Antihypertensive Agents
Antihypertension
Hypertension is a consequence of many diseases. Diseases of components of the central and peripheral nervous systems, which regulate blood pressure, and abnormalities of the hormonal system.
- Diseases of the kidney and peripheral vascular network, which affect blood volume, can create a hypertensive state in humans.
- Hypertension is generally defined as mild when the diastolic pressure is between 90 and 104 mm Hg, moderate when it is 105 to 114 mm Hg, and severe when it is above 115 mm Hg.
Antihypertensive Drugs Classification
Antihypertensive drugs are a class of drugs that are used to treat hypertension.
Antihypertensive Drugs For Hypertension
- Adrenergic receptors antagonist
- For Example. Timolol.
- Angiotensin Converting Enzyme (ACE inhibitors)
- For Example. Captopril, Lisinopril, Enalapril, Benazepril hydrochloride, Quinapril hydrochloride.
- Centrally-acting adrenergic drugs
- For Example. Methyldopate hydrochloride, Clonidine hydrochloride, Guanabenz acetate
- Vasodilators
- For Example Sodium nitroprusside, Hydralazine hydrochloride, Diazoxide, and Minoxidil.
- Adrenergic neuron-blocking agents
- For Example Guanethidine monosulphate, and Reserpine.
- Diuretics
- For Example. Furocemide, thiazides.
- Calcium channel blockers
- For Example. Amlodipine, Felodipine, Verapamil.
“Understanding antihypertensive drug classification through FAQs: Q&A explained”
Adrenergic receptor antagonists (β-blockers)
β-Blockers are among the most widely employed antihypertensives and are also considered the first-line treatment for glaucoma. Most of the β-blockers are in the chemical class of aryloxypropanolamines.
Read and Learn More Medicinal Chemistry II Notes
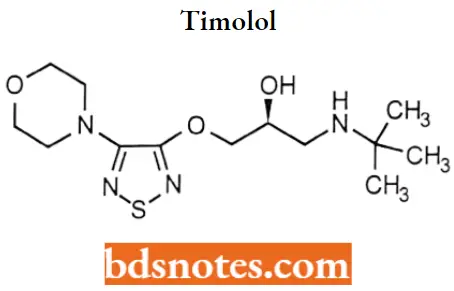
Timolol
“Importance of studying antihypertensive agents for pharmacology students: Questions explained”
Timolol IUPAC name: l-(tert-butylamino)-3-{[4-(morpholin-4-yl)-l/2/5-thiadiazol-3-yl]oxy}propan-2-ol.
Timolol MOA: Like propranolol and nadolol, timolol competes with adrenergic neurotransmitters such as catecholamines for binding at β(1)-adrenergic receptors in the heart and vascular smooth muscle and β(2)-receptors in the bronchial and vascular smooth muscle.
- β(l)-receptor blockade results in a decrease in resting and exercise heart rate and cardiac output, a decrease in both systolic and diastolic blood pressure, and, possibly, a reduction in reflex orthostatic hypotension.
- β(2)-blockade results in an increase in peripheral vascular resistance. The exact mechanism whereby timolol reduces ocular pressure is still not known. The most likely action is by decreasing the secretion of aqueous humor.
Timolol Metabolism: Primarily hepatic (80%) via the cytochrome P450 2D6 isoenzyme. Timolol and its metabolites are primarily excreted in the urine.
Timolol Uses: In its oral form it is used to treat high blood pressure and prevent heart attacks, and occasionally to prevent migraine headaches. In its ophthalmic form, it is used to treat open-angle and occasionally secondary glaucoma.
“Common challenges in understanding antihypertensive mechanisms effectively: FAQs provided”
Angiotensin Converting Enzyme Inhibitors (ACE inhibitors)
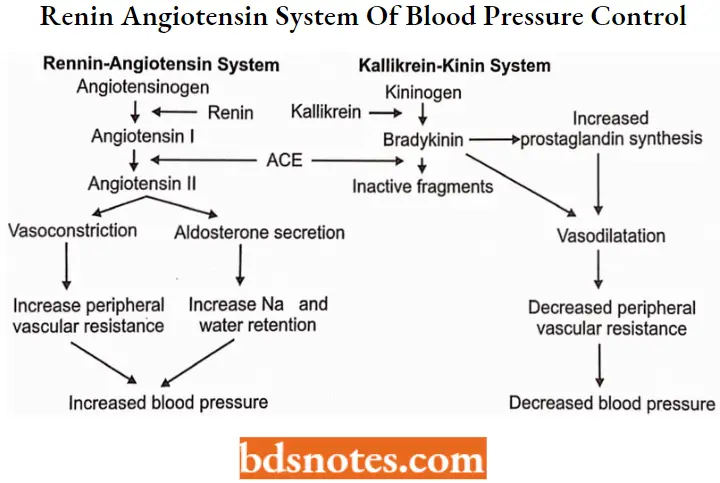
The Renin Angiotensin System and Hypertension
The renin-angiotensin system is a hormonal system that plays a central role in the control of sodium excretion and body fluid volume.
- It interacts closely with the sympathetic nervous system and aldosterone secretion in the regulation of blood pressure.
- Renin, an aspartyl protease cleaves the Leu-Val bond from the aspartic acid end of angiotensinogen polypeptide molecule to release the decapeptide angiotensin 1.
- The cleavage of a dipeptide from the carboxyl terminal] of angiotensin 1 by angiotensin-converting enzyme (ACE) to form the octapeptide angiotensin 2.
- It is a potent vasoconstrictor that increases total peripheral resistance through a variety of mechanisms: direct vasoconstriction, enhancement of both catecholamine release and neurotransmission with the peripheral nervous system, and increased sympathetic discharge.
- The result of all these actions is a rapid pressure response. Additionally, angiotensin 2 slows pressor response, resulting in long-term stabilization of arterial blood pressure.
This long-term effect is accomplished by the regulation of renal function. Angiotensin 2 directly increases sodium reabsorption in the proximal tubule.
- It also alters renal hemodynamics and causes the release of aldosterone from the adrenal cortex.
- causes Angiotensin 3 to be formed by the removal of the N-terminal aspartate residue of angiotensin 2, a reaction catalyzed by glutamyl aminopeptidase.
- In contrast to angiotensin 2, angiotensin 3 has a less potent but significant regulatory effect on sodium excretion by the renal tubules.
- This is primarily results from the effect angiotensin 3 has in stimulating aldosterone secretion, potent mineralocorticoids.
“Factors influencing success with antihypertensive agent knowledge: Q&A”
The regulatory action of the renin-angiotensin system in controlling sodium and potassium balance and arterial blood pressure is modified by vasodilators called kinins.
