Anti-Arrhythmic Agents
Arrhythmia
Arrhythmia is an alteration in the normal sequence of electrical impulse rhythm that leads to the contraction of the tire myocardium.
- It is manifested as an abnormality in the rate, in the site from which the impulses originate, or in tire conduction through the myocardium.
- The rhythm of the heart normally is determined by a pacemaker site called the SA node, which consists of specialized cells that undergo spontaneous generation of action potentials at a rate of 100 to 110 action potentials (“beats”) per minute.
- This intrinsic rhythm is strongly influenced by the vagus nerve, overcoming the sympathetic system at rest. This “vagal tone” brings the resting heart rate down to a normal sinus rhythm of 60 to 100 beats per minute.
Sinus rates below this range are termed “sinus bradycardia,” and sinus rates above the tins range are termed “sinus tachycardia.” The sinus rhythm normally controls both atrial and ventricular rhythm.
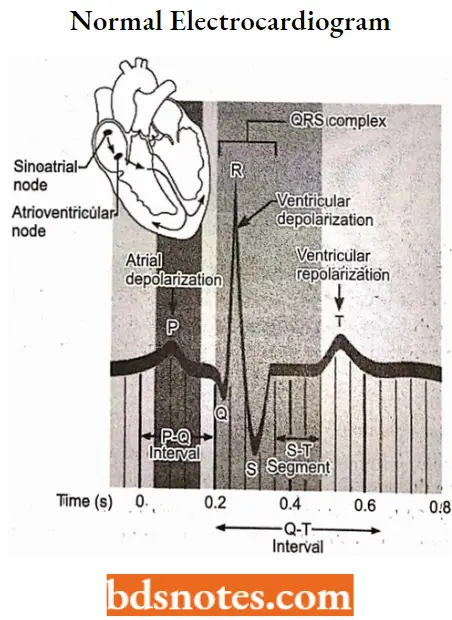
- Action potentials generated by the SA node spread throughout tire atria, depolarizing this tissue and causing atrial contraction. The impulse then travels into the ventricles via the AV node.
- Specialized conduction pathways within the ventricle rapidly conduct the wave of depolarization throughout the ventricles to elicit ventricular contraction.
- Therefore, Normal cardiac rhythm is controlled by the pacemaker activity of the SA node.
- Abnormal or irregular cardiac rhythms (heartbeats) can occur when the SA node fails to function normally, when other pacemaker sites (For Example., ectopic pacemakers) trigger depolarization, or when dysfunction occurs along the normal conduction pathways.
Normal Physiologic Action
Normal cardiac contractions largely are a function of the action of a single atrial pacemaker, a fast and usually uniform conduction in predictable pathways and a normal duration of the action potential and refractory period.
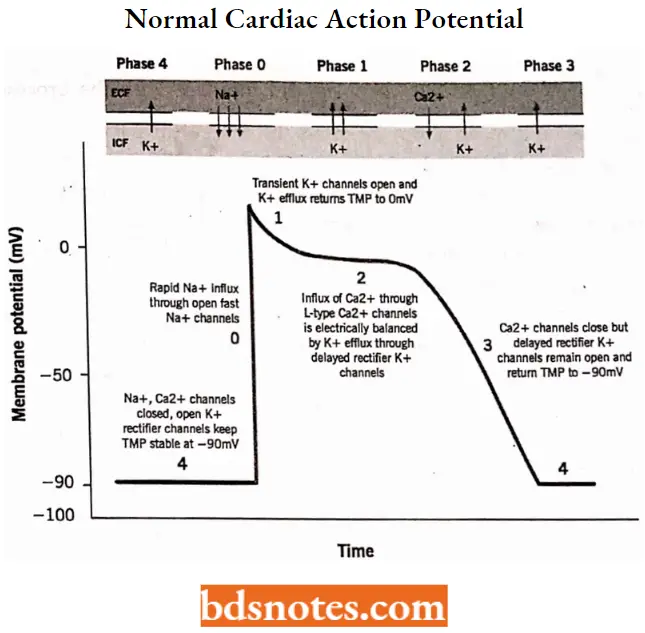
- The depicts a normal cardiac action potential from a Purkinje fiber. The resting cell has a membrane potential of approximately -90 mV, with the inside of the cell being electronegative relative to the outside of the cell.
