Antihistaminic Agents
Histamine
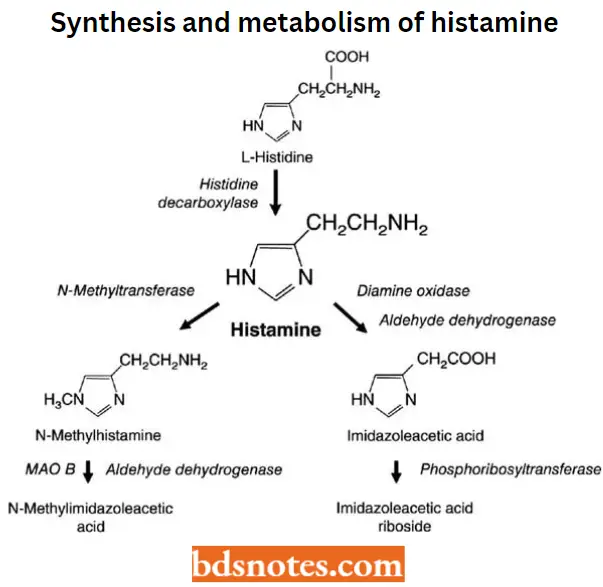
Histamine or β-imidazolyl ethylamine is synthesized from L-histidine by histidine decarboxylase, an enzyme that is expressed in many mammalian tissues including gastric mucosa parietal cells, mast cells, basophils, and the central nervous system (CMS).
- As a result, histamine plays an important role in human physiology including regulation of the cardiovascular system, smooth muscle, exocrine glands, the immune system, and central nerve function.
- It is also involved in embryonic development, the proliferation and differentiation of cells, hematopoiesis, inflammation, and wound healing.
- The involvement of histamine in the mediation of immune and hypersensitivity reactions and the regulation of gastric add secretion has led to the development of important drug classes useful in the treatment of symptoms associated with allergic and gastric hypersecretory disorders.
Biosynthesis And Metabolism Of Histamine
Histamine is synthesized in the Golgi apparatus of its principal storage cells, mast cells, and basophils.
- Histamine is formed from the naturally occurring amino acid L-histidine (S-histidine) via the catalysis of either the pyridoxal phosphate-dependent enzyme histidine decarboxylase or Laromatic amino acid decarboxylase.
- Released histamine is rapidly inactivated by metabolism via two pathways as shown. One pathway involves NT-methylation via the enzyme histamine N-methyltransferase.
- This enzyme is widely distributed in mammalian tissues and catalyzes the transfer of a methyl group from S-adenosyl-L-methionine to the ring tele-nitrogen of histamine, producing NT– – methylhistamine and S-adenosyl-L-homocysteine.
- The other pathway of catabolism involves oxidative deamination by diamine oxidase, yielding imidazole acetaldehyde, which is further oxidized to imidazole acetic acid by aldehyde dehydrogenases.
“What is medicinal chemistry of antihistamines? A detailed notes and Q&A guide”
Read and Learn More Medicinal Chemistry II Notes
Similarly, NT-methylhistamine is converted by both diamine oxidase (DAO) and monoamine oxidase (MAO), followed by ALD-DH to N-methyl imidazole acetic acid. All of these metabolites are devoid of histamine receptor agonist activity.
“Understanding antihistamine agents through FAQs: Medicinal chemistry explained”
Storage And Release Of Histamine
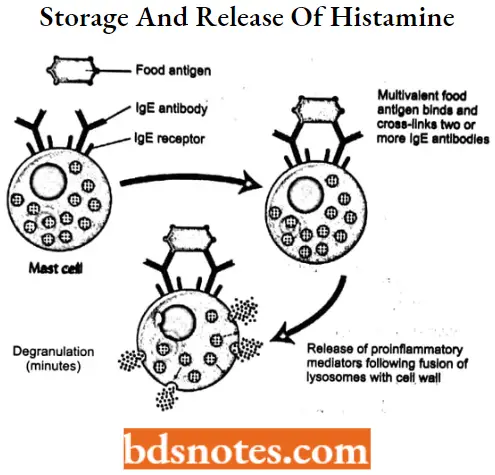
Most histamine is biosynthesized and stored as protein complexes in mast cells (complexed with heparin) and basophilic granulocytes (complexed with chondroitin).
- Protein-complexed histamine is stored in secretory granules and released by exocytosis in response to a wide variety of immune (antigen and antibody) and nonimmune (bacterial products, xenobiotics, physical effects, and cholinergic effects) stimuli.
- The release of histamine as one of the mediators of hypersensitivity reactions is initiated by the interaction of an antigen-IgE complex with the membrane of a histamine storage cell.
- This interaction triggers the activation of intracellular phosphokinase C (PKC), lending to the accumulation of inositol phosphates, diacylglycerol, and calcium.
- Exocytootic Release of histamine follows the degranulation of histamine storage cells.
- Degranulation also results in the release of other mediators of inflammation including prostaglandins, leukotrienes, platelet-activating factors, kinds, etc.
Histamine is released from mast cells in the gastric mucosa by gastrin and acetylcholine. Neurochemical studies also suggest that histamine is stored in and released from selected neuronal tracts in the CNS, and functions as a neurotransmitter.
“How do antihistamines work at the molecular level? FAQ answered”
Histamine Receptors
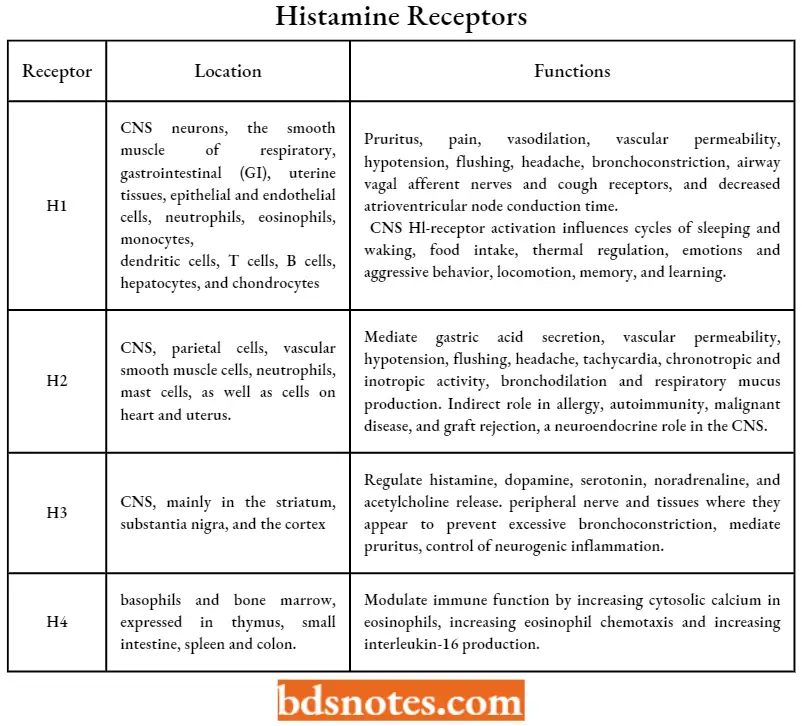
Extensive pharmacological and molecular biology studies have revealed the presence of at least four different histamine receptor subtypes in mammalian systems designated as H1, H2, H3, and H4.
- These receptors have constitutive receptor G-protein-signaling activity that is independent of histamine agonist binding.
- Thus, they exist in two conformations that- are in equilibrium—an active (constitutive) and inactive state. Tire four histamine receptor subtypes differ in their expression, location, and physiologic functions as indicated.
Histamine Receptors
“Importance of studying medicinal chemistry of antihistamines for pharmacy students: Questions explained”
Antihistaminic Agents
The term antihistamine historically has referred to drugs that block the actions of histamine at H1 receptors rather than other histamine receptor subtypes.
- These antihistamines, referred to now as the first-generation or classical antihistamines, are related structurally and include several aminoalkyl ethers, ethylenediamines, piperazines, propylamines, phenothiazines, and dibenzocydoheptenes.
- More recently, several second-generation or “nonsedating” antihistamines have been developed and introduced.
- The second-generation agents are derivatives of several first-generation agents but have been modified to be more specific in pharmacologic action and limited in their tissue distribution or accumulation profiles.
Histamine Classification
Histamine H1 antagonist
Antihistamines are generally categorized as first- and second-generation, which is generally based on whether they cause greater or lesser sedation.
SAR of Histamine H1– antagonists
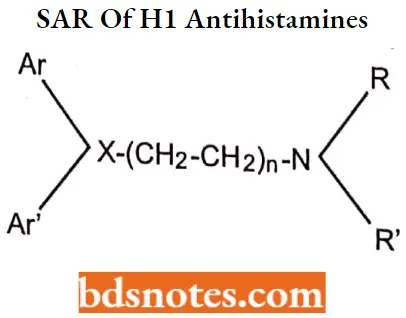
The essential pharmacophore for histamine Hi antagonistic activity is
“Common challenges in understanding antihistamine mechanisms effectively: FAQs provided”
- The nitrogen should be 3° in nature for maximum antihistaminic activity. The ‘N’ may also form a part of heterocyclic moieties like piperidine, piperazine, or diazocine. The nitrogen amine should be separated by 5-6 A° from the aromatic or heteroaromatic group.
- The group present between the nitrogen atom and group X may be saturated unsaturated or substituted.
