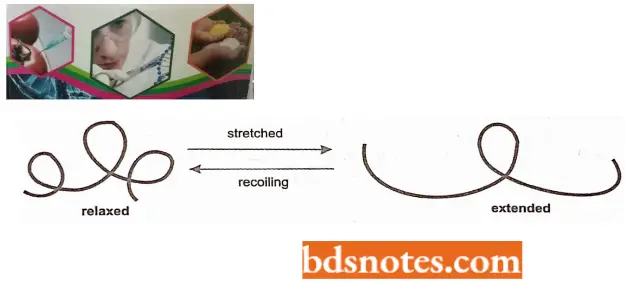
- When DNA molecules in solution are exposed to a driving force (for example, by rotating a cylinder in the solution) they are stretched into an extended conformation.
- When the driving force is removed (for example., by stopping the rotation of the cylinder), the molecules will return to their lowest energy or relaxed state.
- In the solution being analyzed, the recoil time is a function of the size of the DNA molecule. The recoil decays exponentially over long time intervals, and the time constant called the retardation time (T), is a sensitive function of the size of the largest DNA molecules in the solution.
- Thus, even if some of the DNA molecules are broken by shearing forces, the viscoelastometric procedure will permit one to estimate the size of the largest DNA molecules present.
- To minimize the chance of shearing of the molecules when they are released from the cells, the cells can be lysed right in the chamber of the viscoelastometer.
- The largest chromosome in Drosophila melanogaster cells was estimated to have a mass of 4.1 × 1010 daltons by viscoelastometry. The largest chromosomes of Drosophila have been shown to contain about 4.3 × 1010 daltons of DNA by direct microspectrophotometric analysis of metaphase chromosomes.
- Thus, the viscoelastometric estimate of the size of the largest DNA molecules in D. melanogaster nuclei correlated almost exactly with the total amount of DNA present in the largest chromosome.

“Common challenges in understanding eukaryotic genome organization effectively: FAQs provided”
- Autoradiographic evidence. Researchers have also used the technique of autoradiography to attempt to detect chromosome-size DNA molecules from eukaryotic cells in the same way that Cairns was able to visualize the intact E. coli chromosome.
- Similar to viscoelastometry, autoradiography permits one to lyse cells very gently to minimize shearing forces.
- DNA molecules from the lysed cells are permitted to diffuse onto a membrane that is used to pick up the DNA molecules and expose them to the (β-particle – sensitive emulsion. Thus, the breakage of molecules should not be a problem.
- The difficulty with autoradiographic examination of very large DNA molecules is that it is almost impossible to get all segments of the molecule sufficiently spread out on the membrane with no tangles or overlaps so that the entire graphic analysis of DNA molecules from Drosophila has been successful, and the results of these studies also support the concept of chromosome size DNA molecules (Kavenoff et al., 1973).
Packaging The Gaint DNA Molecules Into Chromosomes: DNA compaction ratio. All eukaryotic organisms have elaborate ways of packaging DNA into chromosomes.
- For example, human chromosome 22 contains about 48 million nucleotide pairs. Stretched out end to end, its DNA extends about 1.5 cm. Yet, when it exists as a mitotic chromosome, chromosome 22 measures only about 2 pm in length, giving an end-to-end compaction ratio of nearly 10,000-fold.
- This remarkable feat of compression is performed by proteins that successively coil and fold the DNA into higher and higher levels of organization.
- Although less is dynamic: not only do chromosomes globally condense in accord with the cell cycle, but different regions of the interphase chromosomes condense and decondense as the cells gain access to specific DNA sequences for gene expression, DNA repair, and DNA replication.
- The packaging of chromosomes must therefore be accomplished in a way that allows rapid localized, on-demand access to the DNA.
- When interphase nuclei are broken open very gently and their contents examined under the electron microscope, most of the chromatin is in the form of a fiber with a diameter of about 30nm.
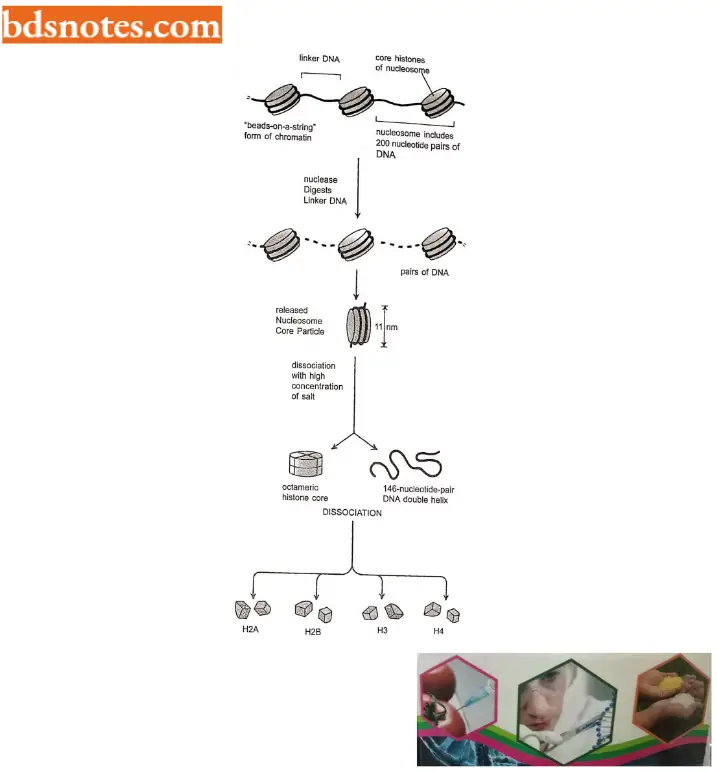
- If this chromatin is subjected to treatments that cause it to unfold partially, it can be seen under the electron microscope as a series of “beads on a string”.
- The string is DNA, and each bead is a “nucleosome core particle” that consists of DNA wound around a protein core formed from histones. The beads on a string represent the first level of chromosomal DNA packing.

- The structural organization of nucleosomes was determined after first isolating them from unfolded chromatin by digestion with particular enzymes (called micrococcal nuclease) that break down DNA by cutting between the nucleosome core particles, and the linker DNA is degraded.
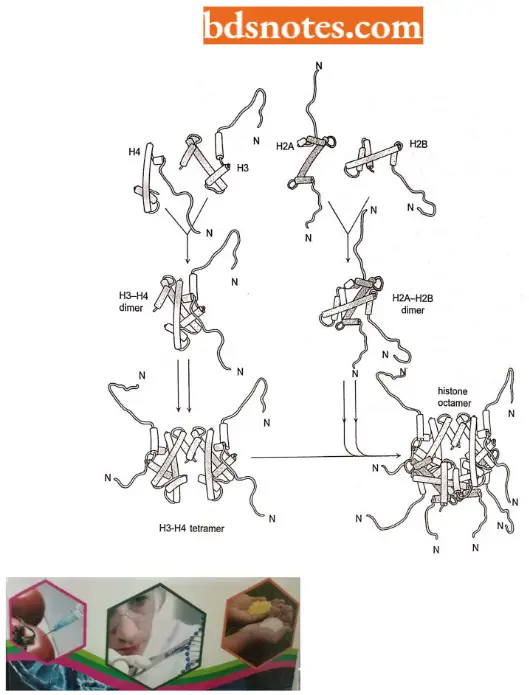
- Each nucleosome core particle consists of an octamer or complex of eight histone proteins – two molecules each of histones H2A, H2B, H3, and H4 and double-stranded DNA (called core DNA) that is 146 nucleotide pairs long.
- The histone octamer forms a protein core (spool according to Tamarin, 2002) around which the double-stranded DNA is bound.

“Why is early learning of genome organization critical for molecular biology? Answered”
- Each nucleosome core particle is separated next by a region of linker DNA, which can vary in length from a few nucleotide pairs up to about 80 (the term nucleosome technically refers to a nucleosome core particle plus one of its adjacent DNA linker, but it is often used synonymously with nucleosome core particle;
- On average, therefore, nucleosome repeats at intervals of about 200 nucleotide pairs. For example, a diploid human cell with 6.4 × 109 nucleotide pairs contains approximately 30 million nucleosomes.
- The formation of nucleosomes converts a DNA molecule into a chromatin thread about one-third of its initial length, and this provides the first level of DNA packing.
- Ultrastructure of nucleosome core particle.
- The high-resolution structure of a nucleosome core particle, solved in 1997 by K.Luger et al., revealed a disc-shaped histone core around which the DNA has tightly wrapped 1.65 turns in a left-handed coil.
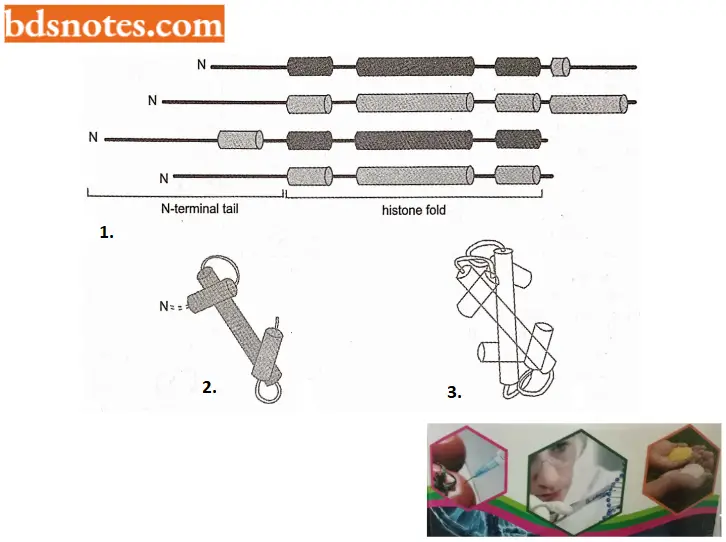
- All four of the histones that make up the core of the nucleosome are relatively small proteins and they share a structural motif, called histone fold, formed from three helices connected by two loops.
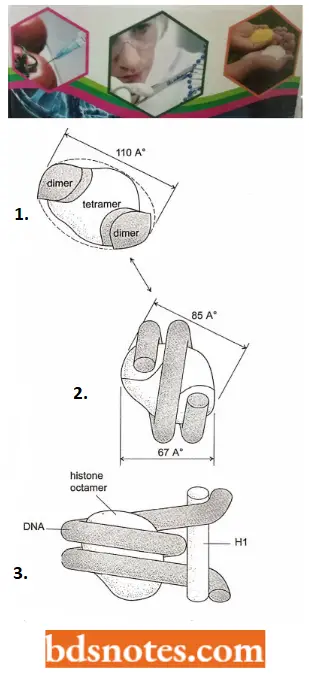
- In assembling a nucleosome, histone folds first bind to each other to form H3 – H4 and H2A – H2B dimers, and the H3 – H4 dimers combine to form tetramers.
- An H3 – H4 tetramer then f further combines with two H2A – y H2B dimers to form the compact octamer core, around which the DNA is wound

“Factors influencing success with eukaryotic genome knowledge: Q&A”
Bonding between histone core and DNA. About 142 hydrogen bonds are formed between DNA and the histone core in each nucleosome. Nearly half of these bonds form between the amino acid backbone of the histones and the phosphodiester backbone of DNA. Numerous hydrophobic interactions and salt linkages also hold DNA and protein together in the nucleosome.
- For example, all the core histones are rich in lysine and arginine (two amino acids with basic side chains), and their positive charges can effectively neutralize the negatively charged DNA backbone.
- These numerous interactions explain in part why DNA of virtually any sequence can be bound on a histone octamer core.
- The path of the DNA around the histone core is not smooth; rather, several kinks (or bends) are seen in the DNA; this is expected from the rough (or nonuniform) surface of the histone core.
“Steps to explain the structure of eukaryotic genome: Chromosomes vs genes: Q&A guide”
- Further, each of the core histones has a long N-terminal amino acid “tail” that extends out from the DNA – histone core. These histone tails are subject to several different types of covalent modifications, which control many aspects of chromatin structure.