- This is termed the “transmembrane resting potential.” On excitation, the transmembrane potential reverses, and the inside of the membrane rapidly becomes positive concerning the outside.
- On recovery from excitation, the resting potential is restored. These changes have been divided into five phases: Phase 0 represents depolarization and reversal of the transmembrane potential, phases 1 to 3 represent different stages of repolarization, and phase 4 represents the resting potential.
Read and Learn More Medicinal Chemistry II Notes
During phase 0, which is also referred to as rapid depolarization, the permeability of the membrane for sodium ions increases, and sodium rapidly enters the cell, causing it to become depolarized.
- Phase 1 results from the ionic shift, which creates an electrochemical and concentration gradient that reduces the rate of sodium influx but favors the influx of chloride and efflux of potassium.
- Phase 2, the plateau phase, results from the slow inward movement of calcium, which is triggered by the rapid inward movement of sodium in phase 0. During this time, there is also an efflux of potassium that balances the influx of calcium, thus resulting in little or no change in membrane potential.
- Phase 3 is initiated by a slowing of the calcium influx coupled with a continued efflux of potassium. This continued efflux of potassium from the cell restores the membrane potential to normal resting potential levels.
- During phase 4, the Na+, K+-ATPase pump restores the ions to their proper local concentrations.
The action potential is a coordinated sequence of ion movements in which sodium initially enters the cell, followed by a calcium influx, and finally, a potassium efflux returns the cell to its resting state. Several antiarrhythmic agents exert their effects by altering these ion fluxes.
Antiarrhythmic Agents
It is widely accepted that most currently available antiarrhythmic drugs can be classified into four categories, which are grouped based on their effects on the cardiac action potential and, consequently, on the electrophysiologic properties of the heart.
Antiarrhythmic Agents Classification
- Class 1 Membrane depressant drugs (Na+ Channel blockers)
- Class 1 (1A) – For Example. Quinidine, Procainamide, Disopyramide.
- Class 1 (1B) – For Example. Lidocaine, Phenytoin, Tocainide, Mexiletine.
- Class 1 (1C) – For Example. Encainide, lorcainide, Moricizine.
- Class 2 β- Adrenergic blocking agents
- For Example. Propranolol.
- Class 3 Repolarization prolongation (K+ Channel blockers)
- For Example. Amiodarone, Bretylium, Sotalol.
- Class 4 Ca++ Channel blockers
- For Example. Verapamil, Diltiazem.
Class 1 membrane depressant drug (Na+ channel blockers)
- Drugs in this class act on the fast Na+ channels and interfere with the process by which the depolarizing charge is transferred across the membrane.
- It is assumed that these drugs bind to the Na+ channel and block its function, preventing Na+ conductance as long as the drug is bound.
Class 1 antiarrhythmic drugs can be subdivided based on the relative ease with which they dissociate from the Na+ ion channel. The action of class 1 local anesthetic-type antiarrhythmic drugs is pH-dependent and may vary with each drug.
- Class 1A-Quinidine, procainamide, and disopyramide are drugs that have an intermediate rate of dissociation from Na+ channels.
- Class 1B-Lidocaine, tocainide, and mexiletine dissociate rapidly from the Na+ channels and thus have the lowest potency as sodium channel blockers. They produce little, if any, change in action potential duration.
- Class 1C– Encainide, lorcainide, and moricizine, are the most potent sodium channel blocking agents. They slowly dissociate from the Na+ channel, causing a slowing of the conduction time of the impulse through the heart.
Class 1A
Quinidine sulphate
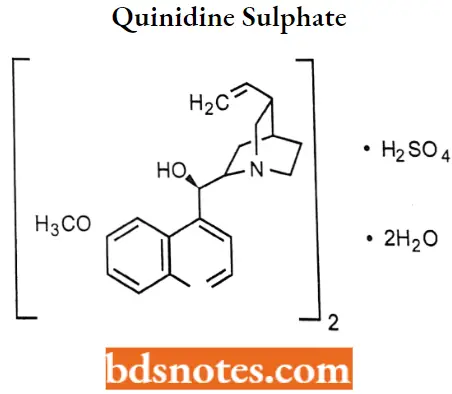
Quinidine sulphate IUPAC name: (5-ethenyl-l-azabicyclo[2.2.2] octan-2-yl) -(6-methoxyquinolin-4-yl) methanol; sulphuric add.