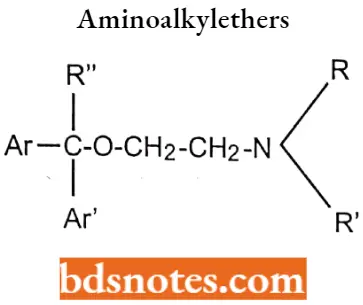
- Ar is aryl (including phenyl, substituted phenyl, and heteroaryl groups such as 2-pyridyl); Ar’ is a second aryl or aryl methyl group.
- The group (X) can be carbon, oxygen, or nitrogen.
Histamine MOA
An antihistamine is not the opposite of a histamine. An antihistamine is a drug that binds to certain sites (receptors on cells) in the body, to prevent histamine from binding.
- Antihistamines act as competitive inhibitors of histamine (agonist) binding. Antihistamines compete for the H1 receptors on blood vessel endothelial cells and smooth muscle cells.
- Some H1 antihistamines can also block histamine release. The concentrations, however, are considerably higher than those required to produce significant histamine receptor blockade.
First generation antihistamines
Aminoalkylethers (Ethanolamines)
For Example. Diphenhydramine hydrochloride, Dimenhydrinate, Doxylamine succinate, Clemastine fumarate, Diphenylphyraline hydrochloride.
- Ethylenediamine
- For Example. Tripelennamine hydrochloride.
- Piperazine (Cyclizines)
- For Example. Chlorcyclizine hydrochloride, Meclizine hydrochloride, Buclizine hydrochloride.
- Propylamines
- For Example. Chlorpheniramine maleate, Triprolidine hydrochloride; Phenidamine tartrate.
- Phenothiazines
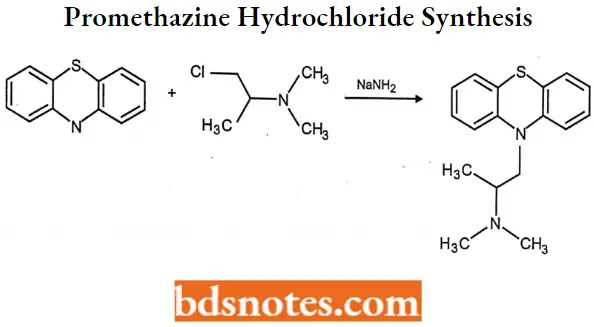
- For Example. Promethazine hydrochloride, Trimeprazine tartrate.
- Dibenzocycloheptenes
- For Example. Cyproheptadine hydrochloride, Azatadine maleate.
Aminoalkylethers (Ethanolamines)
“Why is early learning of antihistamine medicinal chemistry critical for drug design? Answered”
Aminoalkylethers SAR
- The aromatic group may be phenyl or substituted phenyl or heterocyclic for good antihistamine activity. The R and R’ may be hydrogen or methyl.
- If para substitution of Cl, CH3, CH3O, or Br on one of the phenyl rings for better therapeutic profiles and decreased side effects.
- As a result of an asymmetrically substituted benzylic carbon, most of the aminoalkyl ethers are optically active, activity residing predominantly in the S-enantiomers.
- Replacement of one of the phenyl rings of the diphenhydramine with a 2-pyridyl group, as in doxylamine and carbinoxamine, enhances antihistamine activity.
- The diaryl tertiary aminoalkyl ether structure that characterizes these compounds also serves as a pharmacophore for muscarinic receptors i.e. anticholinergic activity.
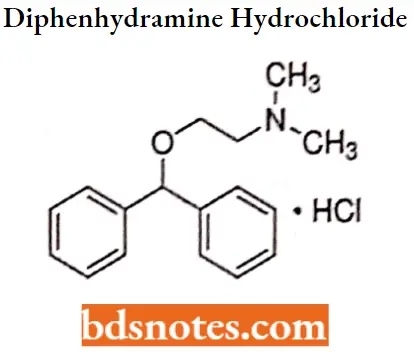
Diphenhydramine hydrochloride
“Factors influencing success with antihistamine medicinal chemistry knowledge: Q&A”
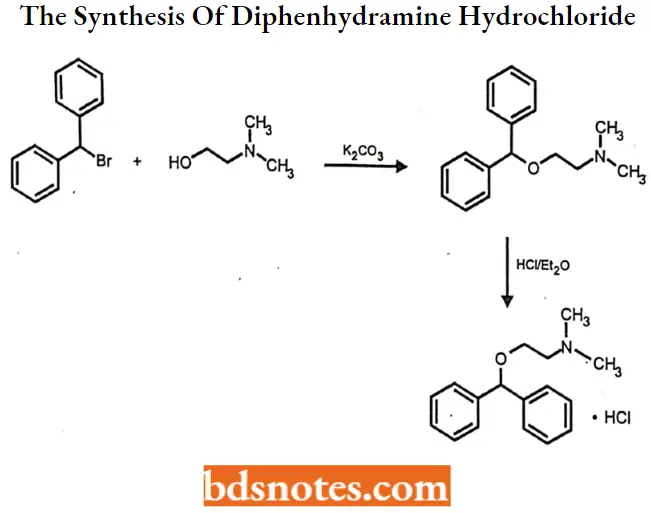
Diphenhydramine hydrochloride IUPAC name: [2-(diphenylmethoxy)ethyl] dimethylamine hydrochloride.
Diphenhydramine hydrochloride MOA: Diphenhydramine competes with free histamine for binding at Hi-receptor sites. This antagonizes the effects of histamine on Hi-receptors, leading to a reduction of the negative symptoms brought on by histamine Hi-receptor binding.
Diphenhydramine hydrochloride Metabolism: It undergoes hepatic and renal metabolism. N- demethylation and subsequent deamination are major pathways for diphenhydramine.
Metabolites are N-desmethyl diphenhydramine and have a shorter half-life than corresponding parent drugs, N, N-didesmethyldiphenhydramine, and Diphenylmethoxyaceticacid.
Diphenhydramine hydrochloride Uses: As an antihistaminic agent, diphenhydramine is recommended in various allergic conditions and, to a lesser as an antitussive and Parkinsonism drug. It is also used in OTC sleep-aid products.
It is administered either orally or parenterally in the treatment of urticaria, seasonal rhinitis (hay fever), and some dermatoses.
Diphenhydramine hydrochloride Side effects: The most common side effect is drowsiness, and the concurrent use of alcohol and other CNS depressants should be avoided.
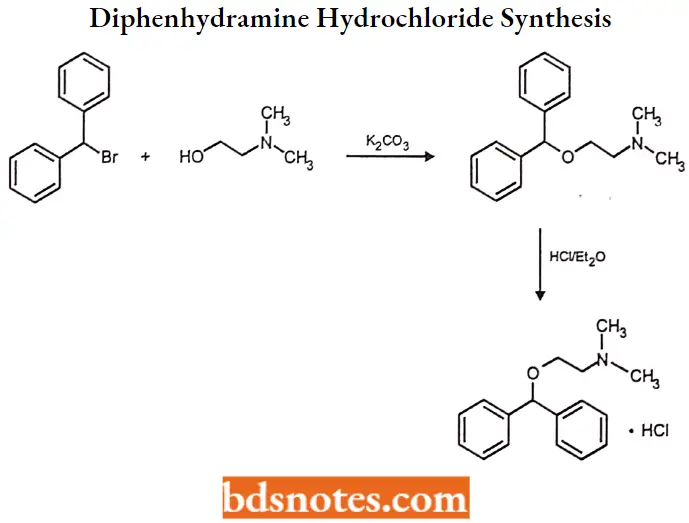
Diphenhydramine hydrochloride Synthesis
“Steps to explain types of antihistamines: First-generation vs second-generation: Notes guide”
Dimenhydrinate
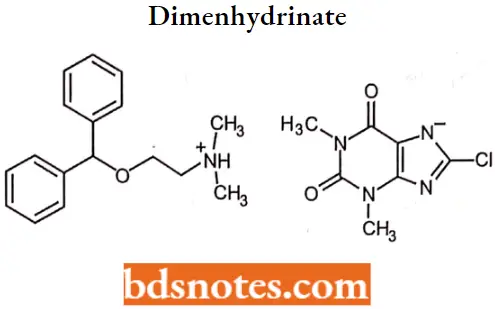
Dimenhydrinate IUPAC name: 8-chloro-l,3-dimethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-7-ide-[2-(Diphenylmethoxy) ethyl]dimethylazanium.
Dimenhydrinate MOA: Dimenhydrinate is a competitive antagonist at the histamine Hi receptor, which is widely distributed in the human brain. Dimenhydrinate’s anti-emetic effect is probably due to Hi antagonism in the vestibular system in the brain.
Dimenhydrinate Uses: Dimenhydrinate is recommended for nausea of motion sickness and hyperemesis gravidarum (nausea of pregnancy).
Dimenhydrinate Side effects: When used in large doses, dimenhydrinate has been shown to cause a “high” characterized by hallucinations, excitement, incoordination, and disorientation.
Doxylamine succinate
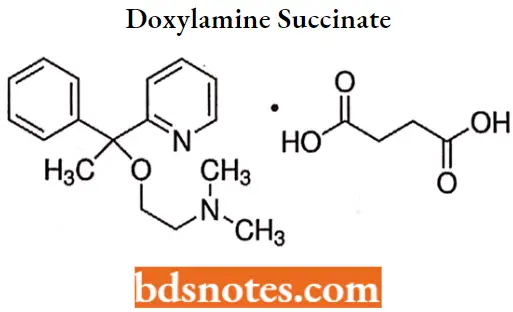
“Role of H1-receptor antagonism in antihistamine action: Questions answered”
Doxylamine succinate IUPAC name: 2-[a-[2-(dimethylamino)ethoxy]- a-methylbenzyl]pyridine succinate.