- Histone variants in sea urchin. Despite the high evolutionary conservation of the core histones, many eukaryotic organisms also produce specialized variants of core histones that differ in amino acid sequences from the main ones.
- For example, the sea urchin has five histone H2A variants, each of which is expressed at a different time during development.
- It is believed that nucleosomes that have incorporated these variant histones differ in stability from regular nucleosomes, and they may be particularly well suited for the high rates of DNA transcription and DNA replication that occur during these early stages of development.
- Bending of DNA in a nucleosome. In principle, nearly every DNA sequence can be folded into nucleoporins, even then the spacing of nucleosomes in the cell can be irregular.
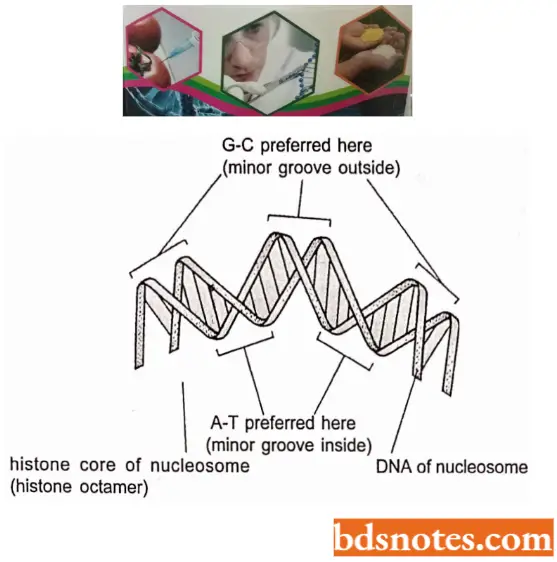
- The following two main influences determine where nucleosomes form in the DNA: One is the difficulty of bending the DNA double helix into two tight turns around the outside of the histone octamer, a process that requires firm compression of the minor groove of the DNA helix.
- Because the A – T rich sequences in the minor groove are easier to compress than G – C rich sequences; each histone octamer tends to “position itself on the DNA to maximize A – T rich minor grooves on the inside of the DNA coil.
- Thus, a segment of DNA that contains short A – T rich sequences spaced by an integral number of DNA turns is easier to bend around the nucleosome than a segment of DNA lacking this feature. The presence of certain other tightly bound proteins in the DNA also influences the position of nucleosomes on DNA molecules.
“Role of non-coding DNA in eukaryotic genome organization: Questions answered”
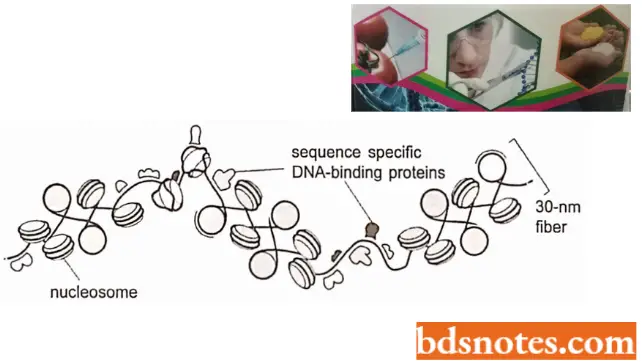
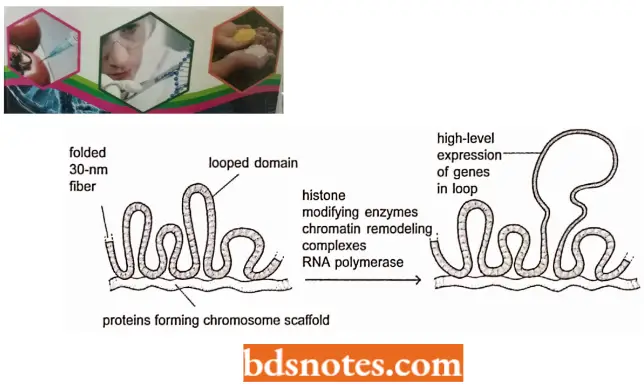
- Packaging of nucleosomes into compact chromatin fiber. The nucleosomes are packed on top of one another generating regular arrays in which the DNA is even more highly condensed.
- Thus, when nuclei are very gently lysed onto an electron microscope grid, most of the chromatin is seen to be in the form of a fiber with a diameter of about 30 nm, which is considerably wider than chromatin in the “beads on a string” form.
- The 30 nm chromatin fiber is found to have a zig-zag structure. The 30 nm structure found in chromosomes is probably a fluid mosaic of the different zig-zag variations. Differences, in linker lengths probably introduce further local perturbations into the zig-zag structure.
- Finally, the presence of other DNA-binding proteins and DNA sequences that are difficult to fold into nucleosome punctuate the 30 nm fiber with irregular features. Several mechanisms probably act together to form the 30 nm fiber from a linear string of nucleosomes.
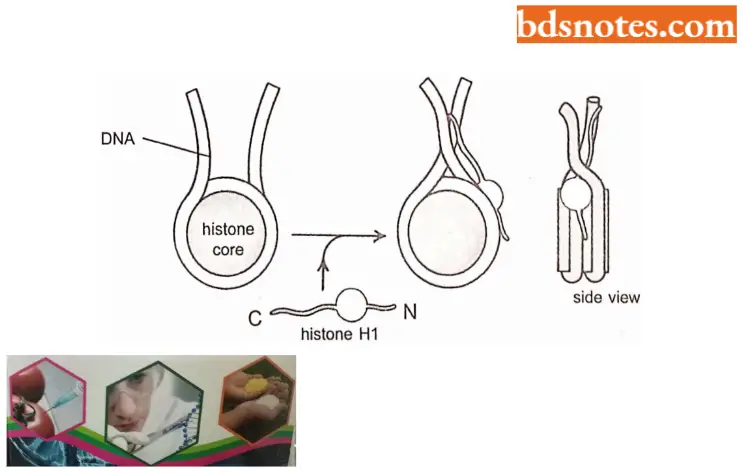
- An additional histone, called histone HI, is involved in this process. HI is larger than the core histones and is considerably less well-conserved. The cells of most eukaryotic organisms make several histone HI proteins of related but quite distinct amino acid sequences.
A single histone HI molecule binds to each nucleosome, contacting both DNA and protein and changing the path of the DNA as it exits from the nucleosome. Histone HI causes a change in the exit path in DNA which is crucial for compacting nucleosomal DNA and ultimately it interlocks to form the 30 nm fiber.
- The second mechanism for forming the 30 nm fiber probably involves the highly flexible tails of the core histones, which, we saw above, extend from the nucleosome.
- It is thought that these tails may help attach one nucleosome to another – thereby allowing a string of them, with the aid of histone HI to condense into the 30 nm fiber.
- This model has been supported by X-ray diffraction analyses and evidence that the histone tails interact with DNA.
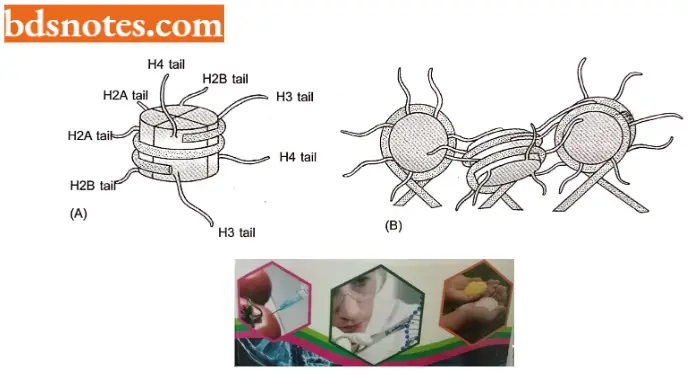
- Roles of histone tails in gene regulation. The N-terminal tails of each of the four core histones are highly conserved in their sequence and perform crucial functions in regulating chromatin structure.
- Each tail is subject to several types of covalent modifications, including acetylation of lysines, methylation of lysines, and phosphorylation of serines synthesized in the cytosol and then assembled into nucleosomes.
“How do histones and nucleosomes organize DNA? FAQ explained”
- Some of the modifications of histone tails occur just after their synthesis but before their assembly.
- For us, those modifications are significant, however, and take place once the nucleosome has been assembled.
- These nucleosome modifications are added and removed by enzymes that reside in the nucleus; for example, acetyl groups are added to the histone tails by histone acetyltransferases (HATs) and taken off by histone deacetylase (HDACs).
- The various modifications of the histone tails have many important outcomes. Although modifications of the tail have little direct effect on the stability of an individual nucleosome, they seem to affect the stability of the 30 nm chromatin fiber and the higher-order structures discussed below.
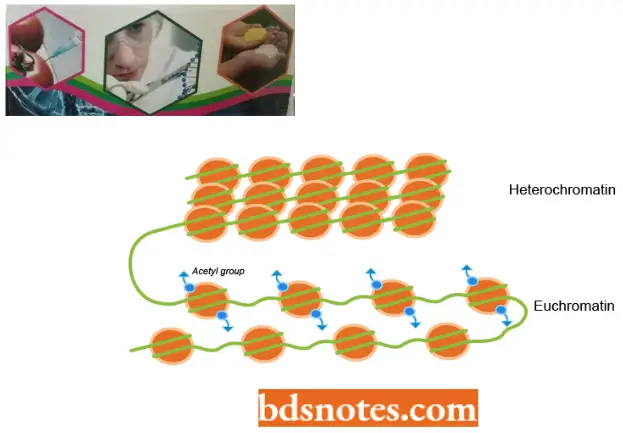
For example, histone acetylation tends to destabilize the chromatin structure, perhaps in part because adding an acetyl group removes the positive charge from the lysine, thereby making it more difficult for histones to neutralize the charges on DNA as chromatin is compacted.
- However, the most acute effect of modified histone tails is their ability to attract specific proteins to a stretch of chromatin that has been appropriately modified.
- Depending on the precise tail modifications, these additional proteins can either cause further compaction of the chromatin or facilitate access to the DNA.
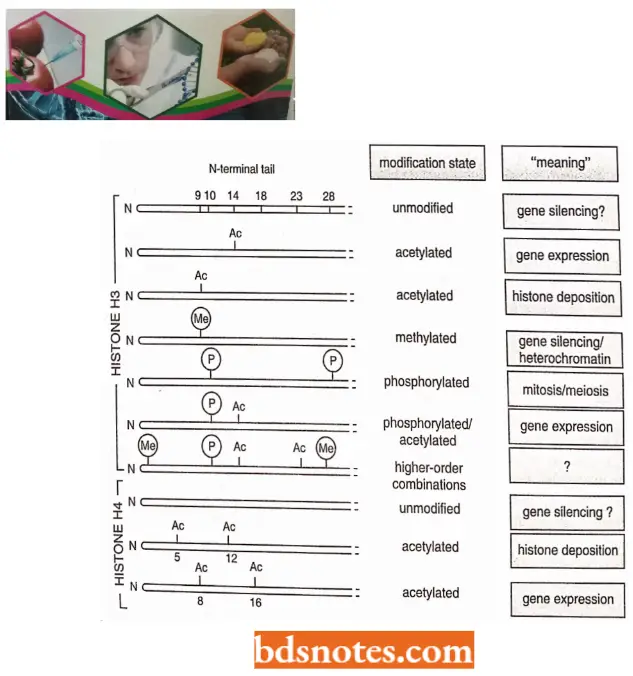
- Histone code hypothesis. This hypothesis was proposed by C.D. Allis and coworkers in 2000 and 2001. According to this hypothesis, histone tails can be marked by different combinations of modifications. Each such marking conveys a specific meaning to the stretch of chromatin on which it occurs.
- Only a few of the meanings of the modifications are known. For example, a doubly acetylated H4 tail is “read” by a protein required for gene expression. H3 tail methylated at lysine 9 is recognized by a set of proteins that creates an especially compact form of chromatin, which silences gene expression.