Quinidine sulfate MOA: It reduces Na+ current by binding the open ion channels. The decreased Na+ entry into the myocardial cell depresses phase 4 diastolic depolarization and shifts the intracellular threshold potential toward zero.
- These combined actions diminish the spontaneous frequency of pacemaker tissues, depress the automaticity of ectopic foci, and, to a lesser extent, reduce impulse formation in the SA node.
- This last action results in bradycardia. During the spike action potential, quinidine’s phase decreases transmembrane permeability to the passive influx of Na+, thus slowing the process of phase 0 depolarization, which decreases conduction velocity.
- This is shown as a prolongation of the QRS complex of electrocardiograms. Quinidine sulfate also prolongs action potential duration, which results in a proportionate increase in the QT interval.
Quinidine sulfate Metabolism: Quinidine is metabolized primarily in the liver by hydroxylation, and a small amount is excreted by the liver.
- The metabolites are hydroxylated derivatives at either the quinoline ring through first-pass O-demethylation or at the quinuclidine ring through oxidation of the vinyl group.
- These metabolites possess only about one-third of the activity of quinidine. Metabolites are 3- hydroxyquinidine and quinidine-N-oxide.
Quinidine sulphate Uses: It is used to treat supraventricular and ventricular ectopic arrhythmias, such as atrial and ventricular premature beats, atrial and ventricular tachycardia, atrial flutter, and atrial fibrillation.
Quinidine sulphate Adverse effects: The most frequent adverse effects associated with quinidine therapy are gastrointestinal disturbances, such as nausea, diarrhea, and vomiting.
Procainamide hydrochloride
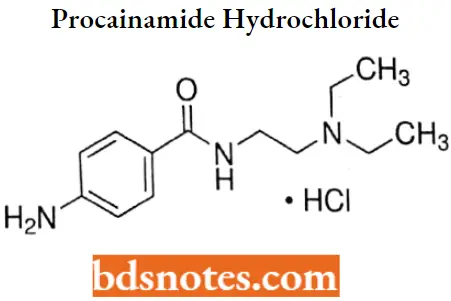
Procainamide hydrochloride IUPAC name: p-amino-N-[2-(diethylamino)ethyl]benzamide monohydrochloride
Procainamide hydrochloride MOA: Procainamide is a sodium channel blocker. It stabilizes the neuronal membrane by inhibiting the ionic fluxes required for the initiation and conduction of impulses thereby affecting local anesthetic action.
Procainamide hydrochloride Metabolism: Procainamide hydrochloride is metabolized through the action of N-acetyltransferase The product of enzymatic metabolism of procainamide hydrochloride is N-acetylProcainamide (NAPA), which possesses only 25% of the activity of the parent compound.
A study of the disposition of procainamide hydrochloride showed that 50% of the drug was exerted unchanged in the urine, with 7% to 24% recovered as NAPA.
Procainamide hydrochloride Uses: For the treatment of life-threatening ventricular arrhythmias.
Procainamide hydrochloride Adverse effects: If therapy is continued, a drug-induced lupus syndrome. These adverse effects which are attributed to the aromatic amino group, are observed more frequently and more rapidly m “slow acetylators.”
As a rule, the symptoms associated with procainamide-induced lupus syndrome subside fairly rapidly after the drug is discontinued.
