Radical Reactions
Formation Of Radicals
The ionization of HBr distributes the two electrons of the single H–Br bond so that the electronegative bromine accepts electrons while hydrogen loses electrons, and the resultant ions are thus H+ and Br−.
- This process is termed heterolytic cleavage, in that the two atoms of the bond suffer different fates and that the two electrons are distributed unevenly. In marked contrast, the two electrons of the single bond can be distributed evenly so that one electron becomes associated with each atom.
- This is termed homolytic cleavage, generating radicals (often termed free radicals). A radical may be defined as a high-energy species carrying an unpaired electron. Note that, to indicate the movement of just one electron, we use a fish-hook curly arrow in mechanisms rather than the normal curly arrow, which denotes the movement of two electrons.
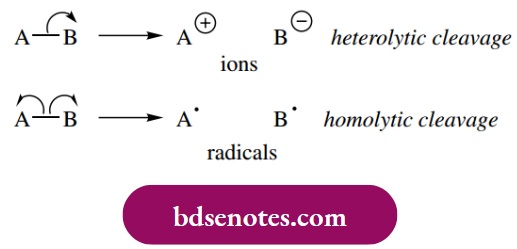
Radicals may be generated in two general ways:
- By homolysis of weak bonds;
- By reaction of molecules with other radicals.
Homolytic cleavage of most σ bonds may be achieved if the compound is subjected to a sufficiently high temperature, typically about 200 ◦C. However, some weak bonds will undergo hemolysis at temperatures a little above room temperature. Bonds of peroxy and azo compounds fall in this category, and such compounds may be used to initiate a radical process. Di-tert-butyl peroxide, dibenzoyl peroxide
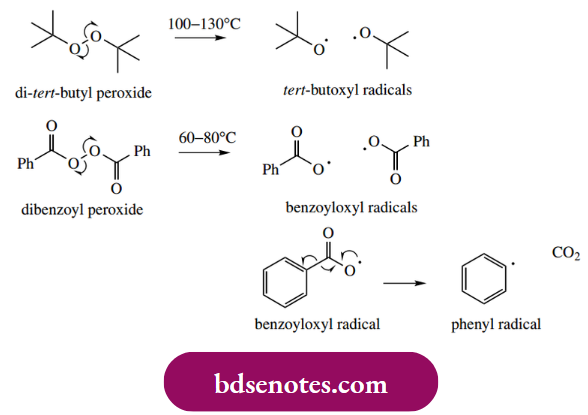
- And azoisobutyronitrile (AIBN) are good source of radicals under typical reaction conditions. At increased temperatures, the peroxide bond is cleaved homolytically, giving radicals. Dibenzoyl peroxide is a diacyl peroxide that cleaves rather more readily than dialkyl peroxide. However, further decomposition occurs in which carbon dioxide is lost, and the phenyl radical is produced. This displacement is favored by the inherent stability of carbon dioxide.
- Homolytic cleavage of diazo compounds such as AIBN is also driven by the stability of a neutral molecule, this time molecular nitrogen, and two alkyl radicals are produced.
An alternative approach to homolytic cleavage is photolysis, the absorption of light energy, especially UV radiation. Thus, halogen molecules are easily photolyzed to generate halogen radicals.
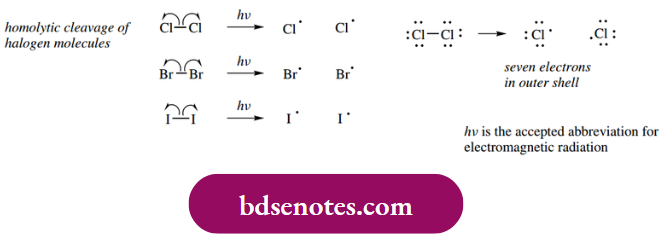
- The halogen molecule is comprised of two halogen atoms each with seven electrons in their outer shell. Sharing of the unpaired electrons creates a stable molecule in which each atom has now acquired an octet of electrons in its outer shell. By absorbing energy, we have removed this stabilization and effectively generated halogen atoms, which are our radicals.
- Radicals formed in one of these initiation reactions may themselves be the means of producing other radicals, by reacting with another molecular species. Abstraction of a hydrogen atom is a particularly common reaction leading to a new radical.