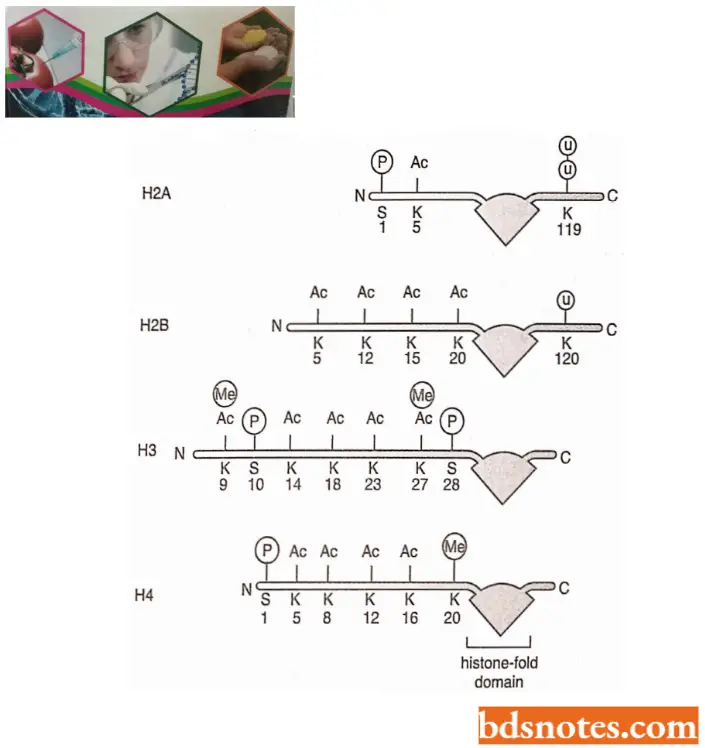
- Likewise, the acetylation of lysine 14 of histone H3 and lysine 8 and 16 of histone H4 – usually associated with gene expression – is performed by the type A histone acetylases (HATs) in the nucleus.
- In contrast, the acetylation of lysins 5 and 12 of histone H4 and lysine of histone H3 takes place in the cytosol, after the histones have been synthesized but before they have been incorporated into nucleosomes; these modifications are catalyzed by the type B histone acetylases.
- These modified histones are deposited onto DNA after DNA replication, and their acetyl groups are taken off shortly afterward by histone deacetylases (HDACs). Thus, the acetylation at these positions signals newly replicated chromatin.
- Further, modification of a particular position in a histone tail can take on different meanings depending on other features of the local chromatin structure.
- For example, the phosphorylation of position 10 of histone H3 is associated not only with the condensation of chromosomes that takes place in mitosis and meiosis but also with the expression of certain genes.
- Lastly, some histone tail modifications are interdependent. For example, methylation of H3 position 9 blocks the phosphorylation of H3 position 10 and vice versa.
Behavior of Nucleosomes During Chromosome Duplication: When DNA is replicated, twice as many nucleosomes are needed since one double helix becomes two.
- Recent studies indicate that a parental nucleosome is partly disassembled during DNA replication and reassembled on one or the other daughter strand, apparently randomly.
- The other DNA strand has a new nucleosome constructed of proteins called chromatin assembly factors which are of the following three types: In fruit flies, a protein complex, called the replication-coupling assembly factor assembles new nucleosomes.
- In addition, a protein complex, called condensin is needed for the condensation of interphase chromosomes to mitotic chromosomes. This complex includes two SMC proteins (for structural maintenance of chromosomes) and two non-SMC proteins.
- SMC proteins also help other chromosomal activities, such as mitotic segregation, sister-chromatid adhesion, dosage compensation, and recombination. Thus, a diverse array of proteins is involved in creating nucleosomes condensing interphase chromosomes, and performing many activities of the chromosome.
- Dosage compensation has recently been associated with a change in nucleosome structure. The activated X chromosome (sex chromatin or Barr body) appears to have a different type of histone present. Histone H2A is replaced by a variant, called mH2A.
Chromatin Remodeling: Nucleosomes play a major role in controlling gene expression; DNA with nucleosomes has a much lower rate of transcription than DNA without nucleosomes. It makes sense that the positions of nucleosomes can provide or prevent access to promoters.
“Early warning signs of gaps in understanding genome organization basics: Common questions”
- There are regions of the DNA, known as nuclease hypersensitive sites, that appear to be nucleosome-free. These sites, usually multiples of a region of about 200 base pairs, are particularly sensitive to digestion by different nuclease enzymes.
- When these regions are isolated they usually have sequences that control functions in replication, transcription, and other activities of DNA. For example, numerous promoter regions in Drosophila, mouse, and human DNA are in nuclease hypersensitive sites.
- Hence, some specific DNA sequences are kept free of nucleosomes. and obese sequences appear to be recognized by various enzymes such as RNA polymerase. In many other cases, however, nucleosomes do appear to cover promoters and repress transcription- For transcription to occur in these cases, some form of chromatin remodeling must take place.
- Two general classes of proteins are involved in chromatin remodeling. First is proteins mat acetylate the N-terminal tails of the histones, a process that may cause the nucleosomes to bind less tightly to the core and thus make it available for attachment of transcription factors. These enzymes are called histone acetyltransferases (HATs).
- Deacetylaling enzymes have the reverse effect. They act to repress transcription. Second, a class of ATP-dependent proteins such as the SWI SNF complex in yeast also affect chromatin remodeling.
- The SWI/SNF complex is a group of eleven proteins involved in transcription activation in many genes, presumably allowing transcription factors to access the promoters by remodeling chromatin.
- These proteins can reposition a nucleosome on DNA by sliding the nucleosome down the DNA.
From the above findings, we can conclude that although nucleosomes serve as a general first-order packing mechanism in eukaryotic DNA, they can be positioned precisely and can diminish transcription. - However, once transcription begins, the RNA polymerase enzyme moves along nucleosome DNA by translocation of the histone by 75 to 80 base pairs without disrupting the nucleosome itself (Studitsky et al., 1994). This seems to be accomplished by the RNA polymerase moving the DNA and then reforming the nucleosome in its wake.
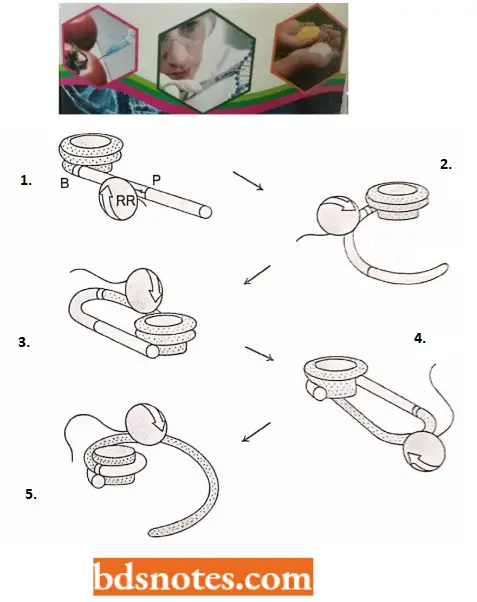
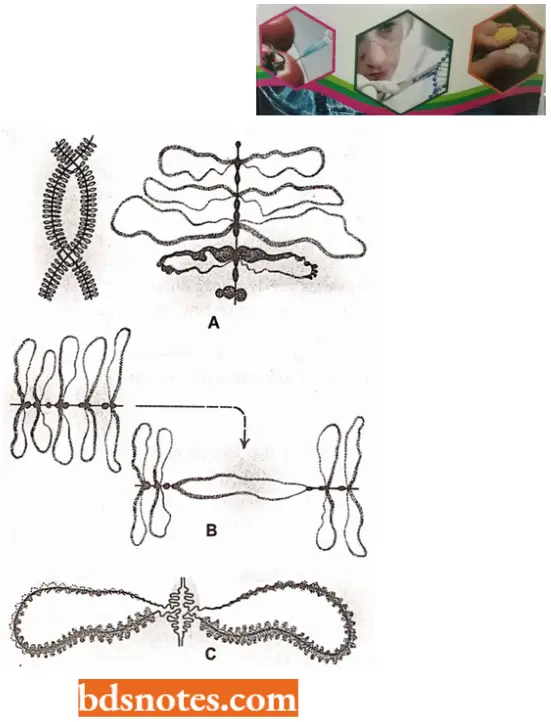
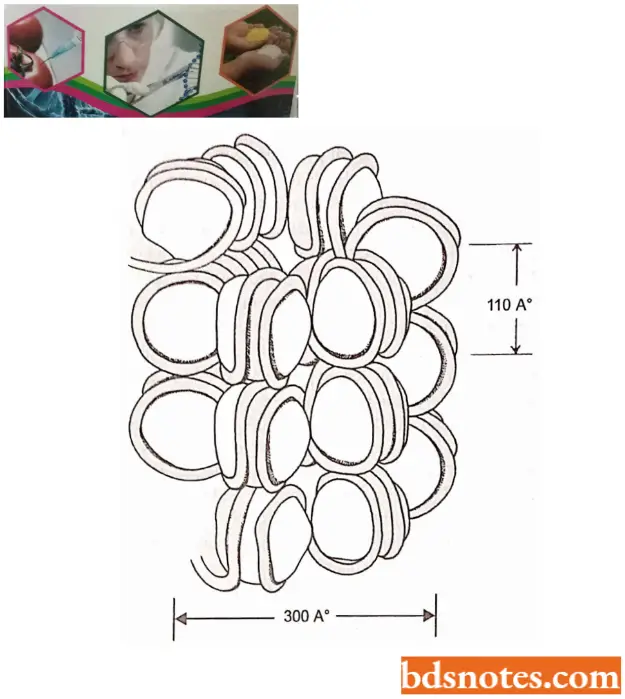
Higher Order Structure of Chromatin: Since the nucleosome has a width of only 110A° and metaphase chromosomes appear to be constructed of a fiber having a diameter of about 2400 A°, several additional levels of chromatin compaction lead to the metaphase chromosome.
“Asymptomatic vs symptomatic effects of ignoring genome organization principles: Q&A”
- Various experiments, which change the ionic strength the chromatin is subject to, indicate that the 110 A° DNA spontaneously forms a 300 A°, solenoidlike fiber with increased ionic strength.
- It seems that this fiber results from the coiling of the nucleosomal DNA. This 300 A° (30nm) fiber is not, however, the final form of the DNA.
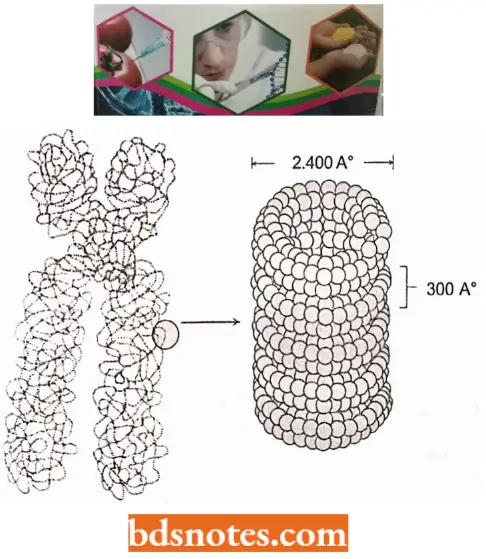
- We can account for the contraction of the 300 A° fiber to the 2,400 A° fiber found in metaphase chromosomes by the formation of a second solenoid-like structure from the winding of the 300 A° fiber.
- When the histones are removed from a chromosome, the DNA billows out. leaving a proteinaceous structure termed a scaffold.
- The scaffold is involved in condensing the 300 A° chromatin fiber into the tightly packed metaphase chromosome.
- The scaffold structure is formed from nonhistone proteins; two of them predominate, namely SCI and SC2. SCI has been identified as topoisomerase II. Several hundred different proteins, many in minute quantities, are believed to be involved in replication, repair, and transcription.

“Can targeted interventions improve outcomes using genome organization knowledge? FAQs provided”
Chromosome Banding
Various chromosomal staining techniques reveal consistent banding patterns. Using these patterns, all of the human chromosomes can be distinguished. Of possibly greater importance is the fact that these staining techniques have provided some insight into the structure of the chromosome.