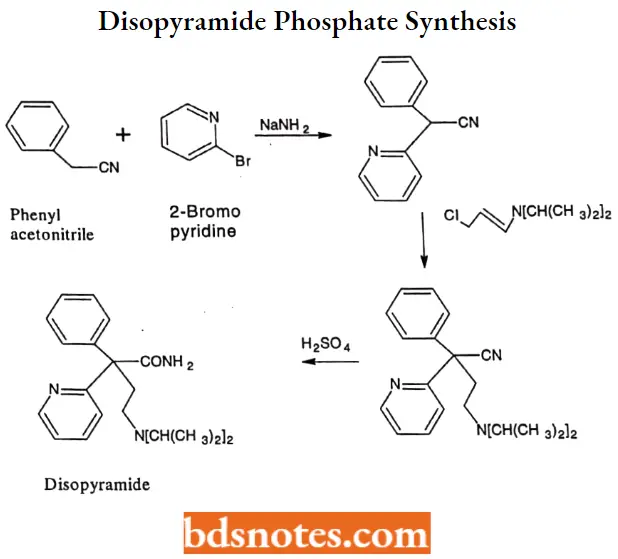
Disopyramide phosphate
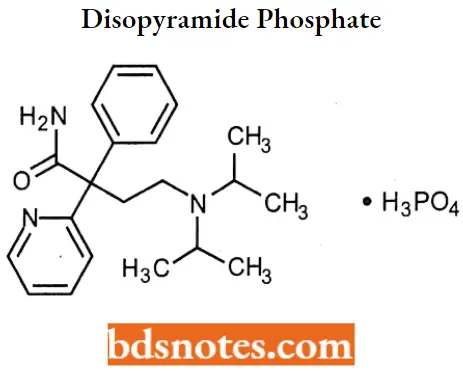
Disopyramide phosphate IUPAC name: α-[2(diisopropylamino)ethyl]- α -phenyl-2-pyridineacetamide phosphate.
Disopyramide phosphate MOA: Disopyramide is a type 1A antiarrhythmic drug (i.e. similar to procainamide and quinidine). It inhibits the fast sodium channels.
- In animal studies, Disopyramide decreases the rate of diastolic depolarization (phase 4) in cells with augmented automaticity, decreases the upstroke velocity (phase 0) and increases the action potential duration of normal cardiac cells.
- Decreases the disparity in refractoriness between infarcted and adjacent normally perfused myocardium, and has no effect on alpha- or beta-adrenergic receptors.
Disopyramide phosphate Metabolism: The drug undergoes hepatic CYP3A4 metabolism, principally to the corresponding N-dealkylated metabolite. This metabolite retains approximately half the antiarrhythmic activity of disopyramide and is also subject to renal excretion.
Disopyramide phosphate Uses: For the treatment of documented ventricular arrhythmias, such as sustained ventricular tachycardia, ventricular pre-excitation, and cardiac dysrhythmias.
Disopyramide phosphate Adverse effects: Adverse effects are primarily anticholinergic and include dry mouth, blurred vision constipation, and urinary retention.
Disopyramide phosphate Synthesis:
Lidocaine hydrochloride
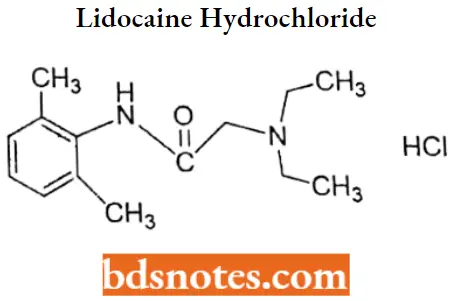
Lidocaine hydrochloride IUPAC name: 2-(diethylamino)-2′,6′-acetoxylidide monohydrochloride.
Lidocaine hydrochloride MOA: Lidocaine stabilizes the neuronal membrane by inhibiting the ionic fluxes required for the initiation and conduction of impulses thereby affecting local anesthetic action.
- Lidocaine alters signal conduction in neurons by blocking the fast voltage-gated sodium (Na+) channels in the neuronal cell membrane that are responsible for signal propagation.
- With sufficient blockage the membrane of the postsynaptic neuron will not depolarize and will thus fail to transmit an action potential.
- This creates the anesthetic effect by not merely preventing pain signals from propagating to the brain but by aborting their birth in the first place.
Lidocaine hydrochloride Metabolism: Hepatic metabolism is rapid (plasma half-life, ~15 to 30 minutes) and primarily involves Ndeethylation to yield monoethylglycinexylide, followed by amidase-catalyzed hydrolysis into Nethylglycine and 2,6-dimethylaniline (2,6-xylidine).
Lidocaine hydrochloride Uses: For the production of local or regional anesthesia by infiltration techniques such as percutaneous injection and intravenous regional anesthesia by peripheral nerve block techniques such as brachial plexus and intercostal and by central neural techniques such as lumbar and caudal epidural blocks.