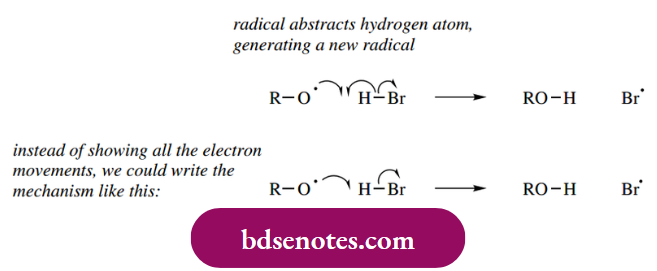
- Thus, the abstraction of a hydrogen atom from HBr generates a bromine radical. Note that, for convenience, we tend not to put in all of the electron movement arrows. This simplifies the representation but is more prone to errors if we do not count electrons.
- Our attacking radical has an unpaired electron, and it abstracts the proton plus one of the electrons comprising the H–Br σ bond, i.e. a hydrogen atom, and the remaining electron from the bond now resides with the bromine in the form of a bromine radical.
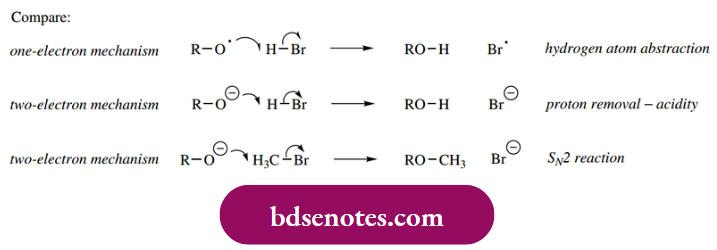
- This is shown as a one-electron mechanism and should be compared with the analogous two-electron mechanisms that account for acidity and SN2 reactions. The only difference is in the number of electrons involved, which we indicate by the fish hook or normal curly arrow.
Compare:
- Another possibility is that we can get radical addition to an unsaturated molecule, for example. an alkene. Writing in all the electron movement arrows, we have one of the double bond π electrons being used to make the new σ bond with the original radical species, whilst the second π electron becomes located on the other end of the double bond, and is now the unpaired electron of the new radical.
- The original radical could potentially have attacked at either end of the double bond; the regiochemistry of addition is governed by the stability of the radical generated (see below).
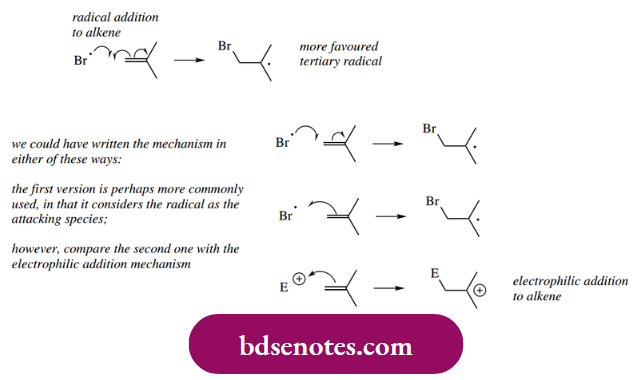
- Note that if we choose not to put in all the curly arrows, we could write the mechanism in two ways: either considering the radical as the attacking species or the double bond as the electron-rich species.
- The first version is perhaps more commonly used, but it is much more instructive to compare the second one with an electrophilic addition mechanism.
- The rationalization for the regiochemistry of addition parallels that of carbocation stability.
Structure And Stability Of Radicals
Most radicals have a planar or nearly planar structure. Carbon is sp2 hybridized in the methyl radical, giving three σ C–H bonds, and the single electron is held in a 2p orbital that is oriented at right angles to the plane of the radical.
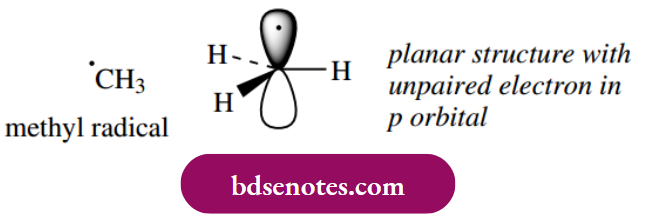
Although a radical is neutral, it is an electron-deficient species that will be very reactive as it attempts to pair off the odd electron. Because radicals are electron deficient, electron-releasing groups such as alkyl groups tend to provide a stabilizing effect. The more electron-releasing groups there are, the more stable the radical. Thus, tertiary radicals are more stable than secondary radicals, which in turn are more stable than primary radicals.