Doxylamine succinate MOA: Doxylamine acts by competitively inhibiting histamine at H1 receptors. It also has substantial sedative and anticholinergic effects.
Doxylamine succinate Uses: It is indicated for the relief of seasonal rhinitis symptoms but is also used as a nighttime sedative. It is also used with antitussives and decongestants for the relief of cough and cold.
Clemastine fumarate
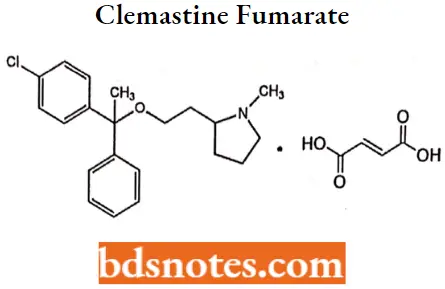
Clemastine fumarate IUPAC name: R, R-2[2[l-(4-chlorophenyl)-l-phenylethoxy]ethyl]-lmethylpyrroIidine hydrogen fumarate.
Clemastine fumarate MOA: Clemastine is a selective histamine H1 antagonist and binds to the histamine H1 receptor. This blocks the action of endogenous histamine, which subseQuestion gently leads to temporary relief of the negative symptoms brought on by histamine.
Clemastine fumarate Metabolism: Antihistamines appear to be metabolized in the liver chiefly via mono- and demethylation and glucuronide conjugation.
Clemastine fumarate Uses
It has antihistaminic activity with anticholinergic and sedative effects.
Diphenylpyraline hydrochloride
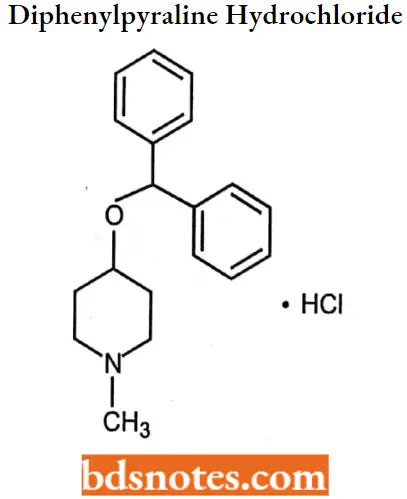
Diphenylpyraline hydrochloride IUPAC name: 4-(diphenylmethoxy)-l-methylpiperidine hydrochloride.
Diphenylpyraline hydrochloride MOA: Diphenylpyraline hydrochloride is used in the treatment of allergy acts by competing with histamine for Hl-receptor sites on effector cells. This reduces the effects of histamine, leading to a temporary reduction of allergy symptoms.
Diphenylpyraline hydrochloride Uses: Use in the treatment of allergic rhinitis, hay fever, and allergic skin disorders.
Ethylenediamine
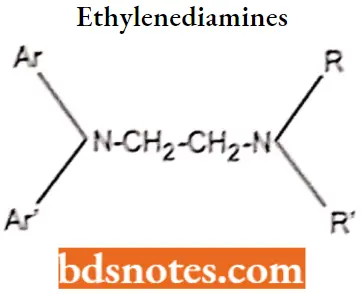
“How does diphenhydramine differ from loratadine? FAQ explained”
Diphenylpyraline hydrochloride SAR
- The ethylenediamine antihistamines are characterized by the presence of a nitrogen-connecting atom (X) and a two-carbon atom chain as the linking moiety between the key diaryl and tertian or amino moieties.
- All compounds in this series are simple diarylethylenediamines except pentazocine, in which the terminal amine and a portion of the carbon chain are included as part of an imidazoline ring system. Because it differs significantly in its pharmacological profile, pentazocine is not always classified as an ethylenediamine derivative.
- Phenbenzamine was the first clinically useful member of this class and served as the prototype for die development of more effective derivatives. Replacement of the phenyl moiety of phenoxybenzamine with a 2-pyridyl system yielded tripelennamine, a significantly more effective histamine receptor blocker.
- Substitution of a para methoxy (pyrilamine or mepyramine), chloro (chloropyramine), or bromo (bromtripelennamine) further enhances activity.
- Replacement of the benzyl group of tripelennamine with a 2-phenylmethyl group provided methapyrilene, and replacement of tripelennamine’s 2-pyridyl group with a pyrimidine moiety (along with p-methoxy substitution) yielded thonzylamine, both of which function as potent H1-receptor antagonists.
Tripelennamine hydrochloride
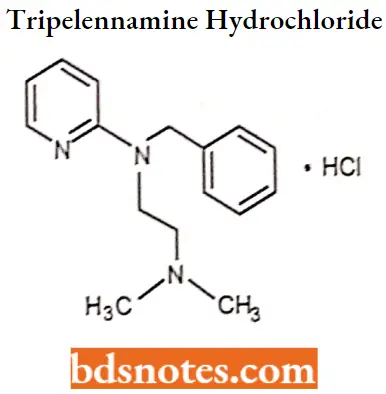
Tripelennamine hydrochloride IUPAC name: N-benzyl-N-[2-(dimelhylamino)ethyl]pyridin-2-amine.
Tripelennamine hydrochloride MOA: Tripelennamine binds to the histamine Hi receptor. This blocks the action of endogenous histamine, which subsequently leads to temporary relief of the negative symptoms brought on by histamine.
Tripelennamine hydrochloride Metabolism: Tripelennamine is metabolized in humans by N-glucuronidation, N-oxidation, and pyridyl oxidation followed by phenol glucuronidation.
Tripelennamine hydrochloride Uses: Tripelennamine is metabolized in humans by N-glucuronidation, N-oxidation, and pyridyl oxidation followed by phenol glucuronidation.
Tripelennamine hydrochloride Side effects: May include clumsiness or unsteadiness, convulsions, drowsiness, dryness of mouth, nose, or throat, feeling faint, flushing or redness of the face, hallucinations, muscle spasms (especially of the neck and back), restlessness, shortness of breath or troubled breathing, shuffling walk, tic-like movements of head and face, trembling and shaking of hands and trouble in sleeping.
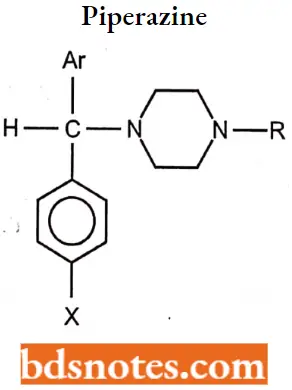
Piperazine (Cyclizines)
The piperazines or cyclizines can also be considered ethylenediamine derivatives or cyclic ethylenediamines.
“Early warning signs of gaps in understanding antihistamine SAR basics: Common questions”
Piperazine (Cyclizines) SAR
- The connecting moiety (X) is a CHN group, and the carbon chain, terminal amine functionality, and the nitrogen atom of the connecting group are all part of a piperazine moiety.
- Both nitrogen atoms in these compounds are aliphatic and thus display comparable basicities.
- The primary structural differences within this series involve the nature of the para aromatic ring substituent (H or Cl) and, more importantly, the nature of the terminal piperazine nitrogen substituent.
Chlorcyclizine hydrochloride
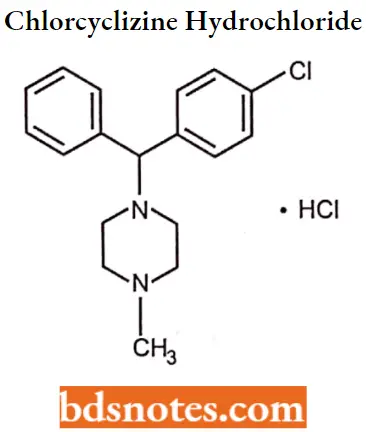
Chlorcyclizine hydrochloride IUPAC name: l-(p-chloro-a-phenylbenzyl)-4-methyl piperazine monohydrochloride.
Chlorcyclizine hydrochloride MOA: Along with H1 antagonists, these agents exhibit peripheral and central antimuscarinic activity, thereby diminishing vestibular stimulation and acting on the medullary chemoreceptor trigger zone.
Chlorcyclizine hydrochloride Metabolism: The primary pathways involve N-oxidation and N-demethylation (norchlorcyclizine), and both of these metabolites are devoid of antihistaminic activity.
Chlorcyclizine hydrochloride Uses: Chlorcydizine is indicated for the symptomatic relief of urticaria, hay fever, and certain other conditions.
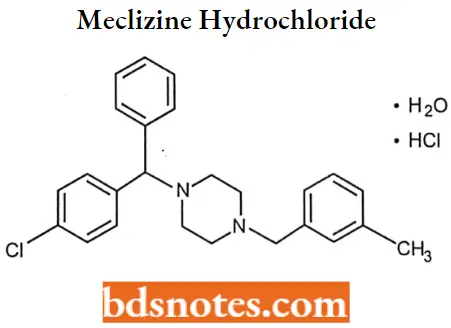
Meclizine hydrochloride
“Asymptomatic vs symptomatic effects of ignoring antihistamine principles: Q&A”
Meclizine hydrochloride IUPAC name: l-(p-chloro-a-phenylbenzyl)-4-(m-methylbenzyl)piperazine dihydrochloride monohydrate.