Lidocaine hydrochloride Adverse effects: The adverse effects of lidocaine include emetic and convulsant properties that predominantly involve the central nervous system and heart. The central nervous system effects can begin with dizziness and paresthesia and, in severe cases, ultimately lead to epileptic seizures.
Phenytoin sodium
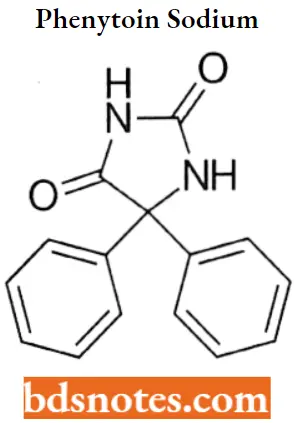
Phenytoin sodium IUPAC name: 5,5diphenyl-2,4-imidazolidinedione.
Phenytoin sodium MOA: Phenytoin acts on sodium channels on the neuronal cell membrane, limiting the spread of seizure activity and reducing seizure propagation.
- By promoting sodium efflux from neurons, phenytoin tends to stabilize the threshold against hyperexcitability caused by excessive stimulation or environmental changes capable of reducing membrane sodium gradient.
- This includes the reduction of post-tetanic potentiation at synapses. Loss of post-tetanic potentiation prevents cortical seizure foci from detonating adjacent cortical areas.
Phenytoin sodium Metabolism: Phenytoin metabolism is relatively slow and predominantly involves aromatic hydroxylation by the CYP2C family of enzymes to p-hydroxylated inactive metabolites.
- Phenytoin also induces its metabolism and is subject to large inter-individual variability.
- The major metabolite, 5-phydroxyphenyl5-phenylhydantoin, accounts for approximately 75% of a dose. This metabolite is excreted through the kidney as the b-glucuronide conjugate.
Phenytoin sodium Uses: For the control of generalized tonic-clonic (grand mal) and complex partial (psychomotor, temporal lobe) seizures and prevention and treatment of seizures occurring during or following neurosurgery.
Tocainide hydrochloride
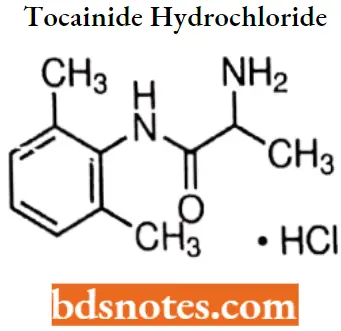
Tocainide hydrochloride IUPAC name: 2-amino-2′,6,-propionoxyxylidide hydrochloride.
Tocainide hydrochloride MOA: Tocainide acts on sodium channels on the neuronal cell membrane, limiting the spread of seizure activity and reducing seizure propagation.
Tocainide binds preferentially to the inactive state of the sodium channels. The antiarrhythmic actions are mediated through effects on sodium channels in Purkinje fibers.
Tocainide hydrochloride Metabolism: Negligible first pass hepatic degradation. Tocainide hydrochloride is hydrolyzed like that of lidocaine. No active metabolites have been found.
Tocainide hydrochloride Uses: For the treatment of documented ventricular arrhythmias, such as sustained ventricular tachycardia, those, in the judgment of the physician, are life-threatening.
Tocainide hydrochloride Adverse effects: Adverse effects associated with tocainide are like those observed with lidocaine —specifically, gastrointestinal disturbances and central nervous system effects.
Mexiletine hydrochloride
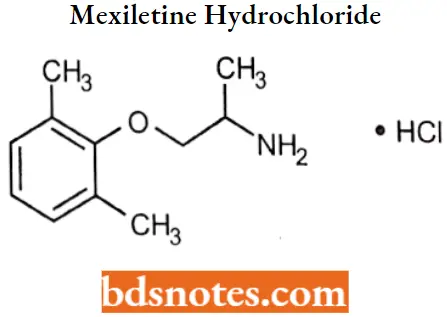
Mexiletine hydrochloride IUPAC name: 1-methyl-2-(2,6-xylyloxy)ethylamine hydrochloride.
Mexiletine hydrochloride MOA: Mexiletine, like lidocaine, inhibits the inward sodium current required for the initiation and conduction of impulses, thus reducing the rate of rise of the action potential, Phase 0.