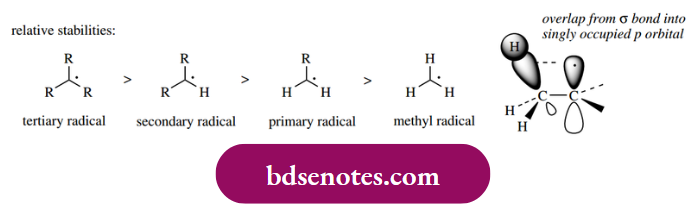
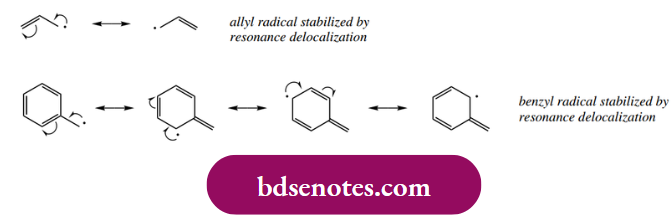
The order of stability is thus the same as with carbocations, another electron-deficient species, and for the same reason. There is favorable delocalization of the unpaired electron through the overlap of the σ C–H (or C–C) bond into the singly occupied p orbital of the radical. The similarity continues, in that resonance delocalization also helps to stabilize a radical so that the allyl radical and the benzyl radical are more stable than an alkyl radical.
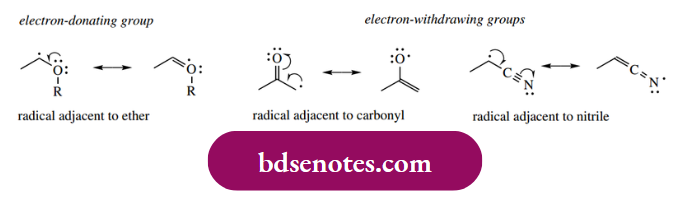
- Electron-donating functional groups, for example, ethers, also stabilize radicals via their lone pair orbitals. However, electron-withdrawing groups can also stabilize radicals, so that radicals next to carbonyl or nitrile are more stable than even tertiary alkyl radicals.
- This is because these groups possess a π electron system and the unpaired electron can take advantage of this. It transpires that features that stabilize an anion, for example. an electron-withdrawing group features that stabilize a carbocation, for example. electron-donating groups, or features such as conjugation that may stabilize either, will all stabilize a radical.
- There is a significant difference between carbocations and radicals when we are thinking about stability, however. One of the more confusing aspects relating to carbocations was their ability to rearrange, either by migration of an alkyl group or of hydride, when a more stable system might be attained by this means.
- We related this trend to the enhanced stability of, say, a tertiary or allylic carbocation over secondary or primary carbocations. Now, although we also find tertiary or allylic radicals are more favorable than secondary or primary radicals, we do not encounter rearrangements with radicals, even if the product radical is more stable.
- This comes from an increased energy barrier to rearrangement in radicals compared with carbocations, which in turn relates to the extra unpaired electron in the radical, which has to occupy a higher energy orbital in the transition state.
Radical Substitution Reactions Halogenation
Halogenation reactions of alkanes provide good examples of radical processes, and may also be used to illustrate the steps constituting a radical chain reaction. Alkanes react with chlorine in the presence of light to give alkyl chlorides, for example. for cyclohexane, the product is cyclohexyl chloride.
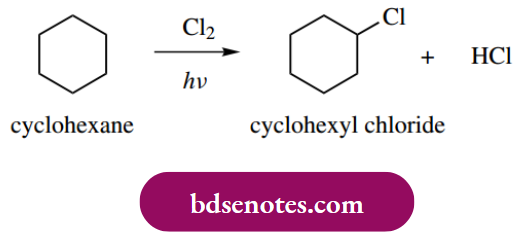
The initiation step is the light-induced formation of chlorine atoms as radicals. Only a few chlorine molecules will suffer this fate, but these highly reactive radicals then rapidly interact with the predominant molecules in the system, namely cyclohexane.