- G-bands. These bands are obtained with Gicmsa stain, a complex of stain specific for the phosphate groups of DNA. Treatment of fixed chromatin with trypsin or hot salts brings out the G -bands. Giemsa stain enhances banding, which is already visible in mitotic chromosomes.
- The banding pattern is caused by the arrangement of chromosomes. Under careful observation, the major G-bands prove to consist of many smaller chromosomes.
- G-banding appearance has led Comings (1978) to suggest the mechanism of chromosome folding.
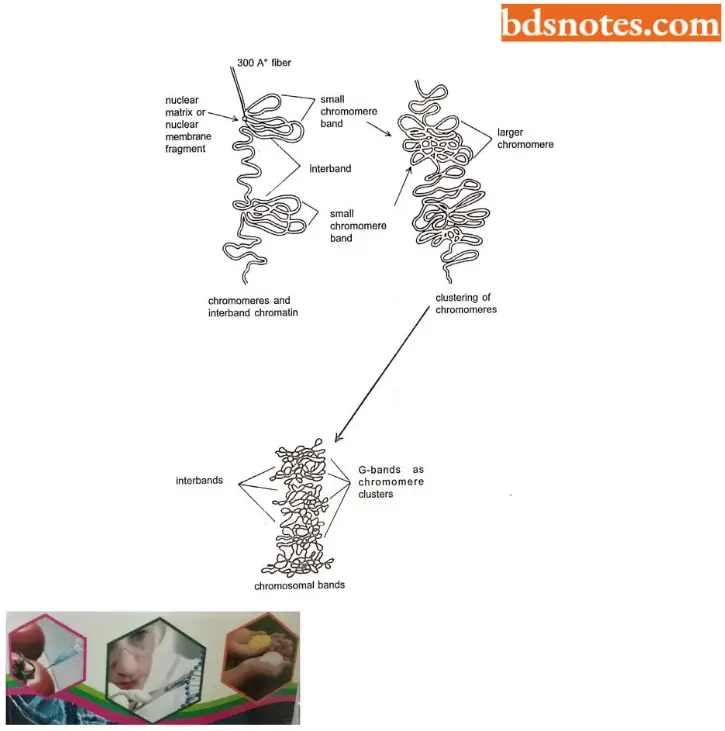
- C-bands. These are giemsa-stained bands after the chromosomes are treated with NaOH. The C is for “centromere” because these bands represent constitutive heterochromatin surrounding the centromers. The DNA is also usually satellite-rich.
- Satellite DNA. This type of DNA differs in buoyant density from the major portion of cellular DNA. When eukaryotic DNA is isolated and centrifuged in CsCl, forming a density gradient, the majority of the DNA forms one band in the gradient at a single buoyant density. The buoyancy is determined by the G – C content of the DNA.
- However, smaller secondary bands are also usually present, indicating regions of DNA having sequences different from the majority of the cell’s DNA.
- DNA isolated in this way is referred to as satellite DNA because of the secondary, or satellite, bands formed in the density gradient.
- In a chromosome, the satellite DNA is found primarily around the centromeres and consists of numerous repetitions of a short sequence.
- R- bands. These bands are visible with a technique that stains the regions between G-bands. The chromosomes are fixed, stained with Giemsa, and observed under the phase contrast microscope.
- Since the dark-light pattern is the opposite of the G-band pattern, these bands are called reverse bands.
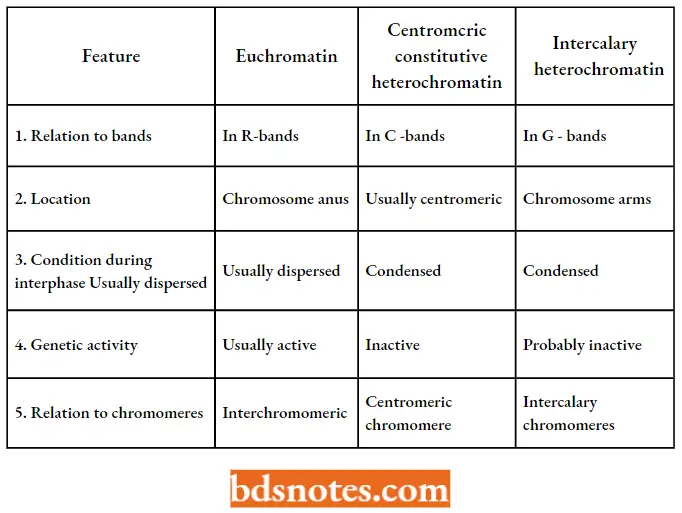
- Based on the information obtained from these staining techniques, Comings (1978), distinguished between three basic chromatin types: euchromatin, constitutive heterochromatin, and intercalary heterochromatin.
- The only chromatin involved in transcription is euchromatin. Constitutive heterochromatin surrounds the centromere and is rich in satellite DNA.
- Intercalary heterochromatin is found throughout the chromosome. Thus, it becomes apparent that the eukaryotic chromosome is a relatively complex structure.
The Three Major Types Of Chromatin In Eukaryotic Chromosome:
“Differential applications of coding vs non-coding regions: Questions answered”
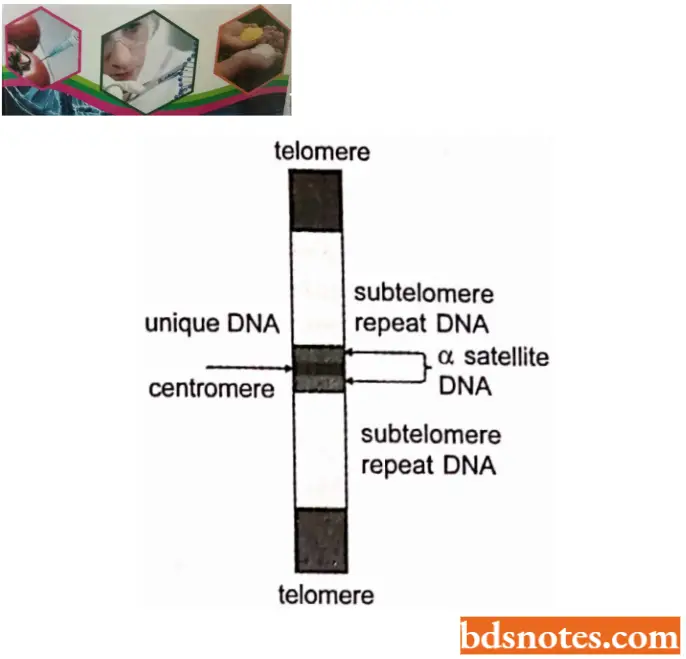
DNA Of Centromere
The centromere is a specific region of the eukaryotic chromosome. It is involved in the movement of the chromosome during mitosis and meiosis.
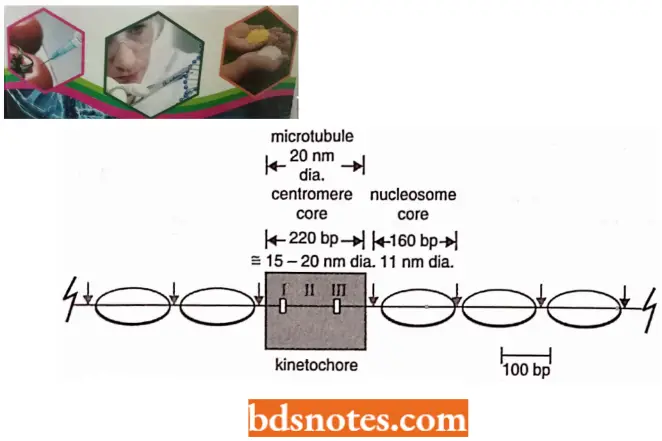
- As we know, the terms centromere and kinetochore, while occasionally used interchangeably, are distinct. The kinetochore is the interface between the visible constriction in the chromosome (the centromere) and the microtubules of the spindle.
- The kinetochore of higher organisms (for example, mammals) contains protein and some RNA. Microscopically it is a trilaminar structure, attached to chromatin at the inner layer and to microtubules at the outer layer.
- Most of the genetical and molecular biological information of centromere has been obtained by studies in the fungus, yeast (Saccharomyces cerevisiae), and the plant, Arabidopsis thaliana. Cells did not maintain most artificially created yeast plasmids because they were lost during mitosis.
- However, some plasmids were isolated that did replicate normally during cell division. Presumably, they contained centromeres, allowing them to replicate and move in synchrony with the host’s chromosomes. Genetic engineering also made it possible to isolate smaller and smaller regions that could serve as centromeres.
- For example, after sequencing the centromeres of fifteen or sixteen yeast chromosomes, it was possible to conclude that the centromere from yeast is about 250 base pairs long with three consensus regions.
- In the light of current molecular biological data, the centromere can be defined as a sequence of DNA, called the C EN locus or C E N region. This region may contain a single modified nucleosome associated with region 2.
- The 250 base-pair length of the CEN regions of yeast chromosomes is about 200 A°, the same as the diameter of a microtubule, indicating that only one microtubule attaches to each centromere during mitosis or meiosis in a yeast cell. This region is called a point centromere.

Higher eukaryotes have large centromeric regions each of which attaches more microtubules, for example., 4 to 7 in rat fetus and 70 to 150 in Plant Haemanthus. These regions are referred to as regional centromeres. Regional centromeres range from 19 to 100 kb, with unique and satellite (repeated sequence). DNA is heterochromatic and may include expressed genes.
- Arabidopsis centromeres span 0.9 – 1.2 Mb of DNA (Box 3.1) and each one is made up largely of 180bp repeat sequences. In humans, the equivalent sequences are 171 bp and are called alphoid DNA.
- DNA of centromere of Arabidopsis contains multiple copies of genome-wide repeats, along with a few genes, the latter at a density of 7 – 9 per 100 kb compared with 25 genes per 100 bp for the non-centromeric regions of Arabidopsis chromosomes (Copenhaver et al, 1999).
- The discovery that centromere DNA contains genes was a big surprise because it was thought that these regions were genetically inactive.
Unit of length for DNA molecules
Because DNA is double-stranded, the lengths of molecules are described as so many base pairs (bp). A kilobase pair (kb) is 103bp and a megabase pair (Mb) is 106bp. A Giga base pair (GP) is 109 MB. In summary:
1Kb = 1000bp
1Mb = 1000kb = 1000 000 bp
1Gb = 1000mb = 1000 000 bp
The length of RNA molecules cannot be expressed in bp because most RNAs are single-stranded: their lengths are described as so many nucleotides.
- The special centromeric proteins in humans include at least seven that are not found elsewhere in the chromosome. One of these proteins, CENP-A is very similar to histone H3 and is thought to replace this histone in the centromeric nucleosomes.
- Part of the human kinetochore is made up of alphoid DNA plus CENP A and other proteins, but its structure has not been described in detail.
Dna Of Telomeres
It is already known that eukaryotic chromosomes are linear, each has two ends, referred to as telomeres. The telomeres not only mark the termination of the linear chromosome but also have several specific functions.
- For example, telomeres must prevent the chromosomal ends from acting in a “sticky” fashion the way the broken chromosomal ends act.
- In other words, chromosomal ends must also prevent the ends of chromosomes from being degraded by exonucleases and must allow chromosomal ends to be properly replicated.
- Most telomeres isolated so far are repetitions of sequences of five to eight bases. In human beings, the telomeric sequence is T T A G G G, repeated 300 to 5000 times at the end of each chromosome.
- The human telomere was discovered by Robert K. Moyzis and his colleagues (1988) when they probed the highly repetitive segment of human DNA.
- When a probe for this sequence was applied to human chromosomes, the sequence was found at the tip of each chromosome in roughly the same quantity. This is a highly conserved sequence, found in all vertebrates studied as well as in unicellular trypanosomes.
- Similar sequences are found in various other eukaryotes the first sequence was isolated by E.BIackburn and J.Gall in 1978.