- It achieves this reduced sodium current by inhibiting sodium channels. Mexiletine decreases the effective refractory period (ERP) in Purkinje fibers in the heart.
- The decrease in ERP is of lesser magnitude than the decrease in action potential duration (APD), which increases the ERP/APD ratio.
- It does not significantly affect resting membrane potential or sinus node automaticity, left ventricular function, systolic arterial blood pressure, atrioventricular (AV) conduction velocity, or QRS or QT intervals.
Mexiletine hydrochloride Metabolism: Mexiletine hydrochloride is metabolized by oxidative and reductive processes by CYP2D6 in the liver. Its metabolites, p-hydroxymexiletine and hydroxymethylmexiletine, are not pharmacologically active as antiarrhythmic agents.
Mexiletine hydrochloride Uses: For the treatment of ventricular tachycardia and symptomatic premature ventricular beats and prevention of ventricular fibrillation.
Lorcainide hydrochloride
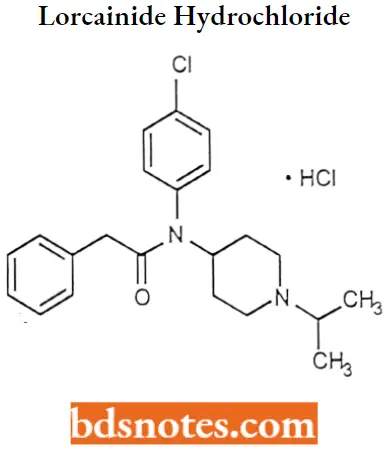
Lorcainide hydrochloride IUPAC name: N-(4-chlorophenyl)-N- (1-isopropylpiperidin-4-yl) -2-phenylacetamide, hydrochloride.
Lorcainide hydrochloride MOA: Irreversibly binding and reducing the fast Na+ influx. Interactions of Lorcainide with Nav1.5 are time and voltage-dependent.
- Class lc drugs have a characteristically slow dissociation rate, which will slow the upstroke duration and amplitude of ventricular myocytes’ action potential and prolong the PR, QRS, and QT intervals of an ECG.
- Lorcainide also increases the fibrillation threshold in a dose-dependent fashion. Overall, Lorcainide ventricular causes a decrease in tachycardiac events but also reduced ventricular contractility ejection fraction.
- The effect on sinus node function is controversial, as some researchers have noted a decreased sinus cycle length and an increase in sinus node recovery.
Lorcainide hydrochloride Metabolism: Noriorcainide, an N-dealkylated derivative, is an active metabolite of Lorcainide. It is as potent as its parent compound with similar antiarrhythmic efficacy, wherein it suppresses chronic premature ventricular complexes.
Lorcainide hydrochloride Uses: It is used to help restore normal heart rhythm and conduction in patients with premature ventricular contractions, ventricular tachycardia, and Wolff-Parkinson-White syndrome.
Class 2 β- Adrenergic blocking agents
(3-adrenergic receptor blocking agents that block the role of the sympathetic nervous system in the genesis of certain cardiac arrhythmias.
- Their dominant electrophysiologic effect is to depress adrenergically enhanced calcium influx through β-receptor blockade.
- Drugs in this class decrease neurologically induced automaticity at normal therapeutic doses.
- At higher doses, these drugs can also exhibit anesthetic properties, which cause decreased excitability, decreased conduction velocity, and a prolonged effective refractory period.
Propranolol
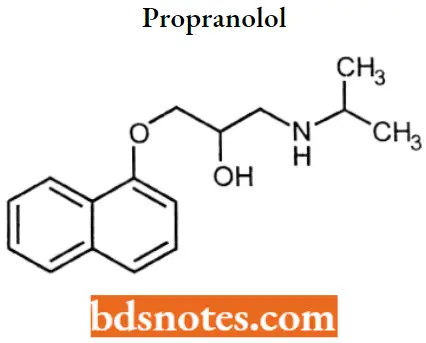
Propranolol IUPAC name: 1-(naphthalen-1-yloxy)-3-[(propan-2-yl)amino]propan-2-ol.