- Proteolytic enzymes that circulate in the plasma form kinins. Kallikrein is activated in plasma by noxious influences, to act on a kinin, callidin, which is converted to bradykinin by tissue enzymes.
- Bradykinin enhances the release of the prostaglandins PGE2 and PGI2 within certain tissues to produce a vasodilatory effect. Bradykinin is converted to inactive products by ACE and other carboxypeptidases.
- Although ACE causes activation of angiotensin and inactivation of bradykinin, actions that appear to be opposite, the balance of the system seems to favor vasoconstriction.
ACE inhibitors
These compounds can be sub-classified into three groups based on their chemical composition:
- Sulfhydryl-containing inhibitors
- For Example. Captopril.
- Dicarboxylate-containing inhibitors
- For Example. Enalapril, Lisinopril, benazepril.
- Phosphonate-containing inhibitors
- For Example. Fosinopril.
Captopril and fosinopril are the alone representatives of their respective chemical subclassifications, whereas the majority of the inhibitors contain the di-carboxylate functionality.
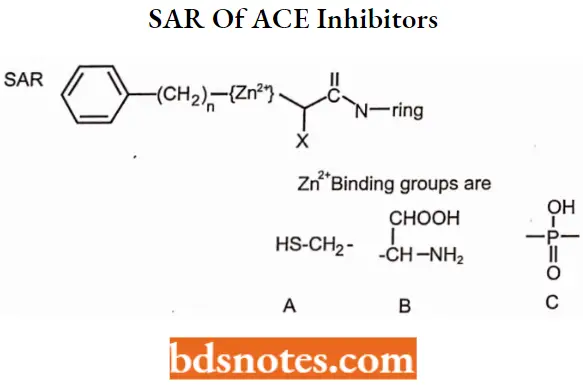
SAR of ACE inhibitors:
“Steps to explain types of antihypertensive agents: ACE inhibitors vs beta-blockers vs diuretics: Q&A guide”
- The N-ring must contain a carboxylic acid to mimic the C-terminal carboxylate of ACE substrates.
- Large hydrophobic heterocyclic rings (i.e., the N-ring) increase potency and alter
pharmacokinetic parameters. - The zinc-binding groups can be either sulfhydryl (A), a carboxylic acid (B), or a phosphinic acid (C).
- The sulfhydryl group shows superior binding to zinc (the side chain mimicking the Phe in carboxylate and phosphinic acid compounds partially compensates for the lack of a sulfhydryl group).
- Sulfhydryl-containing compounds produce a high incidence of skin rash and taste disturbances.
- Sulfhydryl-containing compounds can form dimers and disulfides, which may shorten
the duration of action. - Compounds that bind to zinc through either a carboxylate or phosphinate mimic the peptide hydrolysis transition state and enhance binding.
- Esterification of the carboxylate or phosphinate produces an orally bioavailable prodrug.
- X is usually methyl to mimic the side chain of alanine. Within the dicarboxylate series, when X equals n-butylamine (lysine side chain), this produces a compound that does not require a prodrug for oral activity.
- Optimum activity occurs when the stereochemistry of the inhibitor is consistent with L-amino acid stereochemistry present in normal substrates.
ACE inhibitors MOA: The ACE inhibitors attenuate the effects of the renin-angiotensin system by inhibiting the conversion of angiotensin 1 to angiotensin 2.
- They also inhibit the conversion of angiotensin 1 to angiotensin 3. Inhibitors of ACE increase bradykinin levels that, in turn, stimulate prostaglandin biosynthesis.
- Both of these compounds have been proposed to contribute to the overall action of ACE inhibitors. Additionally, decreased angiotensin 2 levels increase the release of renin and the production of angiotensin 1.
- Because ACE is inhibited, angiotensin 1 is shunted toward the production of angiotensin 1-7 and other peptides. The contribution of these peptides to the overall effect of ACE inhibitors is unknown.
Sulfhydryl-containing inhibitors
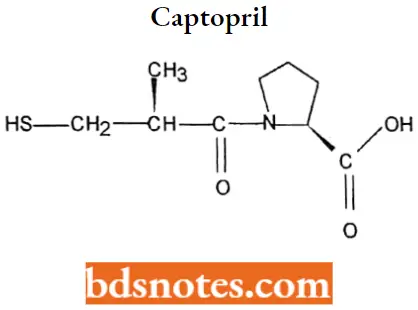
Captopril
“Role of ACE inhibitors in lowering blood pressure: Questions answered”
Captopril IUPAC name: 1-[(2S)-3-mercapto-2-methyl-l-oxopropionyl]proline.
Captopril MOA: It blocks the conversion of angiotensin I (ATI) to angiotensin II (ATII) by inhibiting the converting enzyme. The rational development of captopril as an inhibitor of ACE.
- Captopril also causes an increase in plasma renin activity likely due to a loss of feedback inhibition mediated by ATII on the release of renin and or stimulation of reflex mechanisms via baroreceptors.
- Captopril’s affinity for ACE is approximately 30,000 times greater than that of ATI.
Captopril Metabolism: 25-30% bound to plasma proteins, primarily albumin, half-life is about 2 hours. It undergoes hepatic metabolism.
Major metabolites are captopril-cysteine disulfide and the disulfide dimer of captopril. Metabolites may undergo reversible interconversion.
Captopril Uses: For the treatment of essential or renovascular hypertension (usually administered with other drugs, particularly thiazide diuretics).
- May be used to treat congestive heart failure in combination with other drugs (For Example. cardiac glycosides, diuretics, β-adrenergic blockers).
- May improve survival in patients with left ventricular dysfunction following myocardial infarction. May be used to treat nephropathy, including diabetic nephropathy.
Captopril Adverse effects: Common side effects, skin rashes and taste disturbances (For Example., metallic taste and loss of taste). These side effects usually subsided on dosage reduction or discontinuation of captopril.
Dicarboxylate-containing inhibitors
Enalapril
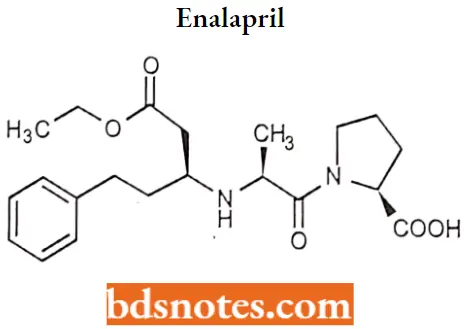
Enalapril IUPAC name: (2SM-[(2S)-2-{[(2S)-l-ethoxy-1-oxo-4-phenylbutan-2-yl]’ amino} propanoyl] pyrrolidine-2-carboxylic acid.