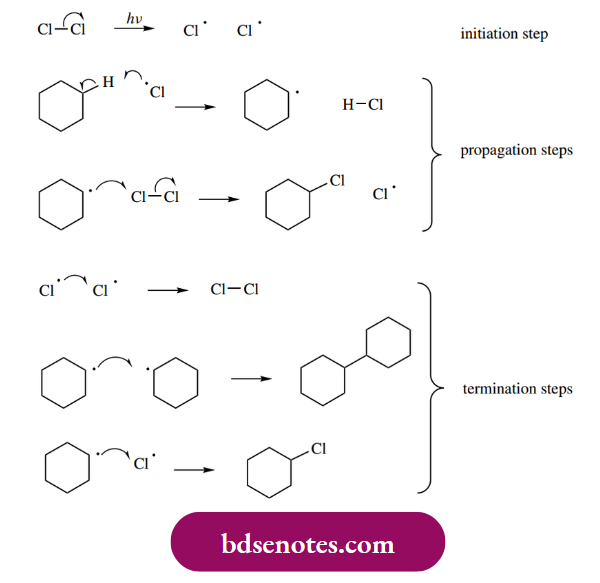
- The chlorine radicals abstract hydrogen atoms from the cyclohexane substrate, producing new radicals, i.e. cyclohexyl radicals. These, in turn, cause further dissociation of chlorine molecules and the production of more chlorine radicals.
- The cyclohexyl radical reacts with a chlorine molecule rather than, say, a further molecule of cyclohexane simply because bond energies dictate it is easier to achieve fission of the Cl–Cl bond than the C–H bond. This results in the production of cyclohexyl chloride and a further chlorine radical.
- The chlorine radical can now abstract hydrogen from another cyclohexane substrate, and we get a repeat of the same reaction sequence, the so-called propagation steps of this chain reaction. During the propagation steps, one radical is used to generate another, so that only one initiation reaction is required to generate a large number of product molecules.
- Finally, when we are running out of cyclohexane, the process terminates with the interaction of two radical species, for example. two chlorine atoms, two cyclohexyl radicals, or one of each species.
- The combination of two chlorine atoms is probably the least likely of the termination steps since the Cl–Cl bond would be the weakest of those possible, and it was the light-induced fission of this bond that started the radical reaction.
- Of course, once we have formed cyclohexyl chloride, there is no reason why this should not get drawn into the radical propagation steps, resulting in various dichloro cyclohexane products, or indeed polychlorinated compounds.
- Chlorination of an alkane will give many different products, even when the amount of chlorine used is limited to molar ratios, and in the laboratory, it is not going to be a particularly useful process.
- However, it is instructive to consider radical chlorination of alkanes just a little further, to appreciate the mechanistic concepts. If we carry out light-induced chlorination of propane, then we obtain two different monochlorinated products, but not in equal amounts. There will also be other products containing more than one chlorine atom. A similar situation pertains if we chlorinate 2-methylpropane.
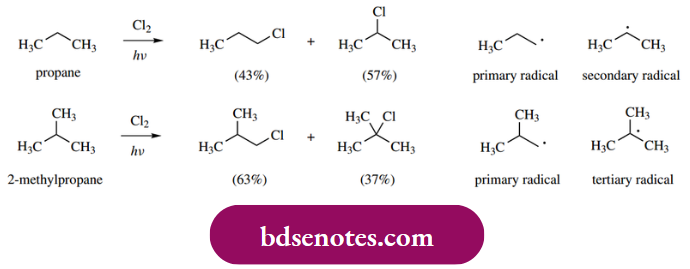
The proportion of each product formed can be rationalized by considering several factors. First, the products from propane are the result of generating either primary or secondary radicals.
- We know that tertiary radicals are more favorable than secondary radicals, which in turn are more favorable than primary radicals. It is also true that tertiary C–H bonds are slightly weaker than secondary C–H bonds, which in turn are slightly weaker than primary C–H bonds.
- It is thus rather easier to break tertiary C–H bonds by the hydrogen abstraction reaction, followed by secondary C–H bonds, and then primary C–H bonds. On the other hand, there is a statistical factor, in that there are six primary hydrogens in propane and only two secondary ones, so it is more likely that a primary C–H bond is attacked by the very reactive radical.
- The net result of these two opposite trends is the slight excess of the secondary halide product. With 2-methylpropane, the statistical factor is even more pronounced (only one tertiary hydrogen to nine primary hydrogens), and hence we get rather more primary product in the reaction mixture, even though tertiary radicals are the more stable and tertiary C–H bond is the weakest.