Meclizine hydrochloride MOA: Along with its actions as an antagonist at H1-receptors, meclizine also possesses anticholinergic, central nervous system depressant, and local anesthetic effects.
Meclizine depresses labyrinth excitability and vestibular stimulation and may affect the medullary chemoreceptor trigger zone.
Meclizine hydrochloride Uses: Although it is a moderately potent antihistamine, meclizine is used primarily as an antinauseant in the prevention and treatment of motion sickness and the treatment of nausea and vomiting associated with vertigo.
Buclizine hydrochloride
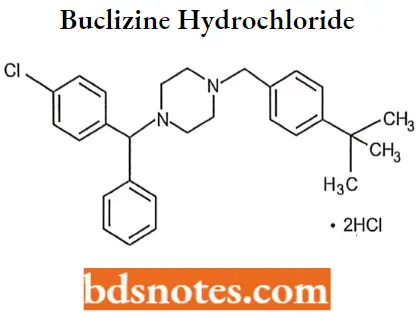
Buclizine hydrochloride IUPAC name: 1(p-tertbutylbenzyl)-4-(p-chloro-a-phenylbenzyl)piperazine dihydrochloride.
Buclizine hydrochloride MOA: The action of buclizine acts to block the histamine receptors in the vomiting center and thus reduce activity along these pathways. Furthermore, since buclizine possesses anti-cholinergic properties as well, the muscarinic receptors are similarly blocked.
Buclizine hydrochloride Uses: For prevention and treatment of nausea, vomiting, and dizziness associated with motion sickness and vertigo.
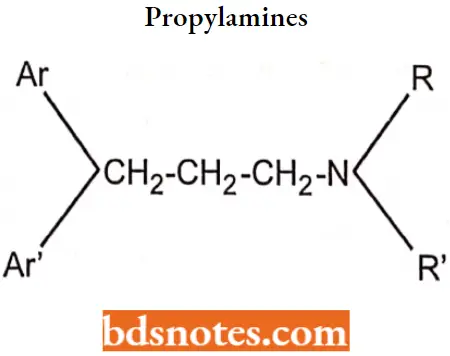
Propylamines (Monoaminopropyl or Alkylamine Derivatives)
“Can targeted interventions improve outcomes using antihistamine medicinal chemistry knowledge? FAQs provided”
Propylamines SAR:
- The propylamine antihistamines are characterized structurally by an sp3 or sp2 carbon-connecting atom with a carbon chain of two additional carbons linking the key tertiary amino and diaryl pharmacophore moieties.
- All of the pheniramines consist of a phenyl a 2-pyridyl aryl group and a terminal dimethylamino moiety.
- The phenyl substituent at the para position: H (pheniramine), Cl (chlorpheniramine), and Br (brompheniramine). The halogenated pheniramines are significantly more potent (20-50 times) and have a longer duration of action than pheniramine.
- All pheniramines are chiral molecules. Antihistaminic activity resides almost exclusively in the S-stereoisomers (200-1,000 times higher Hl-receptor binding affinities).
- The propylamines with an unsaturated connecting moiety include the simple open-chain alkene derivatives pyrrobutarnine and triprolidine and the cyclic alkene analogs dimethindene and phenindamine.
- In the open-chain propylamines, a coplanar aromatic double-bond system appears to be an important factor for antihistaminic activity.
- These compounds are asymmetric and the E-isomers are significantly more potent than the Zisomers.
- The pyrrolidinone group of these compounds is the side-chain tertiary amine that imparts the greatest antihistaminic activity.
Chlorpheniramine maleate
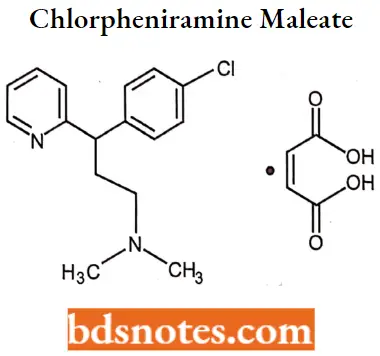
Chlorpheniramine maleate IUPAC name: (±)2-[p-chloro-α-[2-dimethylamino)ethyl]benzyl]pyridine bi-maleate.
Chlorpheniramine maleate MOA: Chlorpheniramine binds to the histamine Hi receptor. These blocks tire action of endogenous histamine, which subseQuestion uently leads to temporary relief of the negative symptoms brought on by histamine.
Chlorpheniramine maleate Metabolism: Undergo hepatic metabolism via cytochrome P450. The primary metabolites for this compound are the mono- and di-N-dealkylation products.
Chlorpheniramine maleate Uses: For the treatment of rhinitis, urticaria, allergy, common cold, asthma, and hay fever.
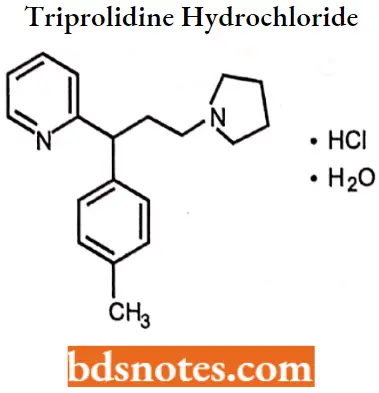
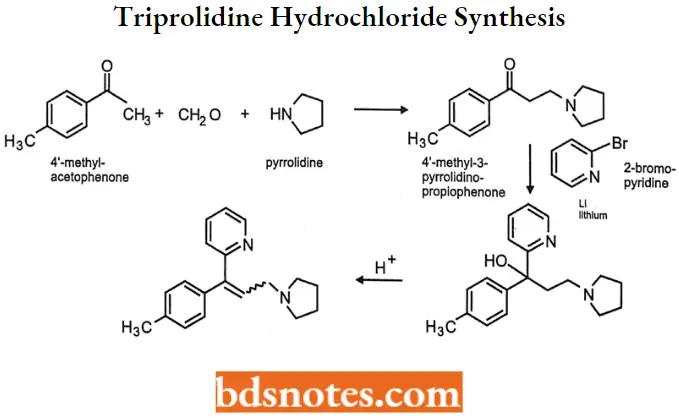
Triprolidine hydrochloride
“Differential applications of sedating vs non-sedating antihistamines: Notes explained”
Triprolidine hydrochloride IUPAC name: (E)-2-[3-(l-pyrrolidinyl)-l-p-tolylpropenyl] pyridine monohydrochloride monohydrate.
Triprolidine hydrochloride MOA: Triprolidine binds to the histamine Hi receptor. This blocks the action of endogenous histamine, which subseQuestion gently leads to temporary relief of the negative symptoms brought on by histamine.
Triprolidine hydrochloride Uses: For the symptomatic relief of seasonal or perennial allergic rhinitis or nonallergic rhinitis; allergic conjunctivitis; and mild, uncomplicated allergic skin manifestations of urticaria and angioedema.
Also used in combination with other agents for the symptomatic relief of symptoms associated with the common cold.
Triprolidine hydrochloride Synthesis
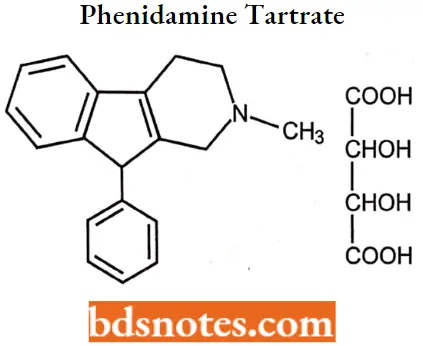
Phenidamine tartrate
“Difference between first-generation and second-generation antihistamines: Notes explained”
Phenidamine tartrate IUPAC name: 2,3,4,9-tetrahydro-2-methyl-9-phenyl-1H-indeno[2, 1-c] pyridine bitartrate.
Phenidamine tartrate MOA: Antihistamines such as phenindamine appear to compete with histamine for histamine Hlreceptor sites on effector cells.
The antihistamines antagonize those pharmacological effects of histamine which are mediated through activation of H1- receptor sites and thereby reduce the intensity of allergic reactions and tissue injury response involving histamine release.
Phenidamine tartrate Uses: Used to treat sneezing, runny nose, itching, watery eyes, hives, rashes, itching, and other symptoms of allergies and the common cold.
Phenidamine tartrate Side effects: It may produce drowsiness and sleepiness; it may also cause a mildly stimulating action in some patients and insomnia when taken just before bedtime.
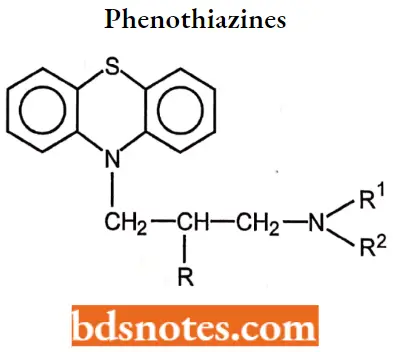
Phenothiazines
Phenothiazines SAR:
- The phenothiazine derivatives that display therapeutically useful antihistaminic actions contain a two- or three-carbon, branched alkyl chain between the ring system and terminal nitrogen atom.
- This differs significantly from the phenothiazine antipsychotic series in which an unbranched propyl chain is required.
- The branched alkyl chain contains a chiral carbon, giving rise to optical isomerism. The enantiomers of the prototype of this class, promethazine, have been resolved and found to possess similar antihistaminic.
- The combination of lengthening of the side chain and substitution of lipophilic groups in the 2- position of the aromatic ring results in compounds with decreased antihistaminic activity and increased psychotherapeutic properties.