Enalapril MOA: It is a long-acting ACE inhibitor. It requires activation by hydrolysis of its ethyl ester to form the diacid enalaprilat.
Enalapril Metabolism: 50-60% of enalaprilat is bound to plasma proteins. ~ 60% of the absorbed dose is extensively hydrolyzed to enalaprilat, primarily by liver esterases.
The average terminal half-life of enalaprilat is 35-38 hours. The effective half-life following multiple doses is 11-14 hours.
Enalapril Uses: For the treatment of essential or renovascular hypertension and symptomatic congestive heart failure. It may be used alone or in combination with thiazide diuretics.
Enalapril Adverse Effects: The most common adverse effects include hypotension, headache, dizziness, and fatigue.
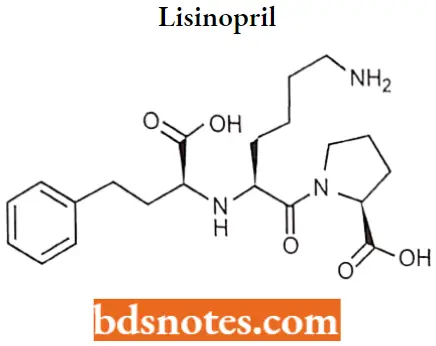
Lisinopril
“Early warning signs of gaps in understanding antihypertensive basics: Common questions”
Lisinopril IUPAC name: (2S)-1-[(2S)-6-amino-2-{[(lS)-1-carboxy-3-phenylpropyl] amino) hexanoyl] pyrrolidine-2-carboxylic acid.
Lisinopril MOA: It is a lysine derivative of enalaprilat, the active metabolite of enalapril. Like all ACE inhibitors, it is an active site-directed inhibitor of the enzyme, with the zinc ion.
Lisinopril Metabolism: Lisinopril does not appear to be bound to serum proteins other than ACE. Does not undergo metabolism, excreted unchanged in urine. The effective half-life of accumulation following multiple dosing is 12 hours.
Lisinopril Uses: For the treatment of hypertension and symptomatic congestive heart failure. May be used in conjunction with thrombolytic agents, aspirin, and or β-blockers to improve survival in hemodynamically stable individuals following myocardial infarction.
May be used to slow the progression of renal disease in hypertensive patients with diabetes mellitus and microalbuminuria or overt nephropathy.
Lisinopril Adverse Effects: The most frequent adverse effects include headache, dizziness, cough, fatigue, and diarrhea.
Benazepril hydrochloride
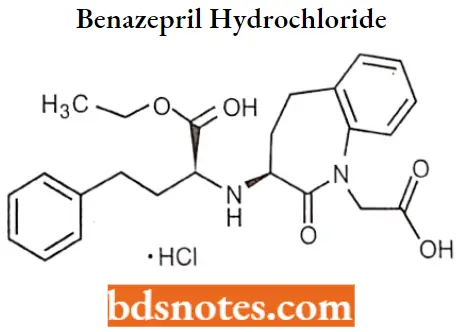
Benazepril hydrochloride IUPAC name: (3S)-3-[[(lS)-1-carbethoxy-3-phenylpropyl]amino]-2,3,4,5tetrahydro-2-oxo-1H-1benzazepine-1-acetic acid 3-ethyl ester hydrochloride.
Benazepril hydrochloride MOA: Benazeprilat, the active metabolite of Benazepril, competes with angiotensin 1 for binding at the angiotensin-converting enzyme, blocking the conversion of angiotensin 1 to angiotensin 2.
- Inhibition of ACE results in decreased plasma angiotensin 2. Angiotensin 2 is a vasoconstrictor and a negative feedback mediator for renin activity.
- Lower concentrations result in a decrease in blood pressure and stimulation of baroreceptor reflex mechanisms, which leads to decreased vasopressor activity and to decreased aldosterone secretion.
- Benazeprilat may also act on kininase 2, an enzyme identical to ACE that degrades the vasodilator bradykinin.
Benazepril hydrochloride Metabolism: Cleavage of the ester group (primarily in the liver) converts benazepril to its active metabolite, benazeprilat.
Benazepril and benazeprilat may be conjugated to glucuronic acid before urinary excretion. Half-life is 10-11 hours.
Benazepril hydrochloride Uses: For the treatment of hypertension. It may be used alone or in combination with thiazide diuretics.
Benazepril hydrochloride Adverse effects: The most common adverse effects include headache, dizziness, fatigue, somnolence, postural dizziness, nausea, and cough.
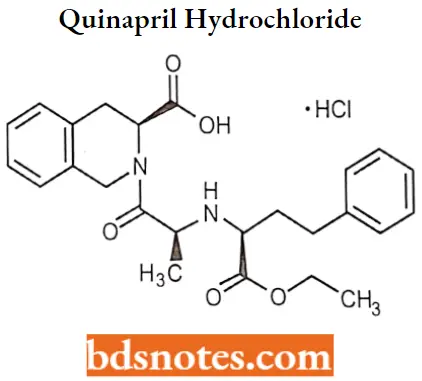
Quinapril hydrochloride
“Asymptomatic vs symptomatic effects of ignoring antihypertensive principles: Q&A”
Quinapril hydrochloride IUFAC name: (S)-[(S)-N-[(S)21-carboxy3-phenylpropyl]alanyl]-l,2,3,4tetrahydro-3-isoquinolinecarboxylic acid 1-ethyl ester hydrochloride.
Quinapril hydrochloride MOA: Quinaprilat, the principal active metabolite of quinapril competes with ATI for binding to ACE and inhibits and enzymatic proteolysis of ATI to ATII.
Decreasing ATII levels in the body decreases blood pressure by inhibiting the pressure effects of ATII. It is more potent than captopril and equipotent to the active form of enalapril.
Quinapril hydrochloride Metabolism: It undergoes hepatic metabolism followed by glucuronide conjugation. Elimination half-life is 2 hours with a prolonged terminal phase of 25 hours.
Quinapril hydrochloride Uses: For the treatment of hypertension and as an adjunct therapy in the treatment of congestive heart failure. May also be used to slow the rate of progression of renal disease in hypertensive individuals with diabetes mellitus and microalbuminuria or overt nephropathy.
Quinapril hydrochloride Adverse effects: The most common adverse effects observed in controlled clinical trials were dizziness, cough, chest pain, dyspnea, fatigue, and nausea or vomiting.
Dicarboxylate-containing inhibitors
Tire phosphinic acid is capable of binding to ACE like enalapril. The interaction of the zinc atom with the phosphinic acid is similar to that seen with sulfhydryl and carboxylate groups.