- Because the chlorine radical is so reactive, the variation in bond strengths is not an especially important factor. It can readily be
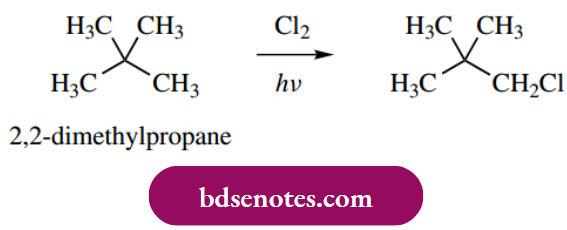
- Appreciated that, even under conditions in which we can maximize monochlorination, it is highly desirable if there is no chance of forming isomers that have to be separated. Substrates that meet these criteria include cyclohexane and 2,2-dimethylpropane.
- Bromine will also halogenate alkanes, but in this case, we find that bromine is considerably less reactive than chlorine. As a result, the reaction becomes much more selective, and the product ratios are more distinctive. Bromination of alkanes is so selective that it is a feasible laboratory process to make alkyl bromides from alkanes.
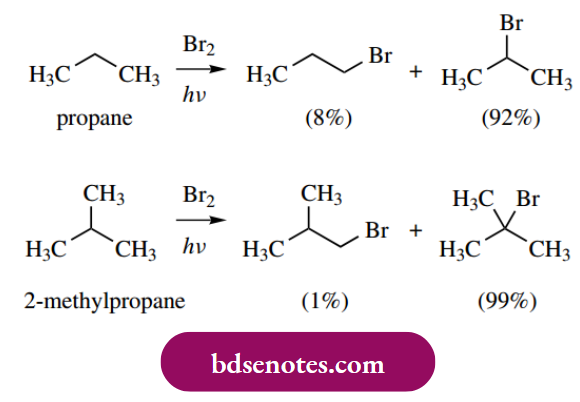
- The product ratios for bromination of propane and 2-methylpropane are quite different from those seen above in the chlorination reaction, in that the more-favored products by far are the secondary and tertiary halides respectively.
- Abstraction of a hydrogen atom by a bromine atom is now much more difficult than with a chlorine atom. The favored product may be rationalized in terms of the relative strength of the C–H bond being broken, and the relative stability of the radical produced, though this is an oversimplification and we ought to consider relative energies of transition states.
Stereochemistry Of Radical Reactions
The planarity of a radical means that, when it reacts with a reagent, there is an equal probability that it can form a new bond with either side of the radical.
- In many cases this is of no consequence; but, should the formation of the product generate a chiral center, we are going to get an equimolar mixture of both possible configurations, i.e. formation of a racemic mixture.
- This outcome has already been noted when a carbocation, another planar system, reacts to produce a chiral center.
- Thus, if we consider radical chlorination of butane, we expect to get a mixture of products, including the monochlorinated compounds 1-chlorobutane and 2-chlorobutane.
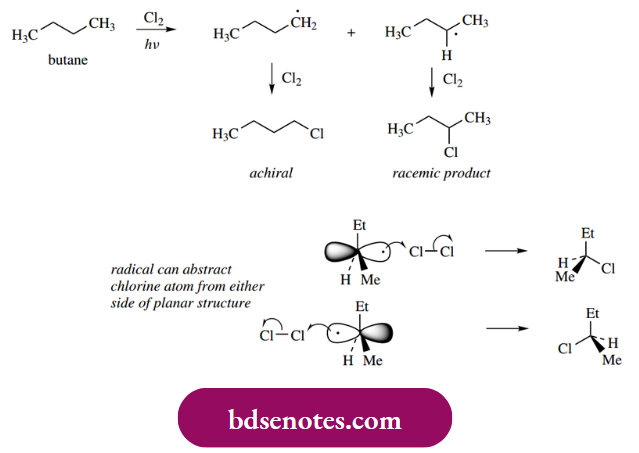
In the formation of 1-chlorobutane, an intermediate primary radical is involved, and there are no stereochemical consequences. However, the secondary radical involved in 2-chlorobutane formation is planar, and when it abstracts a chlorine atom from a chlorine molecule it can do so from either side with equal probability. The result is the formation of a racemic product, (±)-2-chlorobutane.