Telomeric Sequences In Eukaryotes; The G-Rich Strand Of The Double Helix Is Shown:

“Difference between prokaryotic and eukaryotic genome organization: Q&A explained”
- When a linear DNA molecule is replicated, the 3’ – 5’ strand can be replicated up to the end. The 5’ – 3’ strand, however, is replicated with DNA primers that are then degraded, leaving a short gap on the progeny strand.
- It is always the G-rich strand of telomeric DNA that ends up single-stranded, forming a 3’ overhang of twelve to sixteen nucleotides. Thus, the normal replication process of a linear DNA molecule leaves an incomplete terminus.
- Hence, molecular biologists suspected that there would be a unique mechanism for the replication of telomeres.
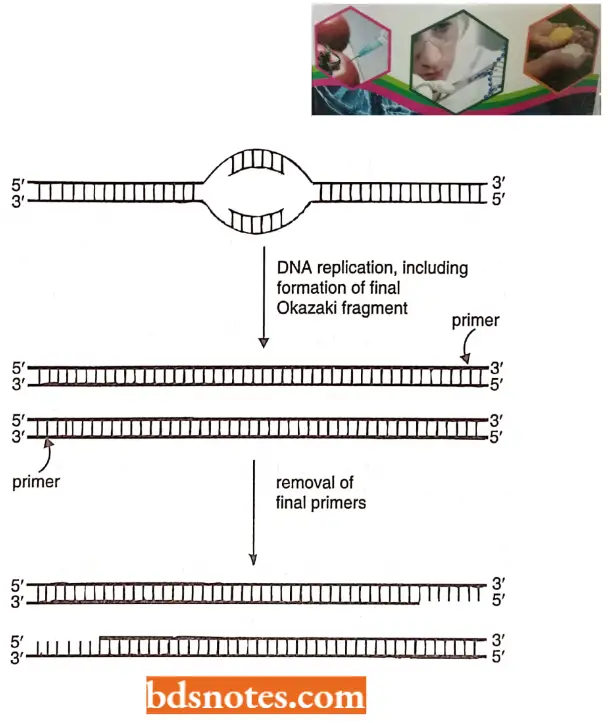
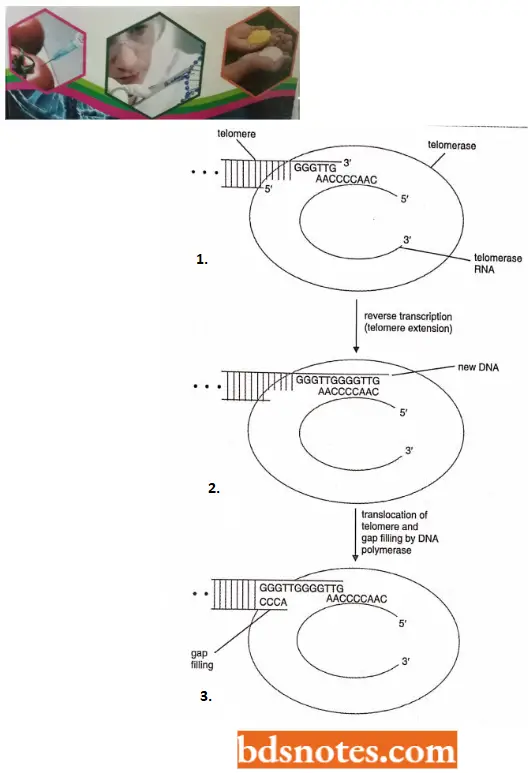
The Telomerase Enzyme: Telomeric sequences appear to be added de novo without DNA template assistance by an enzyme called telomerase. This enzyme was discovered by Blackburn and her colleagues (1990).
- This was seen when telomeres from another species were engineered into yeast cells. After a cell cycle, the yeast telomeric sequence had been added on at the ends of the foreign chromosome, the result presumably of the telomerase enzyme.
- Further, when Blackburn and her colleagues (1992) isolated telomerase, they discovered that a segment of RNA, about 160 nucleotides in length is an integral part of the enzyme. Telomerase RNA has a region that is complementary to the G-rich repeat of the telomeric DNA sequence of the species.
- After careful experimentation, including modifying the gene for the telomerase RNA, Blackburn and her colleagues found that the telomerase enzyme uses its
- RNA as a template for adding telomeric repeats to the ends of chromosomes. Telomerase enzyme is thus a reverse transcriptase, using RNA nucleotides as a template to polymerize DNA nucleotides.
- Telomerase is an unusual enzyme in that it consists of both protein and RNA. In the human enzyme, the RNA component is 450 nucleotides in length and contains near its 5’ end the sequence 5’-CUAACCCUAAC-3’, whose central region is the reverse complement of the human telomere repeat sequence 5’-TTAGGG-3’.
- This enables telomerase to extend the telomeric DNA at the 3’ end of a polynucleotide by the copying, mechanism, in which the telomerase RNA is used as a template for each extension step, the DNA synthesis being carried out by the protein component of the enzyme which is a reverse transcriptase.
Mode of working of telomerase. The first step in, telomerase extension is hybridization of the 3’ end of the telomere with the RNA component of telomerase. Then, with the telomerase RNA as a template, the 3’ end of the telomere is extended.
- Finally, a translocation step takes place that displaces the telomere concerning the RNA, returning to the configuration at the beginning of the process. The single-stranded C-rich strand is then synthesized with DNA polymerase and DNA ligase.
Protection of telomeres. Once the telomeres have been added to the ends of eukaryotic chromosomes, different organisms use any of the three methods known to protect the ends of the chromosomes.
- The guanine-rich DNA can form complex structures. Biochemists have discovered that four guanines can form a planar G-tetraplex, with the four bases hydrogen bonded to each other (Wang and Patel, 1993).
- Proteins have been discovered that bind to the 3’ ends of telomeres. In the ciliate, Oxytricha nova, a protein called the telomerase end binding protein (TEBP) attaches to the 3’ ends of telomeres and protects them (Horvath, 1998).
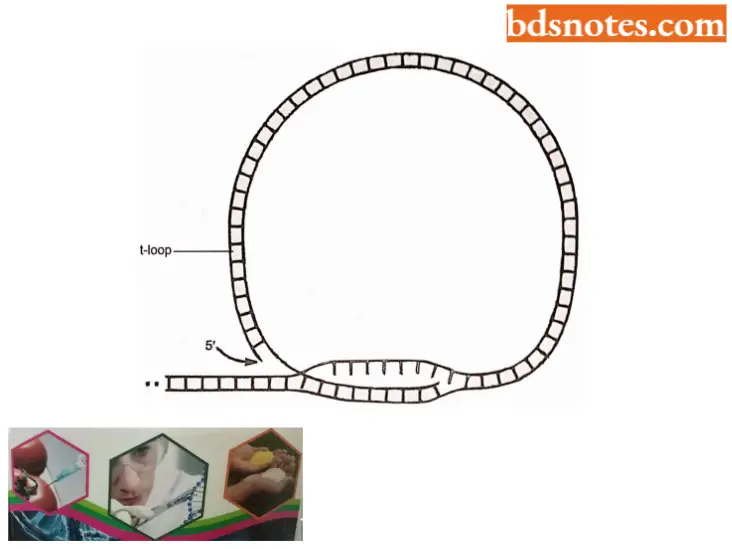
- A novel structure called the t-loop has been discovered at the ends of mammalian telomeres (Griffith et al., 1999).
- The loop forms at the end of chromosomes under the direction of a protein called TRF2 (Telomere Repeat Binding Factor) which causes the 3’ end of the chromosome to loop around and interdigitate into the double helix, forming the loop. A t-loop from a mouse liver cell is about 10,000 bases around (Griffith et al., 1999).
“Most common complications of poorly understood genome organization concepts: FAQs”
- Some telomeric proteins are thought to form a linkage between the telomere and the periphery of the nucleus, the area in which the chromosome ends is localized (Tham and Zakian, 2000).
- How do cells keep track of the number of their telomeric repeats? Proteins have been isolated that bind to telomeres (Rapl in yeast Saccharomyces cerevisiae, Marcand et al., 1997; TRF1 in human beings. Van Steensel et. al., 1997).
- By mutating these proteins or the telomeric sequences, molecular biologists have changed the equilibrium number of telomeric repeats.
- This led to the current model that the cell counts the number of these proteins bound to the telomeres, not the number of telomeres directly, to know whether telomeres should be added.
Telomerase and Aging: In yeast, protozoa, and other single-celled organisms, the telomerase enzyme is active, keeping the ends of the chromosomes at the appropriate length.
- These cells can divide potentially forever. In contrast, in higher organisms, telomere shortening leads to the termination of a cell lineage. For several years biologists have attempted to link this process with cell senescence, a phenomenon originally observed in cell cultures.
- All normal cell cultures have a limited lifetime: after a certain number of divisions, the cells enter a senescent state in which they remain alive but cannot divide. With some mammalian cell lines notably fibroblast cultures (connective tissue cells), senescence can be delayed by engineering the cells so that they synthesize active telomerase (Rcddel 1998).
- In mammals active telomerase is found essential in germ line cells (see KJug and Cummings, 2003). These experiments suggest a clear relationship between telomere shortening and senescence but the exactness of the link has been questioned (Blackburn, 2000).
Telomerase and Cancer: Not all cell lines display senescence. Cancerous cells can divide continuously in culture, their immortality being looked upon as analogous to tumor growth in an intact organism with several types of cancer.
- The absence of senescence is associated with the activation of telomerase, sometimes to the extent that telomere length is maintained through multiple cell divisions, but often in such a way that the telomerase is overactive.
- Human telomerase was isolated from an immortal cell line (HeLa) derived from cervical cancer cells.
- This current information may be exploited for clinical purposes: if telomerase can be deactivated in tumor cells, the cells may stop dividing or die, thereby eliminating the cancer.
- One type of human cancer, dyskeratosis congenital, appears to result from a mutation in the gene specifying the RNA component of human telomerase.
The C-Value Paradox
Why do eukaryotes have so much DNA, and why is there huge variation in the DNA content between species of comparable complexity? These questions define the C-value paradox, in which C refers to the quantity of DNA in a cell.
- For an example of the paradox, although human beings have 3.3 billion base pairs in (the haploid genome, an amoeba has more than 200 billion base pairs.
- Likewise, although an average bony fish has over 300 billion base pairs of DNA in its haploid genome, the Japanese putter fish has less than half a billion base pairs.
- If the basic bony fish pattern can be created with less than half a billion base pairs, why does the average bony fish have over 600 times that much DNA? what is this excess DNA doing? To explain the C-value paradox, researchers examined the repetitiveness of DNA, and more recently probed and sequenced DNA to understand its properties.
Unique DNA and Repetitive DNA: DNA-DNA hybridization is the process of taking DNA from the same or different sources and heating and then cooling it, causing double helices to reform at homologous regions. This technique is useful for determining sequence similarities and degrees of repetitiveness among DNAs.
- R.Britten and Kohnc (1968), using the technique of DNA-DNA hybridization, first systematically analyzed the repetitiveness of the DNA within eukaryotes. When DNA is heated, it cools, it renatures. The rate of renaturation depends on the DNA sequences.
- If the sample contains DNA with repeated sequences, it will hybridize faster than DNA that does not have repeated sequences. From these studies, Britten and Kohnc (1968) found that eukaryotic chromosomes contain regions of unique, moderately repetitive, and highly repetitive DNA.
- Unique DNA is, as its name implies, DNA with unrelated sequences. Repetitive DNA is DNA whose sequences are repeated in the genome. During the last 39 years, hundreds of plants and animals have been analyzed for the relative proportion of repetitive DNA in their genome.
- Satellite DNA, found around centromeres is highly repetitive DNA with a unique repeat length of about two hundred base pairs. Unique DNA makes up most of the transcribed genes of an organism.