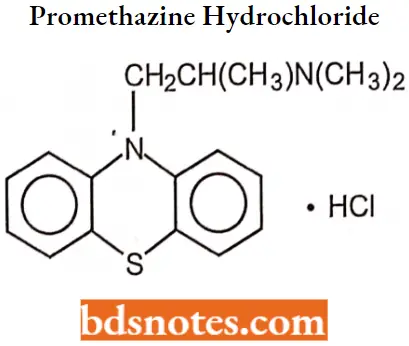
Promethazine hydrochloride
“Most common complications of poorly understood antihistamine medicinal chemistry concepts: FAQs”
Promethazine hydrochloride IUPAC name: (±)10-[2-(dimethylamino)-propyl]phenothiazine monohydrochloride. MOA Like other H1-antagonists, promethazine competes with free histamine for binding at H1-receptor sites in the GI tract, uterus, large blood vessels, and bronchial muscle. The relief of nausea appears to be related to central anticholinergic actions and may implicate activity on the medullary chemoreceptor trigger zone.
Promethazine hydrochloride Metabolism: Promethazine hydrochloride is metabolized in the liver, with the sulfoxides of promethazine and N-desmethyl promethazine being the predominant metabolites appearing in the urine.
Promethazine hydrochloride Uses: For the treatment of allergic disorders and nausea or vomiting.
Promethazine hydrochloride Side effects: It may cause drowsiness and impair the ability to perform tasks reQuestion requiring alertness. Also, concurrent administration of alcoholic beverages and other CNS depressants with the phenothiazines should be avoided.
Promethazine hydrochloride Synthesis:
Trimeprazine tartrate
“Why are antihistamine mechanisms often misunderstood in practice? Questions answered”
Trimeprazine tartrate IUPAC name: (±) 10[3-(dimethylamino)-2-methylpropyl] phenothiazine tartrate.
Trimeprazine tartrate MOA: Trimeprazine competes with free histamine for binding at HA-receptor sites. This antagonizes the effects of histamine on HA-receptors, leading to a reduction of the negative symptoms brought on by histamine HA-receptor binding.
Trimeprazine tartrate Uses: Used to prevent and relieve allergic conditions that cause pruritus (itching) and urticaria (some allergic skin reactions).
Dibenzocycloheptenes
The dibenzocycloheptene and dibenzocycloheptane antihistamines may be regarded as phenothiazine analogs in which the sulfur atom has been replaced by an isosteric vinyl group (cyproheptadine) or a saturated ethyl bridge (azatadine), and the ring nitrogen has been replaced by an sp2 carbon atom.
- The two members of this series are closely related in structure; azatadine is an aza (pyridyl) isostere of cyproheptadine in which the 10,11-double bond is reduced.
- These agents are potent antihistamines with moderate sedation anticholinergic potential and little antiemetic activity.
Cyproheptadine hydrochloride
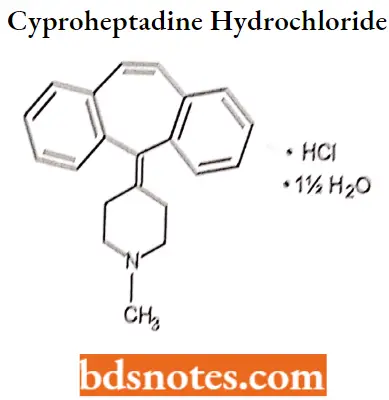
Cyproheptadine hydrochloride IUPAC name: 4-(5H-dibenzo-[a,d]-cyclohepten-5-ylidine)-1-methyl piperidine hydrochloride sesquihydrate.
Cyproheptadine hydrochloride MOA: Cyproheptadine competes with free histamine for binding at HA-receptor sites. This antagonizes the effects of histamine on HA-receptors, leading to a reduction of the negative symptoms brought on by histamine HA-receptor binding. Cyproheptadine also competes with serotonin at receptor sites in smooth muscle in the intestines and other locations. Antagonism of serotonin on the appetite center of the hypothalamus may account for cyproheptadine’s ability to stimulate appetites.
Cyproheptadine hydrochloride Uses: It is Indicated for the treatment of hypersensitivity reactions, perennial, and seasonal allergic rhinitis; vasomotor rhinitis; and allergic conjunctivitis.
- Uncomplicated allergic skin manifestations of urticaria and angioedema; amelioration of allergic reactions to blood or plasma; and cold urticaria.
- It In also used off-label for nightmares associated with posttraumatic stress disorder (PTSD), prevention of migraine, suppression of vascular headaches, and appetite stimulation.
Cyproheptadine hydrochloride Side effects: Sedation is the most prominent side effect, and this is usually brief, disappearing after 3 or 4 days of treatment.
Azatadine maleate
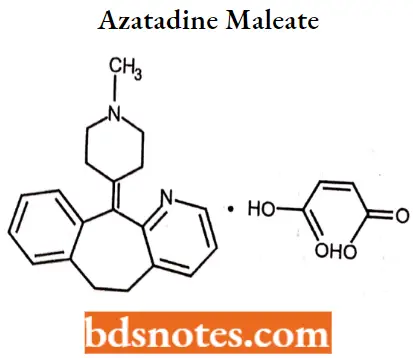
Azntiulluc maleate IUPAC name: 6,11-dihydro11-(1-methyl-4-piperidylidene)-5H-benzo-[5,6]cyclohepta(l,2- b]pyridine maleate.
Azntiulluc maleate MOA: Antihistamines such as azatadine appear to compete with histamine for histamine H1- receptor sites on effector cells.
The antihistamines antagonize those pharmacological effects of histamine which are mediated through activation of H1- receptor sites and thereby reduce the intensity of allergic reactions and tissue injury response involving histamine release.
Azntiulluc maleate Uses: For the relief of the symptoms of upper respiratory mucosal congestion in perennial and allergic rhinitis and the relief of nasal congestion.
Second generation antihistamines
Second-generation antihistamines focused on developing agents with a lower sedation potential and reduced binding affinities for non-target proteins including muscarinic, adrenergic, and serotonergic receptors.
- These efforts led to the introduction of astemizole and terfenadine, derivatives of the first-generation agents with substantially larger N-tertiary amine substituents.
- The new second-generation drugs currently on the market include acrivastine, an alkene-acid derivative of triprolidine.
Cetirizine and levocetirizine, are oxidation metabolites of hydroxyzine; desloratadine, an oxidation-hydrolysis metabolite of loratadine; and fexofenadine, an oxidative metabolite of terfenadine.
Astemizole
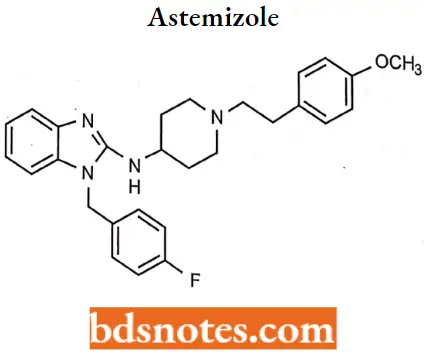
Astemizole IUPAC name: 1-[4-fluorophenylomethyl]-N-{1-[2-(4-methoxyphenyl)ethyl]piperidin-4-yl}-1H-1,3-benzodiazol-2-amine.
Astemizole MOA: Astemizole competes with histamine for binding at H1-receptor sites in the GI tract, uterus, large blood vessels, and bronchial muscle.
- This reversible binding of astemizole to H1-receptors suppresses the formation of edema, flare, and pruritus resulting from histaminic activity.
- As the drug does not readily cross the blood-brain barrier and preferentially binds at HI receptors in the periphery rather than within the brain, CNS depression is minimal. Astemizole may also act on H3-receptors, producing adverse effects.
Astemizole Uses
Astemizole was indicated for use in relieving allergy symptoms, particularly rhinitis and conjunctivitis. It has been withdrawn from the market however due to concerns of arrhythmias.
Loratadine
Loratadine IUPAC name: 4-(8-chloro-5, 6-dihydro- llHbenzo[5,6]-cyclohepta[l,2-b]pyridin-l 1-ylidene-lcarboxylic acid ethyl ester.
Loratadine MOA: Loratadine competes with free histamine and exhibits specific, selective peripheral H1 antagonistic activity.
- This blocks the action of endogenous histamine, which subsequently leads to temporary relief of the negative symptoms (For Example. nasal congestion, and watery eyes) brought on by histamine.
- Loratadine has a low affinity for cholinergic receptors and does not exhibit any appreciable alpha-adrenergic blocking activity in vitro.
- Loratadine also appears to suppress the release of histamine and leukotrienes from animal mast cells, and the release of leukotrienes from human lung fragments.
Loratadine Metabolism: This drug is extensively metabolized by oxidation of the carbamate methylene group, a reaction catalyzed by CYP3A4 and, to a lesser extent, CYP2D6.
This oxidation metabolite readily undergoes O-demethylation and decarboxylation to form the descarboethoxy metabolite (desloratadine), which appears to be responsible for antihistaminic activity.
Loratadine Uses: A self-medication that is used alone or in combination with pseudoephedrine sulfate for the symptomatic relief of pruritus, erythema, and urticaria associated with chronic idiopathic urticaria in patients (not for children under 6 unless directed by a clinician).