Allylic And Benzylic Substitution Halogenation Reactions
The selectivity of radical bromination reactions depends, in part, on the increased stability of secondary or tertiary radical intermediates compared with primary radicals. we noted that allyl and benzyl radicals were especially
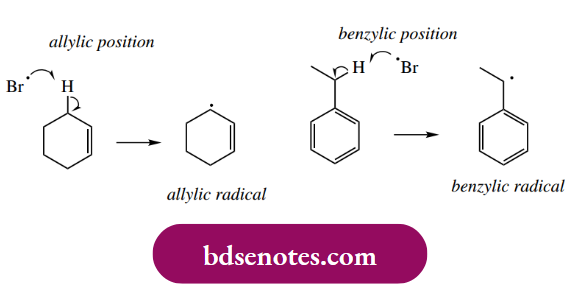
- stabilized by resonance delocalization; indeed, they are even more stable than tertiary radicals. In the presence of a suitable initiator, bromine dissociates to bromine atoms that will selectively abstract an allylic or benzylic hydrogen from a suitable substrate, generating the corresponding allyl and benzyl radicals.
- In the case of cyclohexene, this leads to a resonance-stabilized allylic radical that then reacts with bromine to give the allylic bromide, plus a further bromine atom to continue the chain propagation steps. The symmetry in cyclohexene means that the two resonance structures are identical.
- It does not matter which allylic radical picks up bromine, we get the same product. It is not difficult to appreciate that a mixture of brominated products must result if we start with a non-symmetrical substrate.
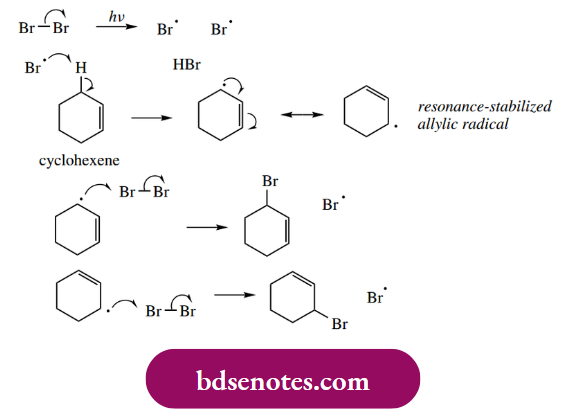
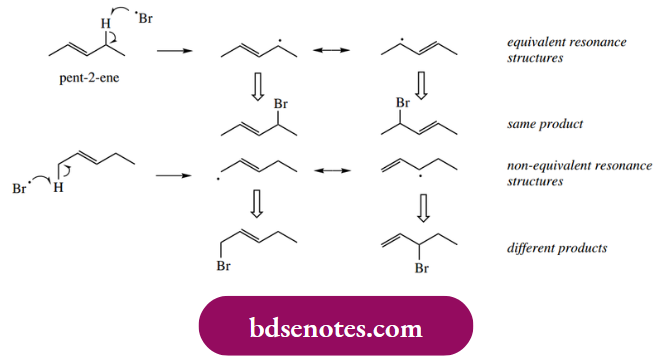
For example, radical allylic bromination of pent- 2-ene must produce a mixture of three products. There are two allylic positions in the substrate, and either can suffer hydrogen abstraction.
- If hydrogen is abstracted from the methylene, then the two contributing resonance structures for the allylic radical are equivalent, and one product results when this captures a bromine atom.
- Abstraction of hydrogen from the terminal methyl gives an allylic radical for which the resonance structures are not equivalent, and hence two different brominated products may be formed. The net result will be a mixture of all three products. If we want to exploit allylic bromination, this means we must choose the substrate carefully if we prefer to get a single product.
Of course, we have also seen that bromine can react with a double bond via electrophilic addition; further, it can add to a double bond via a radical mechanism.
- This could complicate an allylic bromination reaction, and it is necessary to choose conditions that minimize any addition to the double bond. This is achieved by carrying out the reaction in a solvent of low polarity, for example, CCl4, which suppresses the possibility of the polar electrophilic addition, whilst keeping the concentration of bromine very low to suppress radical addition.