- A feature unique to this compound is the ability of the phosphinic acid to more truly mimic the ionized, tetrahedral intermediate of peptide hydrolysis.
- Structural modification to investigate more hydrophobic, C-terminal ring systems, similar to that described earlier for the dicarboxylate compounds, led to a 4-cyclohexylproline analog of the original phosphinic acid.
- This compound, fosinoprilat, was more potent than captopril but less potent than enalaprilat.
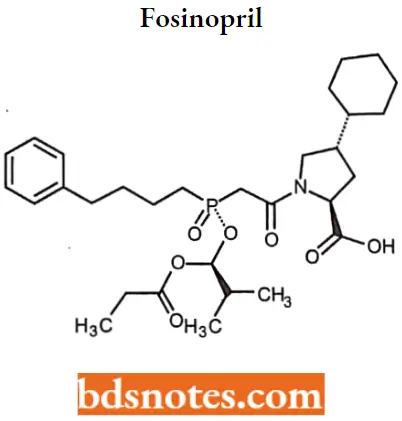
Fosinopril
“Differential applications of ARBs vs ACE inhibitors: Questions answered”
Fosinopril IUPAC name: (4S)-4-cyclohexyl-1-[[[(RS)-1-hydroxy-2-methylpropoxy](4-phenylbutyl) phosphinyl]acetyl]-L-proline.
Fosinopril MOA: Fosinoprilat, the active metabolite of fosinopril, competes with ATI for binding to ACE and inhibits enzymatic proteolysis of ATI to ATIL Decreasing ATII levels in the body decreases blood pressure by inhibiting the pressor effects of ATII.
Fosinopril Metabolism: Since fosinoprilat is not biotransformed after intravenous administration, fosinopril, not fosinoprilat, appears to be the precursor for the glucuronide and p-hydroxy metabolites.
Fosinopril Uses: For treating mild to moderate hypertension, used as an adjunct in treating congestive heart failure, and may be used to slow the rate of progression of renal disease in hypertensive individuals with diabetes microalbuminuria.
Centrally-acting adrenergic drugs
The use of agents that directly affect the peripheral component of the sympathetic nen system represents an important approach to the treatment of hypertension.
- A second approach to modifying sympathetic influence on the cardiovascular system is through inhibition or reduction of CNS control of blood pressure.
- Several widely used medications act by stimulating a2-receptors, which in the CNS reduces sympathetic outflow to the cardiovascular system and produces a hypotensive effect.
Methyldopate hydrochloride
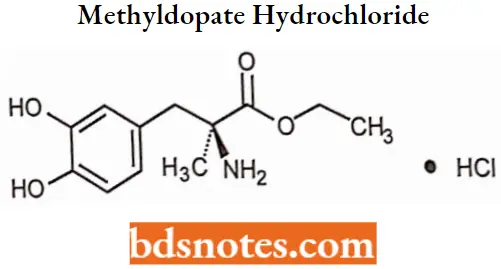
Methyldopate hydrochloride IUPAC name: 3-(3,4-dihydroxyphenyl)-2-methylalanine ethyl ester hydrochloride.
Methyldopate hydrochloride MOA: The current hypothesis concerning the hypotensive activity of methyldopa involves the CNS as the site of action. Methyldopa, on conversion to a-methyl norepinephrine.
- Acts on az-adrenergic receptors to inhibit the release of norepinephrine, resulting in decreased sympathetic outflow from the CNS and activation of parasympathetic outflow.
- A reduction of plasma renin activity can also contribute to the hypotensive action of methyldopa. Postural hypotension and sodium and water retention are also effects related to a reduction in blood pressure.
- If a diuretic is not administered concurrently with methyldopa, tolerance to the antihypertensive effect of the methyldopa during prolonged therapy can result.
Methyldopate hydrochloride Metabolism: If undergoes hepatic, extensively metabolized. The known urinary metabolites are a-methyldopamona-O-sulfate; 3-O-methyl-a-methyldopa; 3,4-dihydroxy phenylacetone; a-methyl dopamine; 3-O-methyl-a-methyldopamine and their conjugates. The plasma half-life of methyldopa is 2 hours.
Methyldopate hydrochloride Uses: Methyldopa is used in the management of moderate to severe hypertension and is reserved for patients who fail to achieve blood pressure goals with stage 2 drugs.
- Methyldopa is also co-administered with diuretics and other classes of antihypertensive drugs, permitting a reduction in the dosage of each drug and minimizing adverse effects while maintaining blood pressure control.
- Methyldopa has been used in the management of hypertension during pregnancy without apparent substantial adverse effects on the fetus and also for the management of pregnancy-induced hypertension.
Methyldopate hydrochloride Adverse effects: The most common adverse effect for methyldopa is drowsiness, which occurs within the first 48 to 72 hours of therapy and can disappear with continued administration of the drug. Sedation commonly recurs when its dosage is increased.
- A decrease in mental acuity, including impaired ability to concentrate, lapses of memory, and difficulty in performing simple calculations, can occur and usually necessitates withdrawal of the drug.
- Patients should be warned that methyldopa can impair their ability to perform activities requiring mental alertness or physical coordination (For Example., operating machinery or driving a motor vehicle).
- Nightmares, mental depression, orthostatic hypotension, and symptoms of cerebrovascular insufficiency can occur during methyldopa therapy and are indications for dosage reduction.
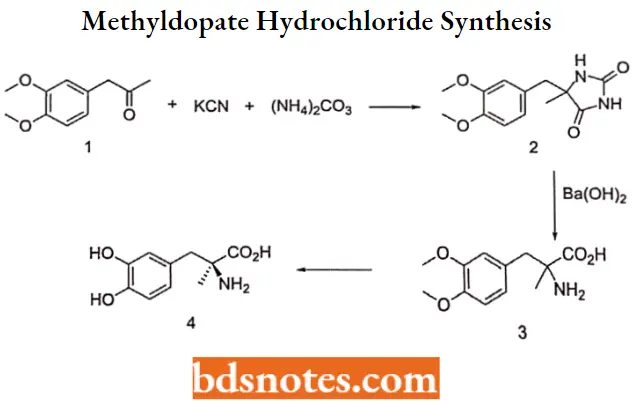
Methyldopate hydrochloride Synthesis:
“Steps to incorporate AI into analyzing antihypertensive drug efficacy: Questions and answers”
Clonidine hydrochloride
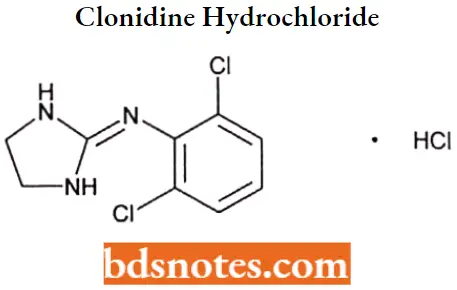
Clonidine hydrochloride IUPAC name: 2[(2,6-dichlorophenyl)imino]imidazolidine monohydrochloride.