Propranolol MOA: P-adrenergic receptor-blocking agents that block the role of the sympathetic nervous system in the genesis of certain cardiac arrhythmias. Their dominant electrophysiologic effect is to depress adrenergically enhanced calcium influx through p -p-receptor blockade.
Propranolol Uses: Its use as an antiarrhythmic is typically for the treatment of supraventricular arrhythmias, including atrial flutter, paroxysmal supraventricular tachycardia, and atrial fibrillation.
Propranolol is also reported to be effective in the treatment of digitalis-induced ventricular arrhythmias.
Class 3 Repolarizatlon prolongation (K+ Channel blockers)
The drugs in this class cause prolongation of the duration of the action potential. This results in a prolongation of the effective refractory period. It is believed that most class 3 antiarrhythmic agents act through phase 3 of the action potential by blocking potassium channels.
Amiodarone
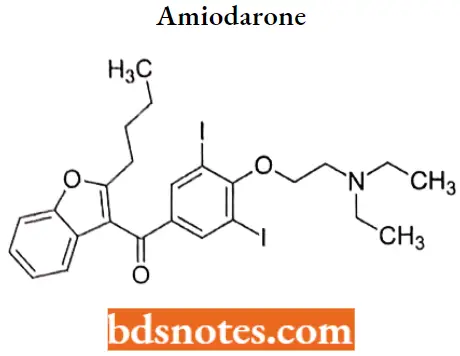
Amiodarone IUPAC name: 2-butyl-3-benzofuranyl-4-[2(diethylamino)ethoxy]-3,5-diiodophenyl ketone.
Amiodarone MOA: It has a unique mechanism of action that involves the alteration of the lipid membrane in which ion channels and receptors are located. Its cardiac effects are not well characterized, but clinical studies indicate that it is primarily a class 3 agent.
Amiodarone Metabolism: Amiodarone is extensively metabolized in the liver via CYP2C8 (under 1% unchanged in urine), and can affect the metabolism of numerous other drugs.
- The major metabolite of amiodarone is desethylamiodarone (DEA), which also has antiarrhythmic properties.
- The metabolism of amiodarone is inhibited by grapefruit juice, leading to elevated serum levels of amiodarone.
Amiodarone Uses: For the treatment of life-threatening ventricular arrhythmias that are refractory to other drugs.
Amiodarone Adverse Effects: Amiodarone has adverse effects involving many different organ systems. It also inhibits the metabolism of drugs cleared by oxidative microsomal enzymes.
- It contains iodine in its molecular structure and, as a result, affects thyroid hormones. Hypothyroidism occurs in up to 11% of patients receiving amiodarone.
- The principal effect is the inhibition of peripheral conversion of T4 to T3. Serum reverse T3 (rT3) is increased as a function of the dose as well as the length of amiodarone therapy.
- As a result, rT3 levels have been used as a guide for judging the adequacy of amiodarone therapy and predicting toxicity.
Sotalol
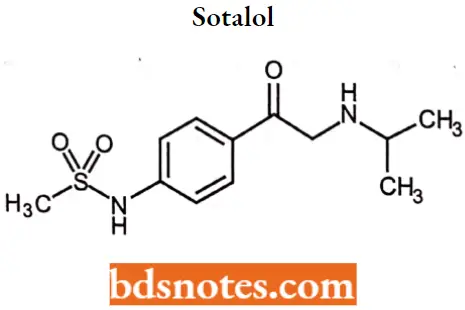
Sotalol IUPAC name: 4′[l-hydroxy-2-(isopropylamino) ethyl]methylsulfonanilide.
Sotalol MOA: Sotalol is an antiarrhythmic drug. Sotalol has both beta-adrenoreceptor blocking and cardiac action potential duration prolongation antiarrhythmic properties.
- Sotalol is a racemic mixture of dand 1-sotalol. Both isomers have similar Class 1 antiarrhythmic effects, while the 1-isomer is responsible for virtually all of the beta-blocking activity.
- Sotalol inhibits response to adrenergic stimuli by competitively blocking β1-adrenergic receptors within the myocardium and β2-adrenergic receptors within bronchial and vascular smooth muscle.
- The electrophysiologic effects of sotalol may be due to its selective inhibition of the rapidly activating component of the potassium channel involved in the repolarization of cardiac cells.