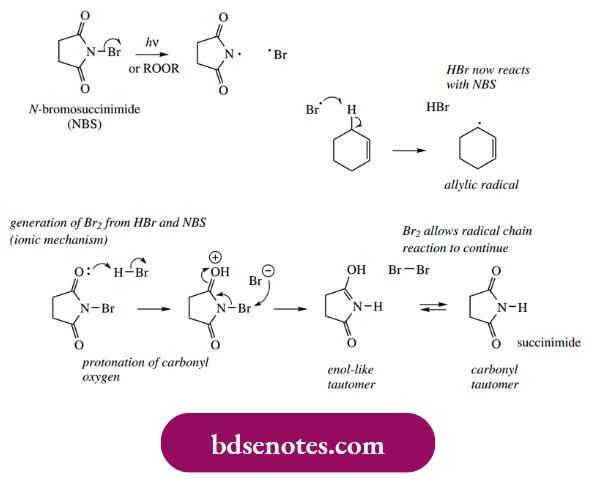
- There is, however, a much better reagent than bromine to brominate at an allylic position selectively. This reagent is N-bromosuccinimide (NBS), and it also reacts via a radical mechanism. The weak N–Br bond in NBS is susceptible to homolytic dissociation initiated either by light or a chemical initiator, such as a peroxide.
- This produces a small amount of bromine radicals, which can then abstract hydrogen from an allylic position on the substrate. The chain reaction continues via a small concentration of molecular bromine, which is generated by an ionic mechanism from NBS and the HBr released as a consequence of the hydrogen abstraction.
- Accordingly, the broad overall reaction is just the same as if we were employing molecular bromine as the reagent. The difference is in the use of NBS to maintain a very low concentration of bromine. Under the conditions used, i.e. in a non-polar solvent in which NBS is not very soluble, and with a very low concentration of bromine produced, there is almost exclusive allylic bromination and very little addition to the double bond.
A benzylic radical is generated if a compound like toluene reacts with bromine or chlorine atoms. Hydrogen abstraction occurs from the side-chain methyl, producing a resonance-stabilized radical. The dissociation energy for the C–H bonds of the aromatic ring system is considerably more than that for the side-chain methyl and relates to the stability of the radical produced.
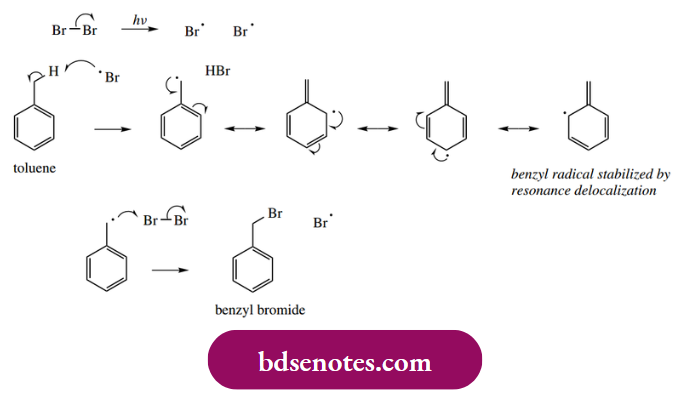
- The typical propagation steps now follow, although all halogenation proceeds in the side-chain; an addition to the ring would destroy the aromaticity and produce a higher energy product.
- Benzyl chloride undergoes further chlorination to give di- and trichloro derivatives, though it is possible to control the extent of chlorination by restricting the amount of chlorine used.
- As indicated above, it is easier to mono-brominate than it is to mono-chlorinate. The particular stabilization conferred on the benzylic radical by resonance is underlined by the reaction of ethylbenzene with halogens.
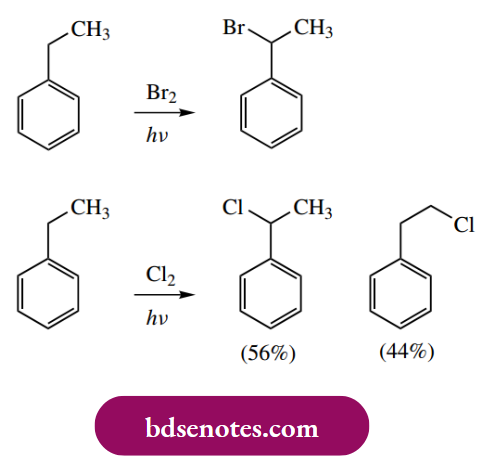
Bromination occurs exclusively at the benzylic position, i.e. adjacent to the benzene ring. The radical formed at this position is resonance stabilized, whereas no such stabilization is available to the primary radical formed by the abstraction of one of the methyl hydrogens.