Relative Proportions (Percent Of Haploid Genome) Of Repetitive Sequences:
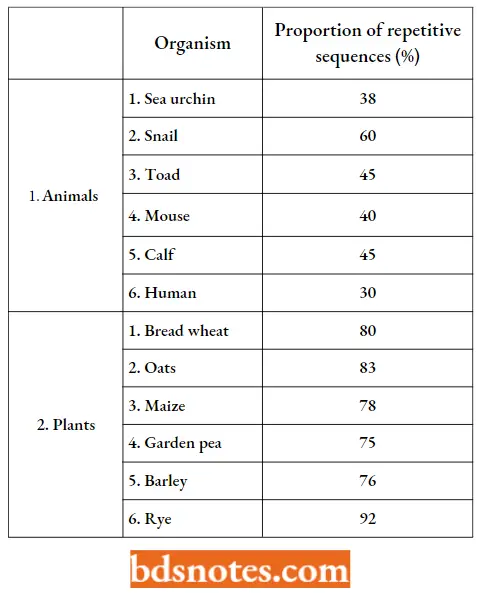
“Why are genome organization mechanisms often misunderstood in practice? Questions answered”
- The rest of the DNA is repetitive DNA in a few to several hundred thousand copies. This repetitive DNA comprises three categories. One is “junk” DNA, DNA that is not useful to the organism, made up of untranscribed and parasitic sequences (selfish DNA).
- Another category is transcribed genes in many copies that have diverged from each other, such as antibody, collagen, and globin genes.
- The term diver gene family has been given to the genes that have arisen by duplication, with or without divergence, from an ancestral gene.
- Finally, the transcribed genes in many copies that are virtually identical such as ribosomal RNA genes and histone genes, make up a third category of repetitive DNA.
Junk DNA: Transposons in prokaryotes are generally viewed as selfish or parasitic: they serve no purpose to the cell. The transposons replicate on their own, increasing in number.
- Eukaryotic transposons are mostly retrotransposons, i.e., transposable elements that move by way of an RNA intermediate. In other words, the retrotransposon is transcribed into RNA and then, by reverse transcription, converted to a cDNA and then inserted into the genome.
- These elements can make up 50 percent of the eukaryotic genome, existing in hundreds of thousands of copies. They generally fall into two categories: LINES and SINES. The term ‘junk DNA’ is falling out of favor of molecular biologists.
- This is partly because the number of surprises resulting from genome research over the last few years has meant that molecular biologists have become less confident in asserting that any part of the genome is unimportant simply because we do not currently know what its function might be.
- One clear thing is that the bulk of the intergenic DNA is made up of repeated sequences of one type or another.
LINES or Long Interspersed Elements. They contain up to seven thousand pairs each and contain genes for reverse transcription, RNA binding, and endonuclease activity. They thus have the ability to jump by way of an RNA intermediate. Human DNA is believed to be composed of about 15 percent LINES.
SINES or Short Interspersed Elements. They are generally derivatives of transfer RNA (tRNA) genes and cannot retrotranspose on their own. That is, in the past, their transcripts were modified, converted to cDNA by reverse transcription, and then reinserted into the host’s genome. They depend on the reverse transcriptase enzyme provided by the genes of LINES or retroviruses.
- One group of SINES not derived from tRNA is derived from RNA of the signal recognition particle; members of this group occur in human beings in about five hundred thousand copies of a three hundred base pair sequence.
- Because these sequences are cleaved by the restriction endonuclease Alu, they are called the Alu family. The human genome is also pervaded by remnants of at least a dozen distinct families of ancient retroviruses scattered throughout our chromosomes.
- At this juncture, some explanation can be forwarded for the C-value paradox. Much eukaryotic DNA is junk. Not harm. In some cases, 97% of the host genome is composed of junk DNA.
- Recent work has explained that gross differences in DNA content between higher organisms may be due to the differing abilities of different species to rid themselves of this parasitic DNA.
- If it builds up without being removed, the DNA content of the species can rise. Thus, wide differences in DNA content among higher eukaryotes mentioned at the beginning of this section have little to do with the complexity of the organism, but rather with the ability of the organism to remove the junk DNA as it forms.
Split Gehes
Split genes mean that the (DNA) sequences containing actual information about the gene (called exons) are interrupted by other sequences (called introns) which are spliced out after transcription.
Discovery of Split Genes: With the discovery of DNA as the genetic material, a gene was regarded as a continuous segment of DNA.
- In 1964, it was also proved that there was co-linearity between the sequence of nucleotides in DNA and the sequence of amino acids in the corresponding protein. Therefore, there was absolutely no doubt about the continuity of nucleotides in a gene represented by a DNA segment. This was proved to be true both for prokaryotes and eukaryotes.
- However, a big surprise came in 1977, when geneticists came to know that in some mammals, birds, and amphibians, a gene may not be represented by a continuous sequence of nucleotides but may be interrupted by some intervening sequences which are not represented in mRNA transcribed from the gene and utilized for the synthesis of proteins.
- Such genes with intervening sequences were called split genes or interrupted genes. The discovery of split genes was made in 1977 by various groups of biologists in a variety of materials:
- Two research groups separately headed by Philip A. Sharp and Richard J. Roberts studied genes of adenovirus
- Research groups of P. Chambon, P.Leader, and R.A. Flavell studied b globin genes, ovalbumin genes, and tRNA genes. In all these cases the genes were found to be interrupted by intervening sequences.
- The credit for the discovery of split genes, however, goes to Philip Sharp and Richard Roberts, who won in 1993 Nobel for Medicine or Physiology for their independent work on split genes.
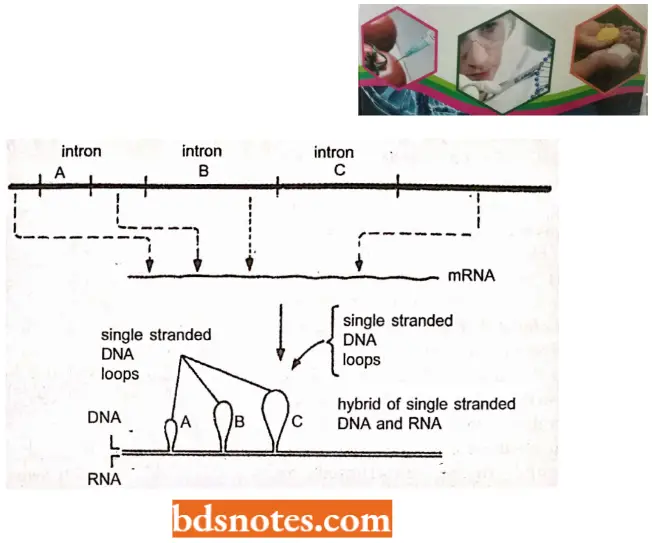
- They analyzed the hybrids of the late mRNA of adenovirus 2 with the adenovirus genomic DNA. When these mRNA-DNA hybrids were examined under an electron microscope, the adjoining sequences of mRNA were found to be hybridized with discontinuous stretches of genomic DNA of adenovirus.
- The intervening DNA sequences were observed as loops and the phenomenon was later described as R-looping.
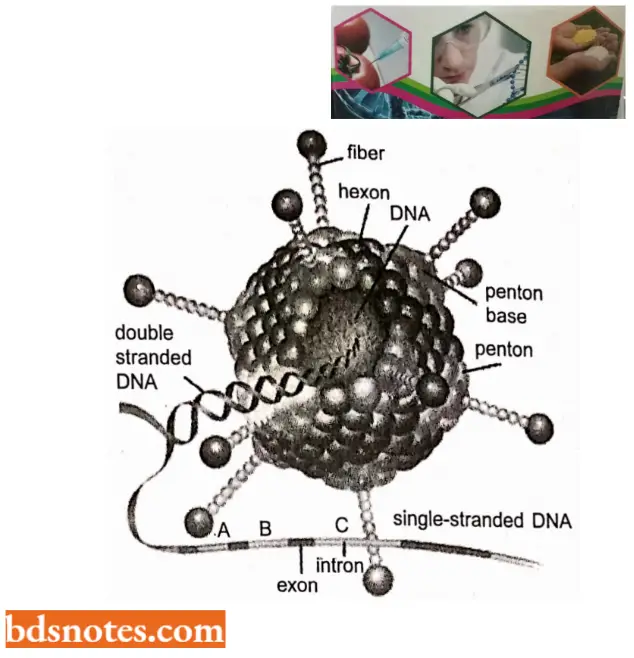
“Cost of ignoring genome organization principles vs benefits of systematic approaches: Q&A”
- Chambon’s group compiled sequences of the boundaries of introns from a large number of protein-coding eukaryotic genes (not ribosomal RNA or tRNA genes), which revealed the presence of consensus sequences at the intron-exon junctions.
- Of these GT (guanine-thymine) was always found at the 5’ side of the intron (left splice junction) and AG (adenine-guanine) at the 3’ side (right splice junction).
- This became popularly known as the GT-AG rule or Chambon’s rule
- Rarely, AT-AC occurs at intron-exon junctions). Some of the diseases (for example, thalassemia) are caused by mutations, which created or abolished these splice junctions.
Methods of Investigations of Split Genes: A detailed study was conducted on the ovalbumin gene found in chickens.
- This ovalbumin gene is responsible for the synthesis of a protein, called ovalbumin consisting of 386 amino acids and synthesized only by highly specialized tubular gland cells of the oviduct at the time when the hen is laying eggs.
- The expression of this ovalbumin gene is controlled by some female sex hormones.
- Chambon and his colleagues synthesized an artificial ovalbumin gene to study its regulation. Such an artificial gene of ovalbumin could be synthesized using ovalbumin mRNA which could give rise to cDNA (complementary DNA) with the help of enzyme reverse transcriptase.
“Is genome organization-related risk reversible if addressed promptly? Answer provided”
- This cDNA was inserted into a plasmid and cloned in E. coli for its multiplication. When this cDNA was compared with corresponding genomic DNA, it was discovered (through DNA hybridization) that the genomic DNA had additional intervening sequences.
- Another ingenious technique used for the study of split genes was the use of restriction enzymes which have the property of cleaving DNA at unique sites. More than 100 such restriction endonuclease enzymes are now available.
- Given this, restriction endonucleases could be used to find out the presence or absence of a certain sequence in a particular gene. For example, when £coRl and Hind III enzymes were used with cDNA for ovalbumin, it was found that no cleavage occurred suggesting that the sequences of six base pairs each recognized by these two restriction enzymes were absent.
- Based on this, it was expected that if the DNA extracted from the oviduct was cleaved by utilizing these two enzymes, the ovalbumin gene would not be broken and the cleavage would occur at other places thus making it possible to isolate the ovalbumin gene from the living cells.
- This DNA segment representing the ovalbumin gene was expected to be separated with the help of hybridization with cDNA artificially synthesized.
- When such hybridization was done with cDNA hybridized with different fragments of DNA rather than with a single fragment.
- Hybridization between the single-stranded DNA having the gene for ovalbumin and its mRNA also showed the formation of distinct loops at specific sites as observed in the electron microscope. Such DNA that exists in the loops is missing in mRNA. This kind of conclusion eventually led to the discovery of split genes in 1977.
- In subsequent years, it could be proved that split genes are present at least in two more cases, i.e., the gene for (3-gIobin (a component of hemoglobin molecule) in rabbit and mouse and immunoglobin gene (antibody gene).
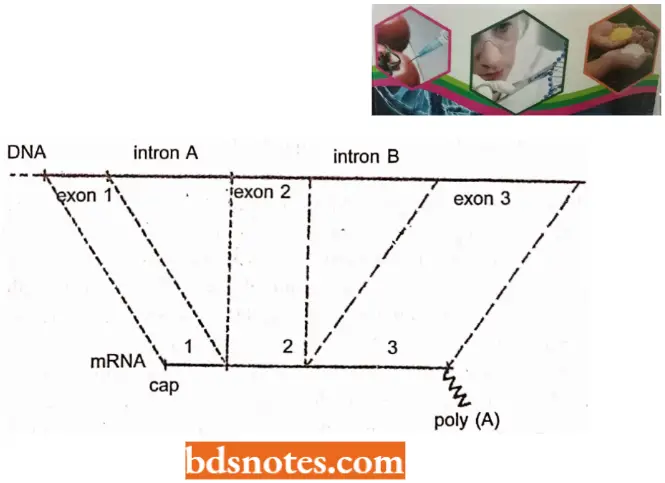
- Later on, split genes were reported to occur in higher organisms as a common phenomenon. To understand the structure of a split gene, we can consider an example of a hypothetical split gene that has a DNA sequence including three pieces, called exons, exon 2, and exon 3..