Clonidine hydrochloride MOA: Clonidine hydrochloride acts by both peripheral and central mechanisms in the body to affect blood pressure.
- It stimulates the peripheral a-adrenergic receptors to produce vasoconstriction resulting in a brief period of hypertension.
- Clonidine Hydrochloride acts centrally to inhibit the sympathetic tone and cause hypotension that is of much longer duration than the initial hypertensive effect.
- Clonidine hydrochloride also acts centrally to cause bradycardia and to reduce plasma levels of renin.
Clonidine hydrochloride Metabolism: The half-life in humans is about 20 hours. Clonidine hydrochloride is metabolized by the body to form two major metabolites, p-hydroxy clonidine and its glucuronide.
p-Hydroxyclonidine does not cross the blood-brain barrier and has no hypotensive effect in humans.
Clonidine hydrochloride Uses: May be used as an adjunct in the treatment of hypertension, as an epidural infusion as an adjunct treatment in the management of severe cancer pain that is not relieved by opiate analgesics alone.
- For differential diagnosis of pheochromocytoma in hypertensive patients, prophylaxis of vascular migraine headaches, treatment of severe dysmenorrhea, and management of vasomotor symptoms associated with menopause.
- Rapid detoxification in the management of opiate withdrawal, treatment of alcohol withdrawal used in conjunction with benzodiazepines, and management of nicotine dependence.
- Topical use to reduce intraocular pressure in the treatment of open-angle and secondary glaucoma and hemorrhagic glaucoma associated with hypertension, and in the treatment of attention-deficit hyperactivity disorder (ADHD).
Clonidine hydrochloride Adverse effects: It has some sedative properties that are undesirable; it also may cause constipation and dryness of the mouth.
Guanabenz acetate
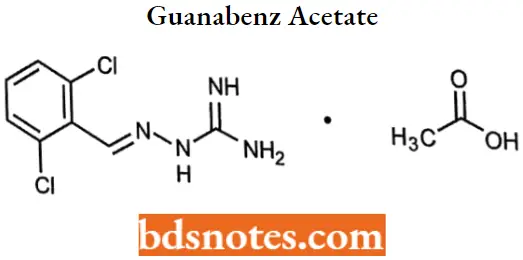
Guanabenz acetate IUPAC name: [(2,6-dichlorobenzylidene)amino]guanidine monoacetate.
Guanabenz acetate MOA: It is a central α2-adrenergic agonist that reduces the release of norepinephrine from the neuron when stimulated. The effect of the drug results in decreased sympathetic tone in the heart, kidneys, and peripheral blood vessels.
Guanabenz acetate Metabolism: Guanabenz is metabolized principally by hydroxylation to its inactive metabolite, 4-hydroxy guanabenz, which is eliminated in the urine as its glucuronide (major) and sulfate conjugates.
Guanabenz acetate Uses: Guanabenz has been used in diabetic patients with hypertension without adverse effects on the control of or therapy for diabetes.
- It has been effective in hypertensive patients with chronic obstructive pulmonary disease, including asthma, chronic bronchitis, or emphysema.
- Guanabenz has been used alone or in combination with naltrexone in the management of opiate withdrawal in patients physically dependent on opiates and undergoing detoxification.
- Guababenz has also been used as an analogic in a limited number of patients with chronic pain.
Guanabenz acetate Adverse effects: Overall, the frequency of adverse effects produced by guanabenz is similar to that produced by clonidine and the other α2-adrenergic agonists, but the incidence is lower.
As with the other centrally active sympatholytic (For Example., Considine), abrupt withdrawal of guanabenz can result in rebound hypertension, but the withdrawal syndrome symptoms appear to be less severe.
Vasodilators
Vasodilator drugs relax the smooth muscle in blood vessels, which causes the vessels to dilate. Dilation of arterial vessels leads to a reduction in systemic vascular resistance, which leads to a fall in arterial blood pressure. Dilation of venous vessels decreases venous blood pressure.
- There are three potential drawbacks to tire use of vasodilators: First, vasodilators can lead to baroreceptor-mediated reflex stimulation of tire heart (increased heart rate and inotropy) from systemic vasodilation and arterial pressure reduction.
- Second, they can impair the normal baroreceptor-mediated reflex vasoconstriction when a person stands up, which can lead to orthostatic hypotension and syncope on standing.
- Third, they can lead to renal retention of sodium and water, increasing blood volume and cardiac output. Antihypertensive agents that produce vasodilation of smooth muscle can be divided into two categories:
Direct acting: include hydralazine hydrochloride, sodium nitroprusside, potassium channel openers, and calcium channel-blocking agents.
Indirect-acting: Include sympatholytic drugs, such as reserpine, a-adrenergic antagonists, such as prazosin hydrochloride, ACE inhibitors, and angiotensin 2 receptor antagonists, such as sralysin.
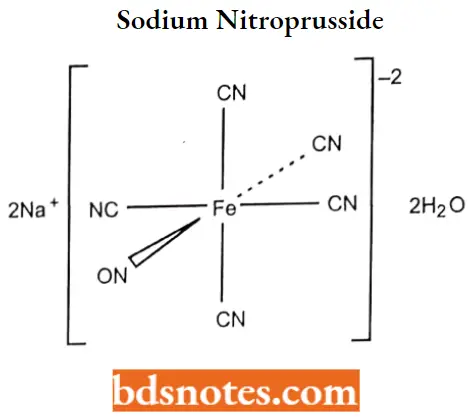
Sodium nitroprusside
“Role of digital tools in improving precision with antihypertensive tracking: FAQs explained”
Sodium nitroprusside IUPAC name: pentacyano(nitroso) ironside.
Sodium nitroprusside MOA: One molecule of sodium nitroprusside is metabolized by combination with hemoglobin to produce one molecule of cyanmethemoglobin and four CN– ions; methemoglobin.
- Obtained from hemoglobin, can sequester cyanide as cyanmethemoglobin; thiosulfate reacts with cyanide to produce thiocyanate; thiocyanate is eliminated in the urine; cyanide not otherwise removed binds to cytochromes.
- Cyanide ion is normally found in serum; it is derived from dietary substrates and tobacco smoke.
- Cyanide binds avidly (but reversibly) to ferric ion (Fe+++), most body stores of which are found in erythrocyte methemoglobin (metHgb) and mitochondrial cytochromes.