Cetrizine
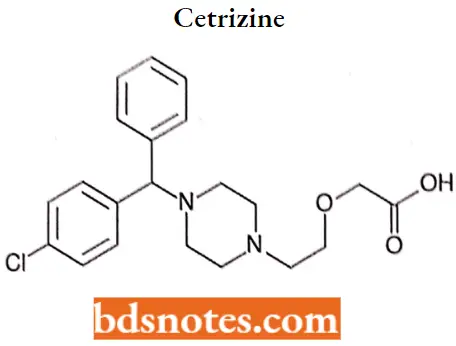
Cetrizine IUPAC name: (±)-[2-[4-[(4-chlorophenyl)phenyhuethyl]-1-piperazinyl]ethoxy]acetic acid.
Cetrizine MOA: Cetirizine competes with histamine for binding at Ht-receptor sites on the effector cell surface, resulting in the suppression of laminomic edema, flare, and pruritus.
The low incidence of sedation can be attributed to reduced penetration of cetirizine into the CNS as a result of the less lipophilic carboxyl group on the ethylamine side chain.
Cetrizine Metabolism: Cetirizine is not extensively metabolized, and more than 60% of a 10-mg oral dose is excreted in the urine (60% as an unchanged drug) and 10% is recovered in the feces. The drug is highly protein-bound (93%) and has a terminal half-life of 8.3 hours.
Cetrizine Uses: Cetirizine is indicated for the temporary relief of runny nose, sneezing, itching of the throat, and or itchy, watery eyes caused by hay fever or other upper respiratory allergies.
It also relieves itching caused by hives (urticaria), but it will not prevent hives or an allergic skin reaction from occurring.
Cetrizine Side effects: The drug has been associated with dose-related somnolence, however, so patients should be advised that cetirizine may interfere with the performance of certain jobs or activities. Other effects of this drug include fatigue, dry mouth, pharyngitis, and dizziness.
Levocetrizine
This is an R-enantiomer that has a 30-fold higher affinity than the S-enantiomer and dissociates more slowly from Hl-receptors.
Levocetrizine MOA: It is an anti-histamine; its principal effects are mediated via selective inhibition of H1 receptors. Levocetirizine has an affinity for the human H1-receptor 2-fold higher than that of cetirizine.
Levocetrizine Uses: Levocetirizine is indicated for the relief of symptoms associated with allergic rhinitis (seasonal and perennial) in adults and children 6 years of age and older and for the treatment of the uncomplicated skin manifestations of chronic idiopathic urticaria in adults and children 6 years of age and older.
Inhibition of histamine release: Mast cell stabilizers: The discovery of the bronchodilator activity of the natural product khellin, obtained from the plant Ammi visnaga, led to the development of the bis (chromones) as compounds that stabilize mast cells and inhibit the release of histamine and other mediators of inflammation. The first therapeutically significant member of this class was cromolyn sodium.
Mast cell stabilizers MOA: Generally, the mast cell stabilizers inhibit activation of, and mediator release from, various inflammatory cell types associated with allergy and asthma, including eosinophils, neutrophils, macrophages; mast cells, monocytes, and platelets.
- In addition to histamine, these drugs inhibit the release of leukotrienes (C4, D4, E4) and prostaglandins. In vitro studies suggest that these drugs stimulate the protein kinase C-mediated phosphorylation of moesin.
- This phosphorylated protein then binds with key proteins on the secretory granule (such as actin) and stabilizes them to prevent exocytosis.
- Other studies suggest that the bis chromones indirectly inhibit calcium ion entry into the mast cell and that this action prevents mediator release.
- In addition to their inhibition of mediator release, some of these drugs also inhibit the chemotaxis of eosinophils at the site of application (i.e., ocular tissue).
Cromolyn sodium
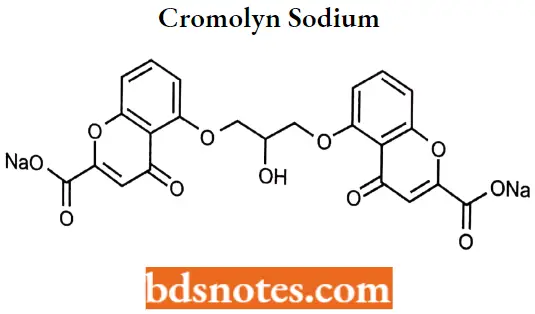
Cromolyn sodium IUPAC name: Disodium l,3bis(2-carboxychromon-5-yloxy)-2-hydroxypropane.
Cromolyn sodium MOA: It inhibits degranulation of mast cells, subsequently preventing the release of histamine and slow-reacting substances of anaphylaxis (SRS-A), mediators of type 1 allergic reactions. It also may reduce tire release of inflammatory leukotrienes. It may act by inhibiting calcium influx.
Cromolyn sodium Uses: Nebulized and aerosol cromolyn have been used for prophylactic management of bronchial asthma and prevention of exercise-induced bronchospasm.
- Cromolyn nasal solution is used for the prevention and treatment of allergic rhinitis, and oral concentrate is used to treat the histaminic symptoms of mastocytosis (diarrhea, flushing, headaches, vomiting, urticaria, abdominal pain, nausea, and itching).
- Topical cromolyn (eye drops) is used to treat allergic conjunctivitis and keratitis. In the treatment of asthma, cromolyn efficacy is manifested by decreased severity of clinical symptoms.
Histamine H2 antagonists: Some of the histamine responses (dilation of capillaries, increasing the rate and force of heart contraction, increasing gastric acid secretion) are exclusively mediated through H2 receptors.
- The classical antihistamines are unable to inhibit these responses hence H2 receptor-blocking drugs were introduced.
- H2-receptor antagonists are drugs used to block histamine H2 receptors reversibly and inhibit H2-receptor-mediated responses. They are used to treat gastric ulcers as they block gastric acid and enzyme secretion.
- Gastric acid secretion by parietal (oxynticell is not blocked by antihistamines acting on Hi receptors. They have little affinity for H1-receptors.
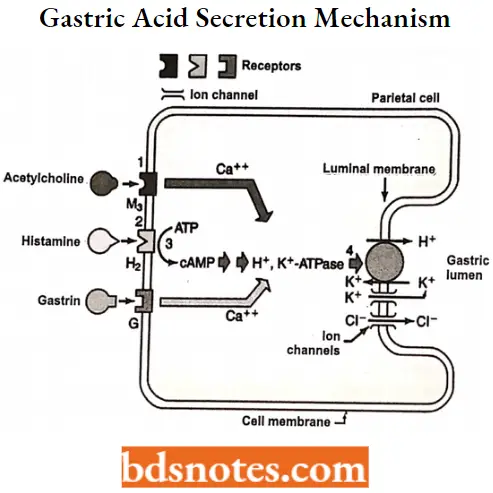
Gastric acid secretion: Since the parietal cell was known to secrete gastric acid, many drugs were developed to target the parietal cell to inhibit the acid secretion.
- Parietal cells contain an H+, K+-ATPase, or hydrogen ion pump that secretes proton (H+) in exchange for the uptake of K+ ion.
- Secretion of acid by gastric parietal cells is stimulated by various mediators at receptors located on the membrane of the basolateral receptor, including histamine agonism of H2-receptors (Cellular), gastrin activity at G receptors (blood), and acetylcholine (ACh) at M2 muscarinic receptors (neuronal).
Gastric and muscarinic receptors modulate acid secretion through calcium-dependent processes, while the H2-receptor is coupled to adenylate cyclase.
“Cost of ignoring antihistamine medicinal chemistry principles vs benefits of systematic approaches: Q&A”
The key player in acid secretion is a H+/K+ ATPase or “proton pump” located in the canalicular membrane. This ATPase is magnesium-dependent, and not inhibitable by ouabain. The current model for explaining acid secretion is as follows:
- Hydrogen ions are generated within the parietal cell from the dissociation of water. The hydroxyl ions formed in this process rapidly combine with carbon dioxide to form bicarbonate ions, a reaction catalyzed by carbonic anhydrase.
- Bicarbonate is transported out of the basolateral membrane in exchange for chloride. The outflow of bicarbonate into the blood results in a slight elevation of blood pH known as the “alkaline tide”. This process serves to maintain intracellular pH in the parietal cell.
- Chloride and potassium ions are transported into the lumen of the canaliculus by conductance channels, and such is necessary for the secretion of acid.
- Hydrogen ion is pumped out of the cell, into the lumen, in exchange for potassium through the action of the proton pump; potassium is thus effectively recycled.
- Accumulation of osmotically-active hydrogen ions in the canaliculus generates an osmotic gradient across the membrane that results in outward diffusion of water – the resulting gastric juice is 155 mM HC1 and 15 mM KC1 with a small amount of NaCl.
Cimetidine
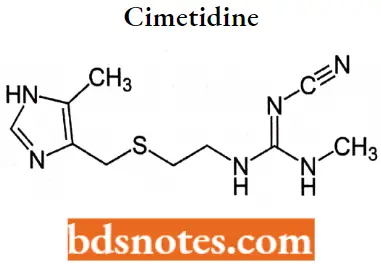
Cimetidine IUPAC name: Nn-cyano-N-methyl-N'[2-[[(5-methyl imidazole-4yl) methyl] thio] ethyl] guanidine.
Cimetidine MOA: Cimetidine binds to an H2 receptor located on the basolateral membrane of the gastric parietal cell, blocking histamine effects. This competitive inhibition results in reduced gastric acid secretion and a reduction in gastric volume and acidity.
Cimetidine Metabolism: Approximately 30% to 40% of a cimetidine dose is metabolized (S-oxidation, 5-CH3 hydroxylation), and the parent drug and metabolites are eliminated primarily by renal excretion.