- These three exons are separated by two long intervening sequences, called intron A and intron B. The terms exon and intron were used by Walter Gilbert (1985) for the first time and have been followed ever since.
- DNA, the ultimate product, i.e., mRNA had only those sequences that correspond to exons, the sequences representing introns being absent. In subsequent years, it is discovered that both exons and introns are first transcribed and this primary transcript is then modified.
- The sequences corresponding to introns are removed from the transcript and the sequences corresponding to exons are joined together, in the correct order to give rise to mRNA.
- A generalization has been made that the order of exons on DNA is the same as the order in which they are found in the processed mRNA.
Examples of split genes or interrupted genes occur in a variety of organisms:
- Nuclear genes for proteins;
- Nuclear genes for rRNAs;
- Nuclear genes for tRNAs;
- Mitochondrial genes in yeast;
- Chloroplast genes in a wide variety of plants;
- Genes in archaebacteria and
- Genes in bacteriophages of E.coli. They were initially believed to be absent in eubacterial genomes but in the 1990s introns were discovered even in eubacteria.
Certain Examples of Split Genes
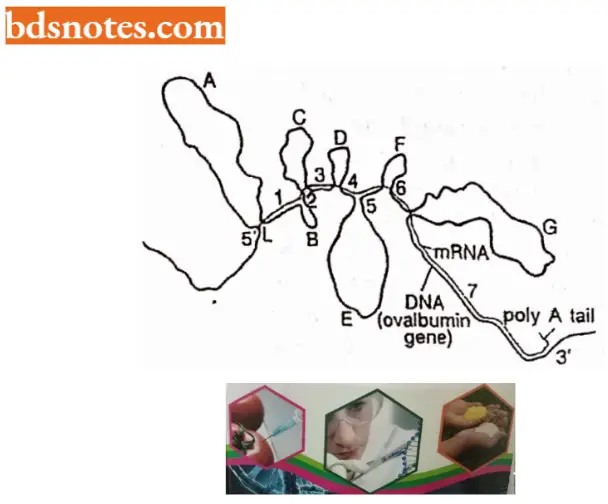
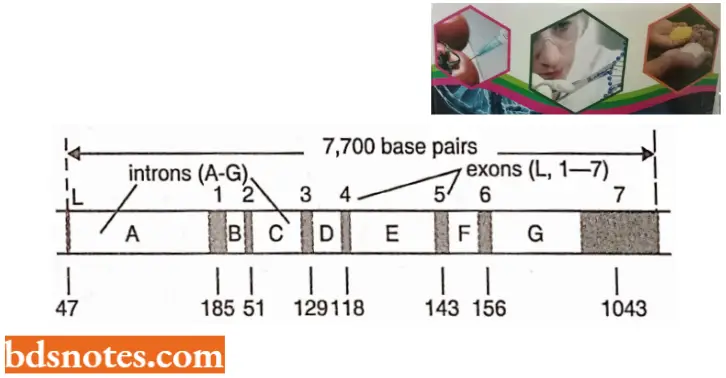
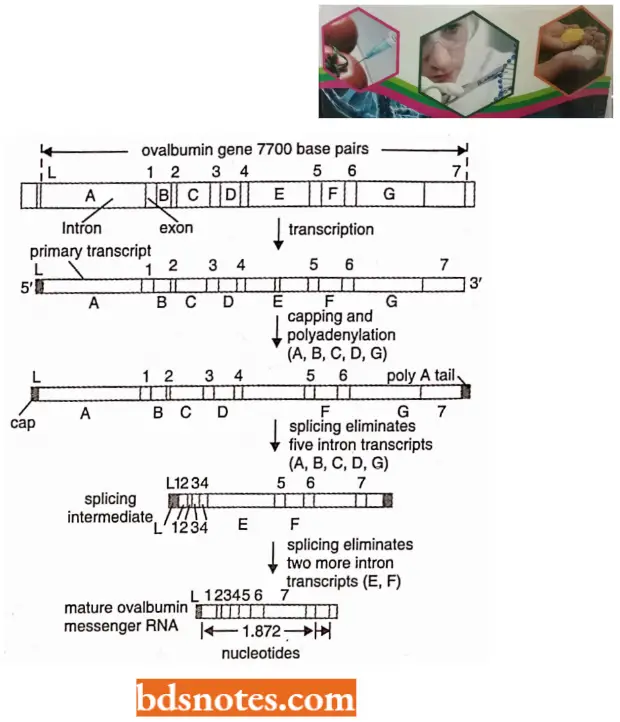
- Structure of ovalbumin split gene of hen (chicken). In the split gene of ovalbumin, there are eight exons and seven introns. The size of the ovalbumin gene is 7,700 base pairs long although the mature mRNA for ovalbumin is only 1,872 nucleotides.
- The entire ovalbumin gene with its 7700 base pairs is first transcribed to a precursor RNA to which a cap consisting of 7 mG is added at the 5’ end and poly-A tail is added at the 3’ end. After the addition of the cap and tail, the introns are excised in the first step and the remaining two in the second step.
- The exons produced due to splicing are then joined with the help of enzyme ligase to produce the mature mRNA of the ovalbumin gene.
- Split genes in humans. In T the human gene for the cystic fibrosis transmembrane regulator, the introns are much longer than the exons. This gene is 250 kb long and is split into 24 exons and 23 introns.
- The average length of the exons is 227 bp, so all the exons added together make up only 2.4% of the gene. These exons are scattered throughout the entire length of the gene, separated by introns that range in size from 2 to 35 kb.
- Split genes in fungal mitochondria. Split genes are also found in the mitochondria. Introns of these split genes in fungal mitochondria are of two types:
- Group 1 introns which are found in a majority of the fungal mitochondria split genes, do not carry any conserved sequence, called internal guide sequence. By internal pairing, this guide sequence brings the two intron-exon junctions together and helps in splicing out the introns.
- These group I introns are also found in the nuclear gene coding for rRNA in Tetrahymena (a ciliate) and Physantm(a slime mold). Features similar to those of group I introns are also reported from introns of phage T4 genes,
- Group 2 introns resemble nuclear genes and have consensus sequences (GT and APy) and a branch sequence that resembles the TACTAAC box. These introns are excised as lariats.
- Split Genes in Chloroplasts Split genes for ribosomal RNA (rRNA), transfer RNAs (tRNA), and some proteins have also been reported in the chloroplast genomes of several plants including Chlamydomonas and Nicotiana. Introns found in chloroplast genes can be classified into three groups based on intron boundary sequences,
- Group 1 introns(eg., in trnL) can be folded in a secondary structure similar to the self-splicing rRNA precursor of Tetrahymena. These can be removed either by self-splicing or by a “maturase” enzyme(as in cytochrome b and cytochrome oxidase mRNA precursors),
- Group 2 introns (for example, majority of genes including tmA and trial) can be folded into a complex secondary structure (as in introns of mitochondrial genes for cytochrome oxidase in maize and yeast),
- Group 3 introns (for example, tmG, tmK, trnV, rpl2, rpsl2, rpsl6, etc.)have conserved sequences at their borders (GTGCGNY at 5’ end, and ATCNRYY(N)YYAY at 3’ end), similar to those in the eukaryotic nuclear genes (R = purine; Y = pyrimidine; N = any nucleotide).
- Although introns are generally absent in protein-coding genes of bacteria, archaebacteria, and some lower eukaryotes, class II type of self-splicing introns have recently been reported even in bacteria. Such a distribution of introns has been used for a study of the role of introns in the evolution of genes or for a study of the origin of genes or for a study of the origin of introns themselves.
Evolutionary Origin of Split Genes: The following three hypotheses/theories have been proposed to explain the origin of split genes:
- Exon (domain) shuffling hypothesis;
- Exon theory; and
- Intron early hypothesis.
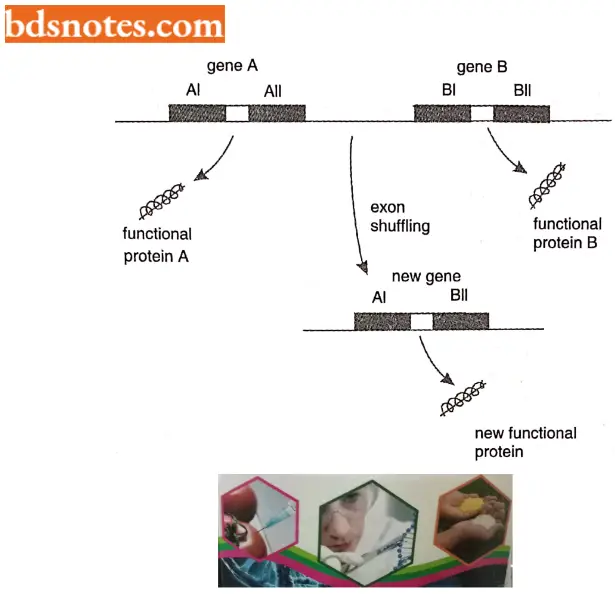
The exon shuffling hypothesis is an interesting idea that holds that each exon of a split gene contains a different subcomponent of the biological information carried by the gene as a whole.
- Individual subcomponents are not sufficient on their own to code for a complete protein. Still, the information they contain is meaningful in that it specifies a recognizable portion of the protein function, for example, enabling the protein to bind to a particular substrate or attach to a specific site in the cell.
- The hypothesis is that during evolution, exons from different discontinuous genes can be “shuffled”, creating new combinations of biological information. Exon shuffling would be more likely to produce new functional proteins than would be entirely random rearrangement of existing genes.
- Domain or exon shuffling is illustrated by tissue plasminogen activator (TPA), a protein in the blood of vertebrates that is involved in the blood clotting response. The TPA gene has four exons, each coding for a different structural domain.
“Success rate of interventions using modern genome organization techniques: FAQ”
The upstream exon codes for a ‘finger’ module that enables the TPA protein to bind to fibrin, a fibrous protein found in blood clots that activates TPA. This exon appears to be derived from a second fibrin-binding protein, fibronectin and is absent from the gene for a related protein, urokinase, which is not activated by fibrin.
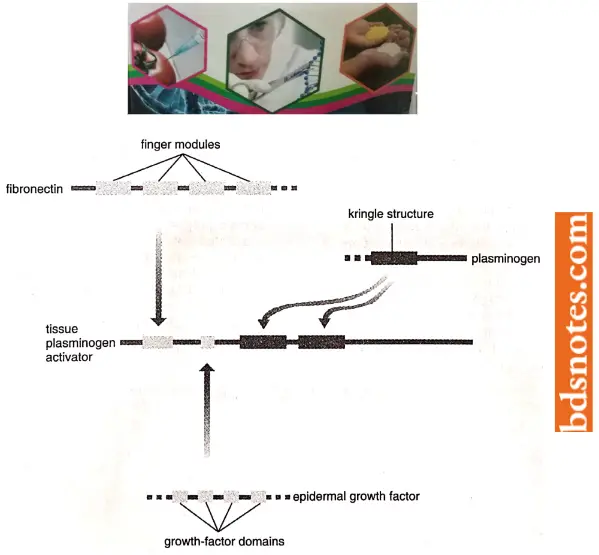
The second TPA exon specifies a growth-factor domain that has been obtained from the gene for epidermal growth factor and which may enable TPA to stimulate cell proliferation. The last two exons code for “Kringle” structures which TPA uses to bind to fibrin clots; these Kringle exons come from the plasminogen gene (Li and Graur, 1991).