- When CN is infused or generated within the bloodstream, essentially all of it is bound to methemoglobin until intraerythrocytic methemoglobin has been saturated.
- Once activated to NO, it activates guanylate cyclase in vascular smooth muscle and increases intracellular production of cGMP.
- cGMP stimulates calcium movement from the cytoplasm to the endoplasmic reticulum and reduces calcium available to bind with calmodulin. This eventually leads to vascular smooth muscle relaxation and vessel dilatation.
Sodium nitroprusside Metabolism: Sodium nitroprusside undergoes a redox reaction that releases cyanide. The cyanide that is produced is rapidly converted into thiocyanate in the liver by the enzyme thiosulfate sulfotransferase (rhodanase) and is excreted in the urine.
The rate-limiting step in the conversion of cyanide to thiocyanate is the availability of sulfur donors, especially thiosulfate. The elimination half-life of thiocyanate is 2.7 to 7.0 days.
Sodium nitroprusside Uses: For immediate reduction of blood pressure of patients in hypertensive crises, reduce bleeding during surgery, and for the treatment of acute congestive heart failure
Sodium nitroprusside Adverse effects: The most clinically important adverse effects of sodium nitroprusside are profound hypotension and accumulation of cyanide and thiocyanate can accumulate in the blood of patients receiving sodium nitroprusside therapy, especially in those with impaired renal function.
- Thiocyanate is mildly neurotoxic at serum concentrations of 60 mg/mL and can be life-threatening at concentrations of 200 mg/mL. Other adverse effects of thiocyanate include inhibition of both the uptake and binding of iodine, producing symptoms of hypothyroidism.
- Sodium nitroprusside can bind to vitamin B12, interfering with its distribution and metabolism, and it should be used with caution in patients having low plasma vitamin B12 concentrations. Excess cyanide can also bind to hemoglobin, producing methemoglobinemia.
Hydralazine hydrochloride
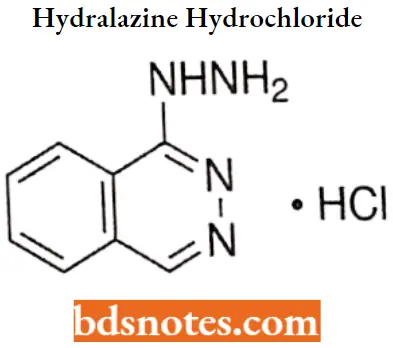
Hydralazine hydrochloride IUPAC name: 1-hydrazinophthalazine monohydrochloride.
Hydralazine hydrochloride MOA: The only drug in this group, hydralazine, does not fit neatly into the other mechanistic classes, in part because its mechanism of action is not entirely clear.
- It seems to have multiple, direct effects on the vascular smooth muscles (VSM). First, it causes smooth muscle hyperpolarization, quite likely through the opening of K+ channels.
- Activation therefore increases the efflux of potassium ions from the cells, causing hyperpolarization of VSM cells and, thus, prolonging the opening of the potassium channel and sustaining a greater vasodilation on arterioles than on veins.
- It can also inhibit the second messenger, the IP3-induced release of calcium from the smooth muscle SR (the PIP2 signal transduction pathway).
- Finally, hydralazine stimulates the formation of NO by the vascular endothelium, leading to cGMP-mediated vasodilation. The arterial vasodilator action of hydralazine reduces systemic vascular resistance and arterial pressure.
- Diastolic blood pressure is usually decreased more than systolic pressure is. The hydralazine-induced decrease in blood pressure and peripheral resistance causes a reflex response, which is accompanied by an increased heart rate.
Hydralazine hydrochloride Metabolism: Hydralazine hydrochloride undergoes benzylic oxidation, glucuronide formation, and N-acetylation by the microsomal enzymes in the tissues.
- Acetylation appears to be a major determinant of the rate of hepatic removal of the drug from the blood and, therefore, of systemic availability.
- Rapid acetylation results in a highly hepatic extraction ratio from blood and greater first-pass elimination.
- The drug is excreted rapidly by the kidneys, and within 24 hours, 75% of the total amount administered appears in the urine as metabolites or unchanged drugs.
Hydralazine hydrochloride Uses: For the treatment of essential hypertension, alone or as an adjunct. Also for the management of severe hypertension when the drug cannot be given orally or when blood pressure must be lowered immediately, congestive heart failure.
Hydralazine hydrochloride Adverse effects: Patients who engage in potentially hazardous activities, such as operating machinery or driving motor vehicles, should be warned about possible faintness, dizziness, or weakness.
Hydralazine should be used with caution in patients with cerebrovascular accidents or with severe renal damage.
Potassium channel opener
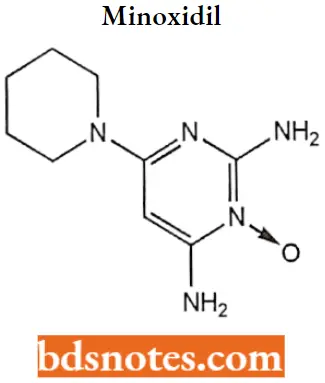
Minoxidil
“Early warning signs of outdated methods in antihypertensive studies: Common questions”
Minoxidil IUPAC name: 2,4-diamino-6-piperidinopyrimidine-3-oxide.
Minoxidil MOA: Potassium channel openers are drugs that activate (i.e., open) ATP-sensitive K+ channels in the VSM.
- By opening these potassium channels, there is increased efflux of potassium ions from the cells, causing hyperpolarization of VSM, which closes the voltage-gated calcium channels and, thereby, decreases intracellular calcium.
- With less calcium available to combine with calmodulin, there is less activation of MLCK (Myosin light chain kinase) and phosphorylation of myosin light chains. This leads to relaxation and vasodilation.
Minoxidil Metabolism: Minoxidil is absorbed from the GI tract and is metabolized to its active sulfate metabolite. Plasma concentrations for minoxidil sulfate peak within 1 hour and then decline rapidly.
- Following an oral dose of minoxidil, its hypotensive effect begins in 30 minutes, is maximal in 2 to 8 hours, and persists for approximately 2 to 5 days.
- The delayed onset of the hypotensive effect of minoxidil is attributed to its metabolism to its active metabolite. The drug is not bound to plasma proteins.
- The major metabolite for minoxidil is its N-Oglucuronide, which, unlike the sulfate metabolite, is inactive as a hypotensive agent.
- Approximately 10% to 20% of an oral dose of minoxidil is metabolized to its active metabolite, minoxidil O-sulfate, and approximately 20% of minoxidil is excreted unchanged.