Sotalol Metabolism: Sotalol is not metabolized, nor is it bound significantly to proteins. Elimination occurs by renal excretion, with more than 80% of the drug eliminated unchanged.
Sotalol Uses: For the treatment of documented life-threatening ventricular arrhythmias.
Anti-Arrhythmic Agents Multiple Choice Questions And Answers
Question 1. What is an arrhythmia?
- Accelerated heartbeat.
- Slow heartbeat.
- Irregular heartbeat.
- A type of heart cancer
Answer: 3. Irregular heartbeat.
Question 2. How do anti-arrhythmic agents work?
- They change the electrical conduction of the heart by targeting the defective myocytes.
- They change the electrical conduction of the heart by targeting the pacemaker
- They change the electrical conduction of the heart by targeting the ion channels.
- They change the electrical conduction of the vasculature by targeting the ion channels.
Answer: 3. They change the electrical conduction of the heart by targeting the ion channels.
Question 3. What is the most common side effect of anti-arrhythmic therapy?
- Pseudo arrhythmia
- Proarrhythmia
- Bradycardia
- Tachycardia
Answer: 2. Proarrhythmia
Question 4. Antiarrhythmic drug: may cause hypothyroidism or hyperthyroidism (frequency-2%-4%) approved for use only in the treatment of serious ventricular arrhythmias, also used for refractory supraventricular arrhythmias.
- Mexiletine
- Tocainide
- Adenosine
- Amiodarone
Answer: 4. Amiodarone
Question 5. Antiarrhythmic drug: 37% iodine by weight, structurally similar to thyroxine.
- Propranolol
- Mexiletine
- Flecainide
- Amiodarone
Answer: 4. Amiodarone
Question 6. Side effects or toxicities of disopyramide
- Negative inotropic- very significant
- Dry mouth
- Urinary hesitancy
- Paradoxical ventricular tachycardia
Answer: 2. Dry mouth
Question 7. The most serious adverse effect with long-term treatment- is rapidly progressive pulmonary fibrosis, with a frequency: of 5%-15%.
- Lidocaine
- Propranolol
- Amiodarone
- Procainamide
Answer: 3. Amiodarone
Question 8. Major non-cardiac side effect of lidocaine (Xylocaine)
- Hepatic
- Renal
- Pulmonary
- CNS
Answer: 4. CNS
Question 9. p-adrenergic receptor blockers.
- Increased AV induction time
- Decreased AV nodal refractoriness
- Both
- Neighter
Answer: 1. Increased AV induction time
Anti-Arrhythmic Agents Short Questions And Answers
Question 1. What is transmembrane resting potential?
Answer:
The resting cell has a membrane potential of approximately -90 mV, with the inside of the cell being electronegative relative to the outside of the cell. This is termed the “transmembrane resting potential.
Question 2. What is arrhythmia?
Answer:
Arrhythmia is an alteration in the normal sequence of electrical impulse rhythm that leads to contraction of the myocardium.
Question 3. On which basis are Class 1 antiarrhythmic agents classified?
Answer:
Class 1 antiarrhythmic drugs can be classified based on the relative ease with which they dissociate from the Na+ ion channel.
- Class 1A drugs that have an intermediate rate of dissociation from Na+ channels.
- Class 1B drugs dissociate rapidly from the Na+ channels and thus have the lowest potency as sodium channel blockers. They produce little, if any, change in action potential duration.
- Class 1C drugs are the most potent sodium channel-blocking agents. They slowly dissociate from the Na+ channel, causing a slowing of the conduction time of the impulse through the heart.
Question 4. Write adverse effects of Amiodarone.
Answer:
Amiodarone has adverse effects involving many different organ systems. It also inhibits the metabolism of drugs cleared by oxidative microsomal enzymes.
- It contains iodine in its molecular structure and, as a result, affects thyroid hormones. Hypothyroidism occurs in up to 11% of patients receiving amiodarone.
- The principal effect is the inhibition of peripheral conversion of T4 to T3. Serum reverse T3 (rT3) is increased as a function of the dose as well as the length of amiodarone therapy.
- As a result, rT3 levels have been used as a guide for judging the adequacy of amiodarone therapy and predicting toxicity.
Leave a Reply