Uses For the treatment and management of acid-reflux disorders (GERD), peptic ulcer disease, heartburn, and acid indigestion.
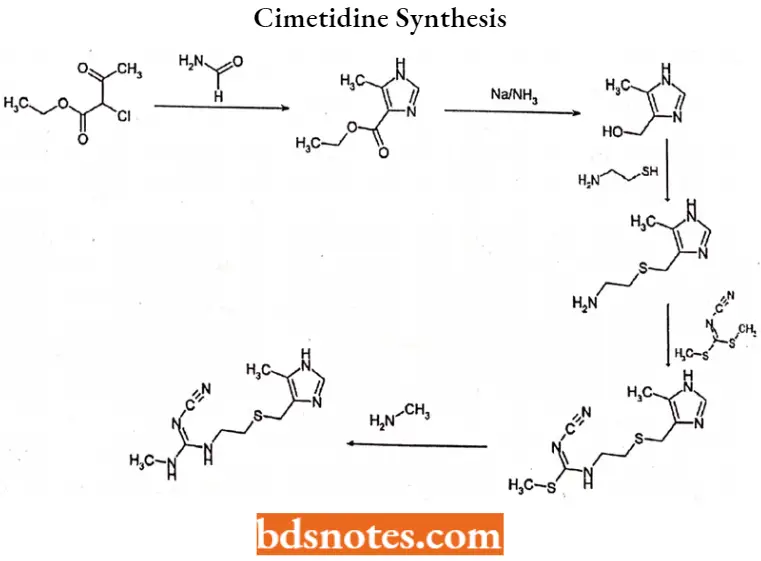
Cimetidine Synthesis
Famotidine
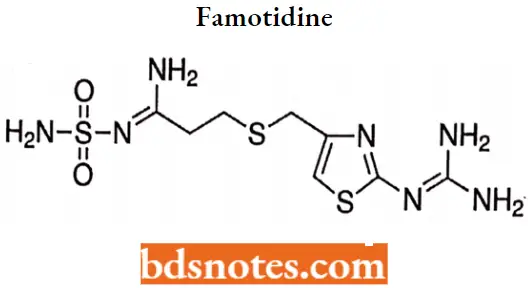
Famotidine IUPAC name: N’-(aminosulfonyl)-3[[[2[(diamnomethylene)amino]-4-thiazolyl]methyl]thio] propanimidamide
Famotidine MOA: Famotidine is a competitive inhibitor of histamine H2 receptors with a potency significantly greater than cimetidine. It inhibits basal and nocturnal gastric secretion as well as secretion stimulated by food and pentagastrin.
Famotidine Metabolism: Famotidine is incompletely absorbed (37%-45% bioavailability).66 The drug is eliminated by renal (65%- 70%) and metabolic (30%-35%) routes. Famotidine sulfoxide is the only metabolite identified in humans.
Famotidine Uses: For the short-term treatment of duodenal and benign gastric ulcers, GERD, pathological hypersecretory conditions (For Example., Zollinger-Ellison syndrome), and heartburn (OTC only).
Ranitidine
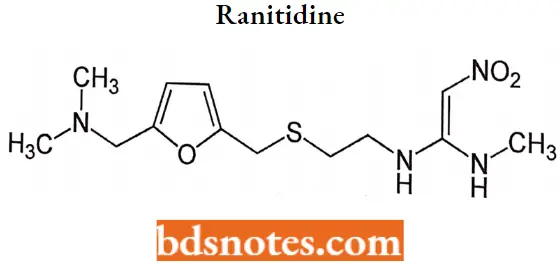
Ranitidine IUPAC name: N-[2-[[[5-(dimethylamino)methyl]thiol] ethyl]-n’-methyl-2nitro-1,1-ethenediamine.
Ranitidine MOA: The H2 antagonists are competitive inhibitors of histamine at the parietal cell H2 receptor. They suppress the normal secretion of acid by parietal cells and the meal-stimulated secretion of acid.
They accomplish this by two mechanisms: histamine released by ECL cells in the stomach is blocked from binding on parietal cell H2 receptors which stimulates acid secretion, and other substances that promote acid secretion (such as gastrin and acetylcholine) have a reduced effect on parietal cells when the H2 receptors are blocked.
Ranitidine Metabolism: The plasma half-life of the drug is 2 to 3 hours, and it is excreted along with its metabolites in the urine. Three metabolites, ranitidine N-oxide, ranitidine S-oxide, and desmethyl ranitidine, have been identified.
Ranitidine Uses: Used in the treatment of peptic ulcer disease (PUD), dyspepsia, stress ulcer prophylaxis, and gastroesophageal reflux disease(GERD).
Proton pump Inhibitor: The final step in acid secretion in the parietal cell is the extrusion or “pumping” of protons into the lumen of the stomach by the membrane H+, K+-ATPase pump as described previously.
Thus, inhibition of this proton pump acts beyond the site of action of second messengers (For Example., calcium and cAMP) and is independent of the action of histamine, gastrin, and acetylcholine. Thus, acid pump inhibitors block both basal and stimulated acid secretion.
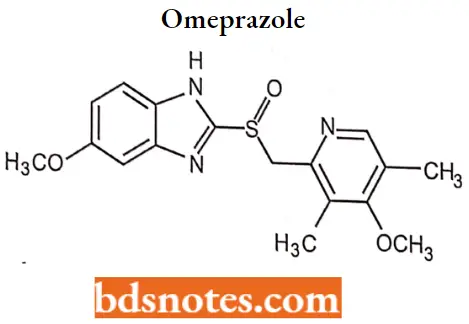
Omeprazole
“Treatment scope in adults vs children for antihistamine therapies: FAQs”
Omeprazole IUFAC name: 5-methoxy-2-9994methoxy-3, 5-dimethyl-2pridiny) methyl) sulfinyl)-lHbenzimidazole
Omeprazole MOA: Omeprazole is a proton pump inhibitor that suppresses gastric acid secretion by specific inhibition of the H+/K+-ATPase in the gastric parietal cell. By acting specifically on the proton pump, omeprazole blocks the final step in acid production, thus reducing gastric acidity.
Omeprazole Metabolism: Most (77%) of an oral dose of omeprazole is excreted in the urine as metabolites with insignificant antisecretory activity. The primary metabolites of omeprazole are 5-hydroxyomeprazole (CYP2C19) and omeprazole sulfone (CYP3A4).
Omeprazole Uses: For the treatment of heartburn, GERD, duodenal ulcer, erosive esophagitis, gastric ulcer, and pathological hypersecretory conditions.
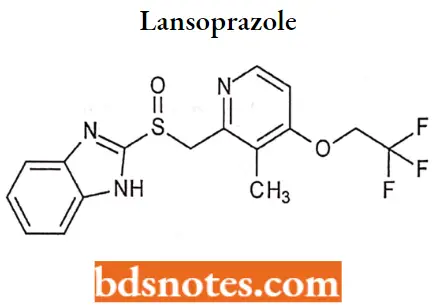
Lansoprazole
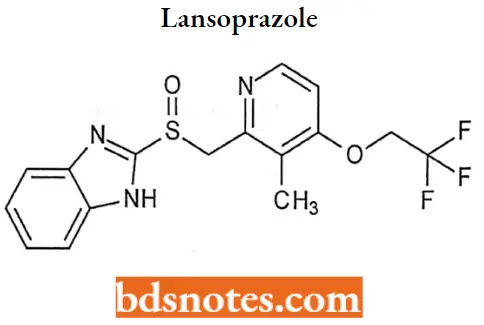
Lansoprazole IUPAC name: 2-[[[3-methyl-4-(2,2,2trifluoroethoxy)-2-pyridyl] methyl] sulfinyl]-lHbenzimidazole.
Lansoprazole MOA: Lansoprazole belongs to a class of antisecretory compounds, the substituted benzimidazoles, that do not exhibit anticholinergic or histamine FL-receptor antagonist properties.
- But rather suppress gastric acid secretion by specific inhibition of the (H+/ K+)-ATPase enzyme system at the secretory surface of the gastric parietal cell.
- Because this enzyme system is regarded as the acid (proton) pump within the parietal cell, Lansoprazole has been characterized as a gastric acid-pump inhibitor, in that it blocks the final step of acid production.
- This effect is dose-related and leads to inhibition of both basal and stimulated gastric acid secretion irrespective of the stimulus.
Lansoprazole Metabolism: The drug is metabolized in the liver (sulfone and hydroxy metabolites) and excreted in bile and mine, with a plasma half-life of about 1.5 hours.
Lansoprazole Uses: For the treatment of acid-reflux disorders (GERD), peptic ulcer disease, H. pylori eradication, and prevention of gastrointestinal bleeding with NSAID use.
Rabeprazole
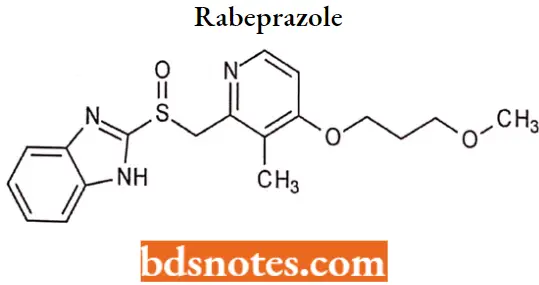
Rabeprazole IUPAC name: 2[[4-(3methoxypropoxy)-3-methyl-2-pyridinyl] methyl] sulfinyl]lH-benzimidazole.