Minoxidil Uses: For the treatment of severe hypertension and in the topical treatment (regrowth) of androgenic alopecia in males and females and stabilization of hair loss in patients with androgenic alopecia.
Minoxidil Adverse effects: Adverse effects include cardiovascular effects associated with hypotension such as sudden weight gain, rapid heartbeat, faintness, or dizziness.
Diazoxide
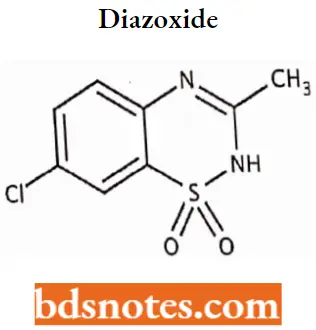
Diazoxide IUPAC name: 7-chloro-3-methyl-2H-1,2,4-benzothiadiazine 1,1-dioxide.
Diazoxide MOA: Diazoxide reduces peripheral vascular resistance and blood pressure by a direct vasodilating effect on the VSM with a mechanism similar to that described for minoxidil by activating (opening) the ATP-modulated potassium channel.
- Thus, diazoxide prolongs the opening of the potassium channel, sustaining greater vasodilation on arterioles than on veins.
- Diazoxide increases blood glucose concentration (diazoxide-induced hyperglycemia) by several different mechanisms: by inhibiting pancreatic insulin secretion, by stimulating release of catecholamines, or by increasing hepatic release of glucose
Diazoxide Metabolism: It undergoes hepatic metabolism. Its half-life is 28+8.3 hours in normal adults in patients with renal impairment, the half-life is prolonged.
Approximately 90% of the diazoxide in the blood is bound to plasma proteins. approximately 20% to 50% of diazoxide of the 3-methyl group to its 3-hydroxymethyl-and 3-carboxyl-metabolites.
Diazoxide Uses: Used parentally to treat hypertensive emergencies. Also used to treat hypoglycemia secondary to insulinoma.
Diazoxide Adverse effects: Diazoxide causes sodium and water retention and decreased urinary output, which can result in expansion of plasma and extracellular fluid volume, edema, and congestive heart failure, especially during prolonged administration.
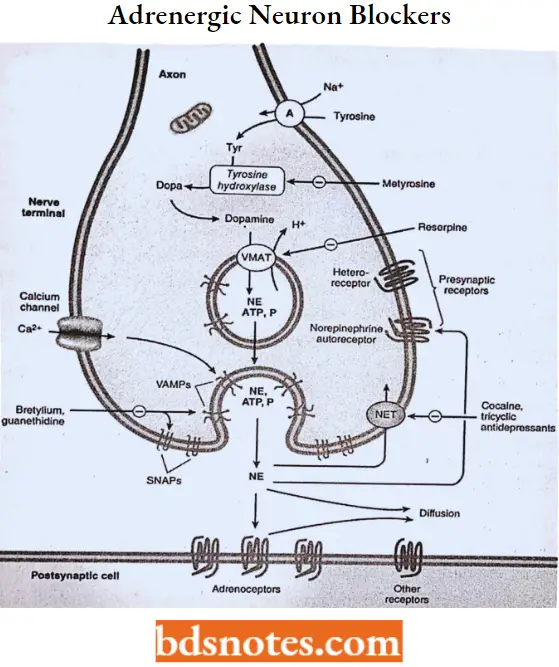
Adrenergic Neuron-Blocking Agents
Drugs that reduce blood pressure by depressing the activity of the sympathetic nervous system have been used as effective agents in the treatment of hypertension. This can be accomplished in several ways:
- Depleting the stores of neurotransmitters,
- Reducing the number of impulses traveling in sympathetic nerves,
- Antagonizing the actions of the neurotransmitter on the effector cells, and
- Inhibiting neurotransmitter release.
“Differential applications of traditional vs cutting-edge antihypertensive techniques: Questions answered”
Guanethidine mono sulphate
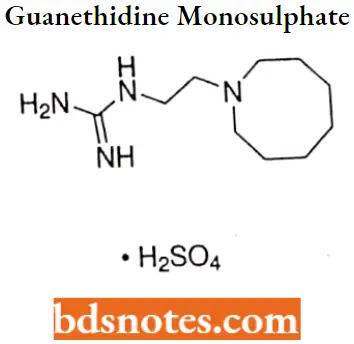
Guanethidine monosulphate IUPAC name: [2-(hexahydro-1 (2H)-azocinyl)ethyl]guanidine sulfate.
Guanethidine monosulphate MOA: Guanethidine is an adrenergic neuronal blocking agent that produces a selective block of peripheral sympathetic pathways by replacing and depleting norepinephrine stores from adrenergic nerve endings, but not from the adrenal medulla.
- It prevents the release of norepinephrine from adrenergic nerve endings in response to sympathetic nerve stimulation. The chronic administration of guanethidine results in an increased sensitivity of these effector cells to catecholamines.
- Following the oral administration of usual doses of guanethidine, depletion of the catecholamine stores from adrenergic nerve endings occurs at a very slow rate, producing a more gradual and prolonged fall in systolic blood pressure than in diastolic pressure.
Guanethidine monosulphate Metabolism: Guanethidine is incompletely absorbed from the GI tract and is metabolized in the liver to several metabolites, including guanethidine N-oxide from flavin mononucleotide.
These metabolites of guanethidine are excreted in the urine and have less than 10% of its hypotensive activity. Guanethidine accumulates in the neurons with an elimination half-life of 5 days.
Guanethidine monosulphate Uses: Guanethidine is used in the management of moderate to severe hypertension and in the management of renal hypertension.
It has been administered as ophthalmic drops in the treatment of chronic open-angle glaucoma and for endocrine ophthalmopathy, ophthalmoplegia, lid lag, and lid retraction.
Guanethidine mono sulfate Adverse effects: Adverse effects of guanethidine frequently are dose-related, including dizziness, weakness, lassitude, and syncope, and resulting from postural or post-exercise hypotension.
- A hot environment(i.e., a hot bath) can aggravate postural hypotension. Patients should be warned about possible orthostatic hypotension and about the effect of rapid postural changes on blood pressure (For Example arising in the morning) that can cause fainting.
- Especially during the initial period of dosage adjustment. Sodium retention (edema) is usually controlled by the co-administration of a diuretic.