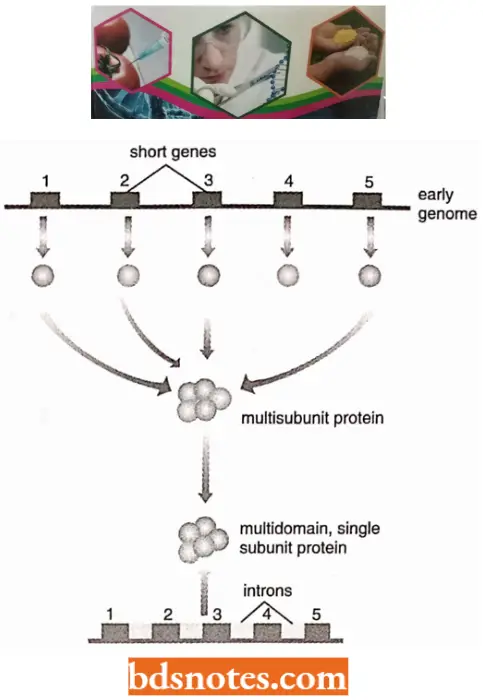
- According to the “early-intron hypothesis” (also called the exon theory of genes by W.Gilbert in 1987), ancestral exons existed as independent genetic units of microgreens and their association with intron helped the generation of diversity in genes through trans-splicing and exon shuffling.
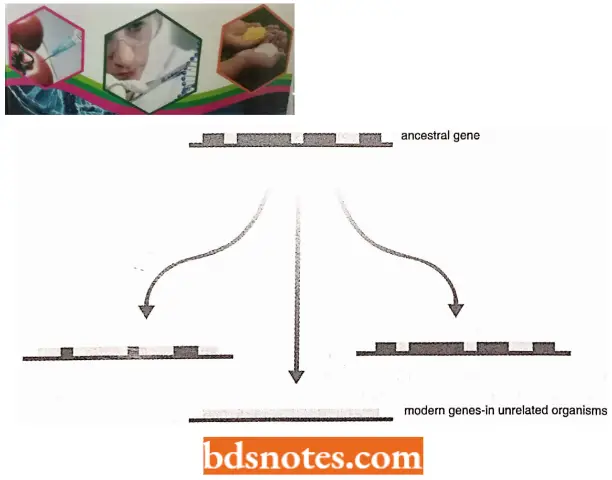
- There are examples, where new genes could be generated by exon shuffling through intron recombination events, According to the ‘late intron hypothesis’ (also called the “insertional theory of intron origin”) evolution of the introns was independent of the evolution of protein-coding genes.
- It is assumed in this hypothesis that the self-splicing introns (class I introns) invaded the genome of eukaryotic ancestors, as endosymbionts, and revolved into mitochondria and chloroplasts containing class 2 introns.
- It is postulated that invasion took place at the mRNA level, which was followed by reverse transcription and recombination with DNA, producing split genes.
- These introns gave other types of introns later. This explains, why introns are absent in bacteria, archaebacteria, and lower eukaryotes. The recent report of introns in bacteria may be the initial stage of this process of evolution of introns.
Overlapping Genes
Bacteriophage ΦX174 contains a single-stranded DNA of approximately 5,400 nucleotides in length. The complete sequence of these 5.400 nucleotides was worked out by Fredrick Sanger and his colleagues.
- The genome of the ΦX174 phage consists of nine cistrons. Amino acid sequences of major proteins coded by the genome of ΦX174 phage have also been worked out and even their molecular weights are known.
- From the information about protein codes, an estimate could be made of the number of nucleotides required. This estimate of several nucleotides exceeds 6,000 which is much higher than the actual number of nucleotides present, i.e., 5,400.
- Thus, it was difficult to explain how these proteins could be synthesized from a DNA segment that is not long enough to code for the required number of amino acids.
- When a detailed study of the system was done then it was discovered that sequences in the same segment could be utilized by two different cistrons coding for different proteins.
- Such overlapping of genes (cistrons) will be theoretically possible if the two cistrons have two functions at different times and their nucleotide sequences are translated in two different reading frames.
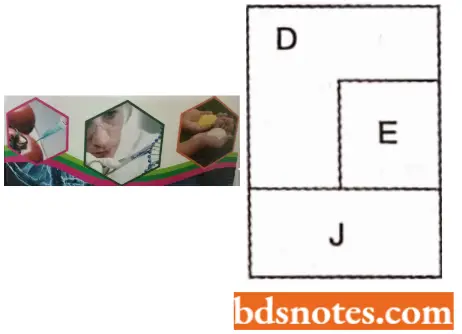
- In 1976 Barrell and his coworkers discovered that in ΦX174 phage which has nine cistrons (A, B, C, D, E, J, F, G H), cistron E is present between D and J and that the cistron E overlaps cistron D. This could be done by working out the nucleotide sequences of DNA segment representing gene D, E and J.
- It could be shown that amber (codon for chain termination) mutations in cistron E lie within cistron D and these amber mutations do not influence the translation of cistron D into its protein.
- Similarly, some nonsense mutations for cistron D lie in cistron E suggesting that cistrons D and E overlap in the DNA sequences and that the cistrons D and E are translated in two different reading frames so that amber codon in mRNA of one cistron will be read as termination of the other cistron.
- Among genes D, E, and J in ΦXl 74 phage, cistron D codes for a simple protein, cistron £ codes for a protein for lysis, and cistron J codes for a particle component.
- It was shown that enough space is not available where these three genes are located, thus, conforming that there was overlapping between D and E cistrons.
- Overlapping has also been suggested to be present in regions of genes A and B.
- Overlapping sequences (=genes) have also been discovered in tryptophan mRNA of E.coli and many other cases. Given wide.
- Overlapping of occurrence of overlapping genes, it seems that the phenomenon of overlapping genes is an economic device to make better use of genetic information in less DNA.
Pseudogenes
In multicellular organisms, a wide variety of DNA sequences are found, which are of no apparent use to the organism. Some of these sequences are defective copies of functional genes and are therefore called pseudogenes.
- Such pseudogenes are found to occur in human beings, mice (for example, globin pseudogenes), Drosophila (for example., histone pseudogenes), etc.
- For example, human α-globin and a-globin pseudogenes, are found in each of the two globin gene clusters.
- The complete nucleotide sequence of alpha globin pseudogene is now known. It has been also reported that both of these genes (α-globin and a-globin) are non-translatable, since they may have mutations in initiation codons and also frame-shift mutations along their length.
Cryptic Genes
Cryptic genes are phenotypically, silent DNA sequences that are not expressed during the normal life cycle of an organism.
- They may be activated in a few individuals of a population, due to events such as mutation, recombination, transposition, and other genetic mechanisms due to specific altered environmental conditions and this leads to their detection.
- For example, genes for α-glucoside utilization in E. coli and Salmonella. Four cryptic genetic systems (bgl, cel, ase, arbT) involved in the a -glucoside utilization have been characterized in E. coli.
Organization Of Genetic Material Questions And Answers
Question 1. What evidence is used to determine the length of DNA associated with a nucleosome? What is a nuclease-hypersensitive site? What functions are associated with these sites?
Answer:
The length of DNA associated with nucleosomes was determined by footprinting, in which free DNA was digested, leaving only those segments protected by nucleosomes. Nucleosome hypersensitive sites are sites not in a nucleosomal state; they seem to be sites involved in initiation, transcription, and other DNA activities.
Question 2. What is a telomere? What are its functions? What is its structure?
Answer:
- Telomeres are repetitive DNA sequences at the chromosomal end. They are repetitions of five or eight-base sequences. Most telomeres are G-rich.
- Telomeres protect the ends of chromosomes and probably provide signals on the senescence of cells.
Question 3. What functions exist in unique, repetitive, and highly repetitive DNAs?
Answer:
- Highly repetitive DNA usually makes up the centromeric and telomeric regions of the chromoUnique DNA, Making up the bulk of structural genes, has a large component that is transcribed.
- Repetitive DNA is composed of dispersed DNA (for example, short and long interspersed elements – SINES and LINES), multiple copies of transcribed DNA (for example., ribosomal RNA, histones), and diverged copies of ancestral genes (for example., globin family genes).
Question 4. What is the C-value paradox, and how is it explained?
Answer:
The C-value paradox involves the issues of the excessive amounts of DNA in eukaryotic species that seem to have similar complexity. It is explained by the large amount of structural DNA in chromosomes as well as the large amounts of short and long interspersed elements(Sines And Lines).
Question 5. What is satellite DNA? What does it signify?
Answer:
Satellite DNA differs in its base sequence from the main quantity of DNA and thus forms a satellite band during buoyant density analysis. It is usually centromeric heterochromatin, composed of a highly repetitive DNA.
Question 6. If chromatin is digested with an endonuclease enzyme to produce two hundred base-pair fragments, and these fragments are then used for transcription experiments, very little RNA is made. Explain this observation.
Answer:
Spaces between the nucleosomes must contain many promoter sequences. For DNA to be digested, it must be unprotected. Since we see little transcription, the promoters must be missing and must have been destroyed.
Question 7. If radioactive probes are made from highly repetitive DNA, these probes hybridize in situ mainly to centromeric and telomeric regions. What does this result suggest about the organization of chromosomes?
Answer:
Highly repetitive DNA must be located in these chromosomal regions. Since most highly repetitive DNA is not transcribed, the results suggest that centromeric and telomeric regions are not transcribed.
Question 8. Why is higher-order chromosomal structure expected in eukaryotes but not in prokaryotes?
Answer:
- The simplest explanation is the difference in the amount of genetic material in prokaryotes and eukaryotes.
- Since the average human chromosome has several centimeters of DNA, that DNA must be contracted to a size in which it can be moved during mitosis and meiosis, without tangling and breaking.
- Nucleosomes form the first order of coiling of the nucleosome DNA bringing it down to a manageable size for nuclear divisional processes.
Question 9. How could comparative DNA studies aid us in understanding the roles of the different kinds of DNA present in the eukaryotic chromosomes?
Answer:
- Comparative DNA studies can help understand the roles of the various types of DNA in the eukaryotic chromosomes if there are cases in which there are substantial differences in the amount of DNA in similar species.
- It can be then inferred that the basic developmental plan of an organism is contained in the one with the lower amount of DNA, and the extra DNA in the species with more DNA may be superfluous.
- We do have cases in which amphibians differ by as much as one hundred times the amount of DNA found in similar species.
- The puffer fish has only one-sixth amount of DNA as other higher eukaryotes.
Organization Of Genetic Material Multiple Choice Question And Answer
Question 1. Chromatin consists of
- RNA
- DNA
- RNA and histones
- DNA and histones
Answer: 4. DNA and histones
Question 2. DNA is associated with highly basic proteins called
- Histones
- Non-histones
- Albumins
- All of these
Answer: 1. Histones
Question 3. A nucleosome is made of
- DNA
- Histone
- Histone wrapped over an octameric core of nucleic acid
- DNA wrapped over an octameric core of histone
Answer: 4. DNA wrapped over an octameric core of histone
Question 4. A solenoid is a structure of
- A nucleosomal organization with a long diameter.
- Condensed chromatin fiber with a 30nm diameter
- A highly condensed form of chromatid with a 300 nm diameter
- Well-organized chromosome with 1400 nm thickness
Answer: 2. Condensed chromatin fiber with 30nm diameter
Question 5. The terminal end of a chromosome is called
- Metamere
- Telomere
- Centromere
- Basal granule
Answer: 2. Telomere
Question 6. A gene that specifies the amino acid sequence of a polypeptide chain is termed as
- Structural gene
- Regulator gene
- Operator gene
- Split gene
Answer: 1. Structural gene
Question 7. The gene not expressing any protein is known as
- Epistatic gene
- Hypostatic gene
- Pseudogene
- None of these
Answer: 3. Pseudogene
Question 8. The intervening sequence of ‘gene’ is known as
- Introns
- Exons
- Cistron
- Codons
Answer: 1. Introns
Question 9. In split genes, the coding sequences are called
- Introns
- Operons
- Exons
- Cistrons
Answer: 3. Exons
Question 10. The Exon part of mRNA has codes for
- Protein
- lipid
- Carbohydrate
- Phospholipid
Answer: 1. Protein
Leave a Reply