Rabeprazole MOA: Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton pump inhibitors) that do not exhibit anticholinergic onc or histamine H2-receptor antagonist properties.
- But suppress gastric acid secretion by inhibiting the gastric H+/K+ ATPase (hydrogen potassium adenosine triphosphatase) at the secretory surface of the gastric parietal cell.
- Because this enzyme is regarded as the acid (proton) pump within the parietal cell, rabeprazole has been characterized as a gastric proton pump inhibitor.
- Rabeprazole blocks the final step of gastric acid secretion. In gastric parietal cells, rabeprazole is protonated, accumulates, and is transformed into an active sulfenamide. When studied in vitro, rabeprazole is chemically activated at pH 1.2 with a half-life of 78 seconds.
Rabeprazole Metabolism: Rabeprazole is extensively metabolized in the liver. The thioether and sulfone are the primary metabolites measured in human plasma resulting from CYP3A oxidation.
Additionally, desmethyl rabeprazole is formed via the action of CYP2C19. Approximately 90% of the drug is eliminated in the urine, primarily as thioether carboxylic acid and its glucuronide and mercapturic acid metabolites.
Rabeprazole Uses: For the treatment of acid-reflux disorders (GERD), peptic ulcer disease, H. pylori eradication, and prevention of gastrointestinal bleeds with NSAID use.
Pantoprazole
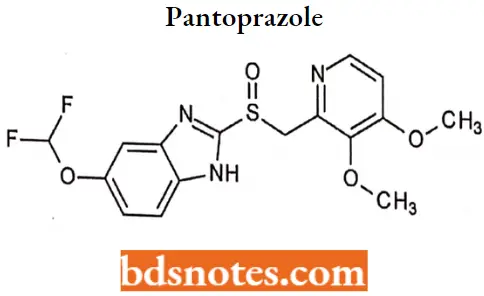
Pantoprazole IUPAC name: 6-(difluoromethoxy)-2-[(3,4-dimethoxypyridin-2-yl) methanesulfinyl]-1H-l,3-benzodiazole.
Pantoprazole MOA: pantoprazole is a proton pump inhibitor (PPI) that suppresses the final step in gastric acid production by forming a covalent bond to two sites of the (H+, F+)-ATPase enzyme system at the secretory surface of the gastric parietal cell.
This effect is related and leads to the inhibition of both basal and stimulated gastric acid secretion irrespective of the stimulus.
Pantoprazole Metabolism: Pantoprazole is extensively metabolized in the liver through the cyp system, including O-demethylation (CYP2C19), with subsequent sulfation. Other metabolic pathways include sulfur oxidation by CYP3A4. There Is no evidence that any of the pantoprazole metabolites have significant pharmacological activity.
Pantoprazole Uses: Short-term (UP to 16 weeks) treatment of erosive esophagitis.
Side Effects Of Antihistamines
- Central nervous system reactions include drowsiness, sedation, dizziness, faintness, disturbed coordination, lassitude, confusion, restlessness, excitation, tremor, seizures, headache, insomnia, euphoria, blurred vision, hallucinations, disorientation, disturbing dreams or nightmares, schizophrenic-like reactions, weakness, vertigo, hysteria, nerve pain, and
convulsions. Overdoses may cause involuntary movements. - Gastrointestinal problems include increased appetite, decreased appetite, nausea, vomiting, diarrhea, and constipation.
- Hematologic reactions are rare but may be severe. These include anemia, or breakdown of red blood cells; reduced platelets; reduced white cells; and bone marrow failure.
- A large number of additional reactions have been reported. Not all apply to every drug, and some reactions may not be drug-related. Some of the other adverse effects are chest tightness; wheezing; nasal stuffiness; dry mouth, nose, and throat; sore throat; respiratory depression; sneezing; and a burning sensation in the nose.
Antihistaminic Agents Multiple Choice Questions And Answers
Question 1. Which of the following statements about histamine is correct?
- Histamine is stored in peripheral nerve endings
- Histamine is released from mast cells following an allergic challenge
- Histamine is a vasoconstrictor
- Histamine is an essential amino acid
Answer: 2. Histamine is stored in peripheral nerve endings
Question 2. Which of the following activities occurs following the stimulation of H2 receptors?
- Vasodilation
- Uterine contraction
- Bronchial smooth muscle contraction
- Enhanced secretion of hydrochloric acid in the stomach
Answer: 4. Enhanced secretion of hydrochloric acid in the stomach
Question 3. Which of the following statements about histamine is not correct?
- It is bronchodilator
- Large-scale release of it may cause a fall in blood pressure
- Large-scale release of it may cause a fall in blood volume
- Histamine release contributes to the symptoms of anaphylaxis
Answer: 1. It is bronchodilator
Question 4. A 43year ship captain complains of seasonal allergies. Which one of the following would be indicated?
- Cyclizine
- Doxepin
- Doxylamine
- Fexofenadine
Answer: 4. Fexofenadine
Question 5. Which one of the following agents could significantly impair the ability to drive an automobile?
- Ergotamine
- Fexofenadine
- Diphenhydramine
- Ranitidine
Answer: 3. Diphenhydramine
“Is antihistamine-related risk reversible if addressed promptly? Answer provided”
Question 6. Which of the following is the most frequent side effect of H1 antihistamines that is less common with second-gen antihistamines?
- Decreased appetite
- Sedation
- Bradycardia
- Hypertension
Answer: 2. Sedation
Question 7. Histamine is released in response to various stimuli and effects of binding to one of four types of receptors. Which group of receptors are widely expressed and targeted by clinically useful drugs?
- H1 and H4
- H1 and H2
- H2 and H4
- H3 and H1
Answer: 2. H1 and H2
Question 8. Histamine stimulates what type of cell in the stomach?
- Alveolar cell
- Intestinal cell
- Gastric acid
- Parietal cell
Answer: 4. Parietal cell
Question 9. H1 antihistamines used for motion sickness and nausea include
- Diphenhydramine and meclizine
- Toradol and Meclizine
- Ephedrine and Loratidine
- Loratidine
Answer: 1. Diphenhydramine and meclizine
Question 10. What is one of the benefits of the pK of many oral antihistamines?
- Allows for twice daily dose
- Allows for once-daily dosing
- Allows for subcutaneous injection
- Allow to divide into many smaller doses
Answer: 2. Allows for once-daily dosing
Antihistaminic Agents Short Questions And Answers
Question 1. Explain the importance of Histamine.
Answer:
Histamine plays an important role in human physiology including regulation of the cardiovascular system, smooth muscle, exocrine glands, the immune system, and central nerve function.
- It is also involved in embryonic development, the proliferation and differentiation of cells, hematopoiesis, inflammation, and wound healing.
- The involvement of histamine in the mediation of immune and hypersensitivity reactions and the regulation of gastric acid secretion.
Question 2. Enlist histamine receptor and their location.
Answer:
H1 receptor- CNS neurons, the smooth muscle of respiratory, gastrointestinal (GI), uterine tissues, epithelial and endothelial cells, neutrophils, eosinophils, monocytes, dendritic cells, T cells, B cells, hepatocytes, and chondrocytes.
- H2 receptor- CNS, parietal cells, vascular smooth muscle cells, neutrophils, mast cells, as well as cells in the heart and uterus.
- H3 receptor- CNS, mainly in the striatum, substantia nigra, and the cortex.
- H4 receptor- basophils and bone marrow, expressed in the thymus, small intestine, spleen, and colon.
Question 3. Discuss the SAR of H1 Antihistamines.
Answer:
The essential pharmacophore for histamine H1 antagonistic activity is:
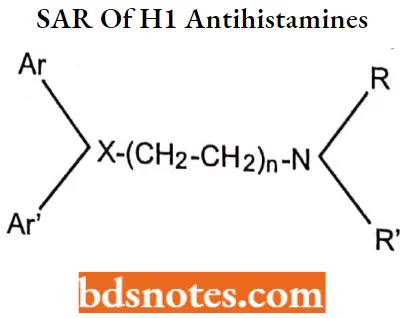
- The nitrogen should be 3° in nature for maximum antihistaminic activity. The ‘N’ may also form a part of heterocyclic moieties like piperidine, piperazine, or diazocine. The nitrogen amine should be separated by 5-6 A° from the aromatic or heteroaromatic group.
- The group present between the nitrogen atom and group X may be saturated unsaturated or substituted.
- Ar is aryl (including phenyl, substituted phenyl, and heteroaryl groups such as 2-pyridyl): Ar is a secondary aryl or aryl methyl Group.
- The group (X) can be carbon, oxygen, or nitrogen.
“Success rate of interventions using modern antihistamine medicinal chemistry techniques: FAQ”
Question 4. Outline the synthesis of Diphenhydramine hydrochloride.
Answer:
Question 5. Draw the structure and IUPAC name of Tripelennamine hydrochloride.
Answer:
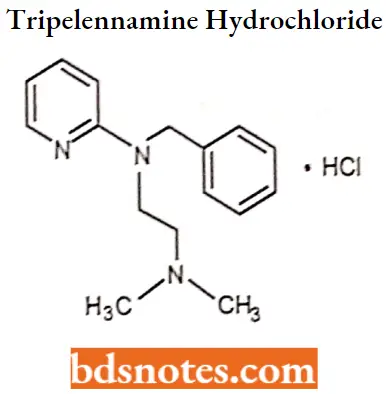
IUPAC name. N-benzyl-N-[2-(dimethylamino)ethyl]pyridin-2-amine.
Leave a Reply