Reserpine
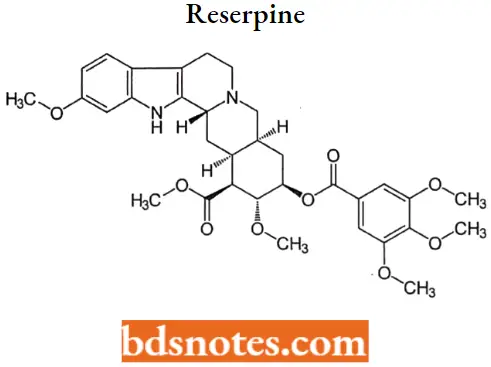
Reserpine MOA: Reserpine acts to replace and deplete the adrenergic neurons of their stores of norepinephrine by inhibiting the active transport Mg-ATPase responsible for sequestering norepinephrine and dopamine within the storage vesicles.
- The norepinephrine and dopamine that are not sequestered in vesicles are destroyed by MAO. As a result, the storage vesicles contain little neurotransmitter, adrenergic transmission is dramatically inhibited, and sympathetic tone is decreased, leading to vasodilation.
- Reserpine has the same effect on epinephrine storage in the adrenal medulla. Reserpine readily enters the CNS, where it also depletes the stores of norepinephrine and serotonin. The CNS neurotransmitter depletion led to the use of reserpine in treating certain mental illnesses.
Reserpine Metabolism: The elimination of reserpine appears to be biphasic, with a plasma half-life averaging 4.5 hours during the first phase and approximately 11.3 days during the second phase.
Reserpine is metabolized to unidentified inactive compounds. Unchanged reserpine and its metabolites are excreted slowly in urine and feces, with an average of 60% reserpine recovered in feces within 96 hours after oral administration of 0.25 mg of radiolabeled reserpine.
Reserpine Uses: Reserpine has been used in the management of mild to moderate hypertension, but because of very significant CNS adverse effects and its cumulative action in the adrenergic neurons, reserpine is rarely used.
Reserpine has been used in the symptomatic treatment of agitated psychotic states, such as schizophrenic disorders, although other antipsychotic agents have generally replaced reserpine and alkaloids.
Reserpine Adverse effects: The common adverse CNS effects of reserpine include drowsiness, fatigue, and lethargy.
- Mental depression is one of the most serious potential adverse effects for reserpine, which can be severe enough to require hospitalization or result in suicide attempts.
- Reserpine-induced depression can persist for several months after the drug is discontinued.
“Can bioinformatics revolutionize antihypertensive classification? FAQs provided”
Antihypertensive Agents Multiple Choice Questions And Answers
Question 1. Clinical use of clonidine:
- Analgesic
- Antihypertensive
- Both
- Neither
Answer: 3. Both
Question 2. β-blockers adverse effects:
- Bronchoconstriction
- Bradycardia
- Depression
- Mask hypoglycemia
Answer: 1. Bronchoconstriction
Question 3. Name of the drug contraindicated with digoxin.
- Captopril
- Atenolol
- Furosemide
- Losartan
Answer: 3. Furosemide
Question 4. Which one will cause heart failure when combined with verapamil?
- Captopril
- Atenolol
- Aliskiren
- Furosemide
Answer: 2. Atenolol
Question 5. This hypertension drug is the first choice for diabetic and renal failure pts.
- K sparing diuretics
- ACE inhibitors
- Loop diuretics
- Calcium channel blockers
Answer: 3. Loop diuretics
Question 6. Impotence is most commonly caused by which antihypertensive agent.
- Calcium channel blocker
- ACE inhibitors
- ATI receptor antagonist
- Beta-blockers
Answer: 4. Beta-blockers
“Asymptomatic vs symptomatic effects of ignoring new trends in antihypertensive research: Answered”
Question 7. Which drug inactivates bradykinin?
- Lisinopril
- Losartan
- Gaunethedin
- Reserpine
Answer: 1. Lisinopril
Question 8. Hydralazine works on hypertension by:
- Decrease in sympathetic response
- Peripheral vasodilation
- Directly inhibits renin
- Block Ca++ from stimulating muscle wall
Answer: 2. Peripheral vasodilation
Antihypertensive Agents Short Questions And Answers
Question 1. Give the classification of ACE inhibitors.
Answer:
- Sulfhydryl-containing inhibitors For Example. Captopril.
- Dicarboxylate-containing inhibitors For Example. Enalapril, lisinopril.
- Phosphonate-containing inhibitors For Example. Fosinopril.
Question 2. Write the uses of captopril.
Answer:
For the treatment of essential or renovascular hypertension (usually administered with other drugs, particularly thiazide diuretics).
- May be used to treat congestive heart failure in combination with other drugs (For Example. cardiac glycosides, diuretics, p-adrenergic blockers).
- May improve survival in patients with left ventricular dysfunction following myocardial infarction. May be used to treat nephropathy, including diabetic nephropathy.
Question 3. Give MOA and metabolism of Benazepril.
Answer:
MOA– Benazeprilat, the active metabolite of Benazepril, competes with angiotensin 1 for binding at the angiotensin-converting enzyme, blocking the conversion of angiotensin 1 to angiotensin 2. Inhibition of ACE results in decreased plasma angiotensin 2.
- As angiotensin 2 is a vasoconstrictor and a negative feedback mediator for renin activity, lower concentrations result in a decrease in blood pressure and stimulation of baroreceptor reflex mechanisms, which leads to decreased vasopressor activity and decreased aldosterone secretion.
- Benazeprilat may also act on kininase 2, an enzyme identical to ACE that degrades the vasodilator bradykinin.
- Metabolism– Cleavage of the ester group (primarily in the liver) converts benazepril to its active metabolite, benazeprilat. Benazepril and benazeprilat may be conjugated to glucuronic acid before urinary excretion. Half-life is 10-11 hours.
Question 4. Draw the structure and IUFAC name of Clonidine hydrochloride.
Answer:
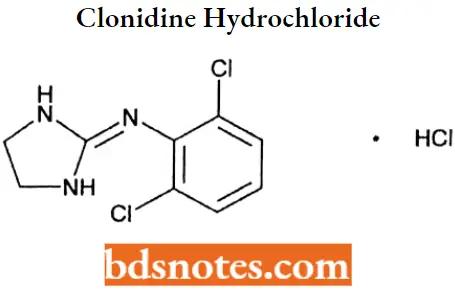
IUPAC name: 2[(2,6-dichlorophenyl)imino]imidazolidine monohydrochloride.
Question 5. What are the adverse effects related to vasodilators?
Answer:
- Vasodilators can lead to baroreceptor-mediated reflex stimulation of the heart (increased heart rate and inotropy) from systemic vasodilation and arterial pressure reduction.
- They can impair the normal baroreceptor-mediated reflex vasoconstriction when a person stands up, which can lead to orthostatic hypotension and syncope on standing.
- They can lead to renal retention of sodium and water, increasing blood volume and cardiac output.
Leave a Reply