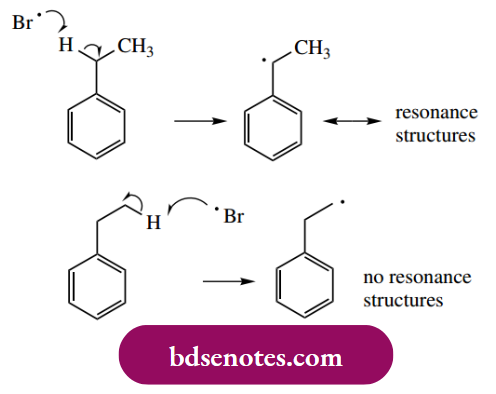
However, with the more reactive chlorine, chlorination can occur at either position, though the major product is the benzylic halide. Benzylic bromination is also efficiently achieved by the use of Nbromosuccinimide as the halogenating species.
Radical Addition Reactions Addition Of HBr To Alkenes
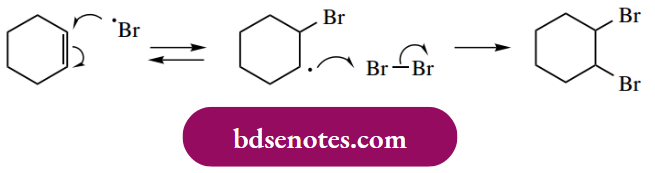
The radical addition of halogen to an alkene has been referred to briefly in. We saw an example of bromination of the double bond in cyclohexene as an unwanted side reaction in some allylic substitution reactions.
- The mechanism is quite straightforward, and follows a sequence we should now be able to predict.
- More relevant to our consideration now is the radical addition of hydrogen bromide to an alkene.
- The radical formation is initiated usually by the homolysis of a peroxide, and the resultant alkoxyl radical may then abstract a hydrogen atom from HBr.
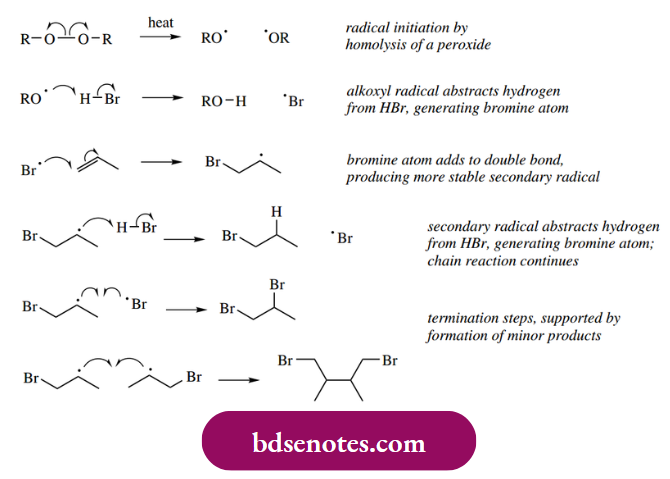
- The bromine atom then adds to the alkene, generating a new carbon radical. In the case of propene, as shown, the bromine atom bonds to the terminal carbon atom. In this way, the more stable secondary radical is generated. This is preferred to the primary radical generated if the central carbon were attacked.
- The new secondary radical then abstracts hydrogen from a further molecule of HBr, giving another bromine atom that can continue the chain reaction. The main product is thus the result of the addition of HBr to the alkene. Minor products detected are consistent with the proposed chain-termination steps.
- This looks quite logical and consistent with what we know about radical reactions. However, remind yourself of the addition of HBr to an alkene, as we discussed under electrophilic reactions in Section 8.1.1. There is a significant difference in the product.
Electrophilic Adition Of HBr:
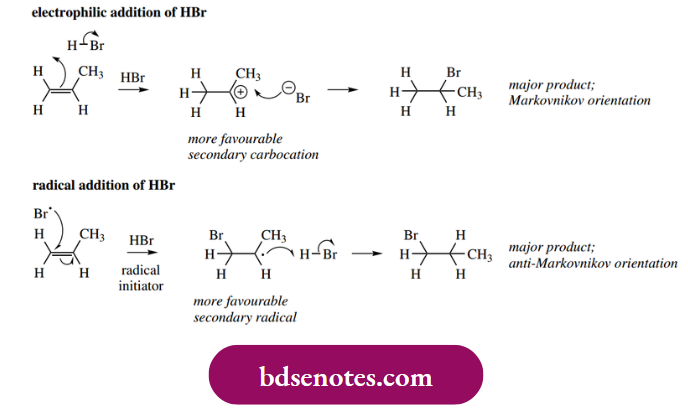
- Electrophilic addition of HBr to propene gives predominantly the so-called Markovnikov orientation; Markovnikov’s rule states that the addition of HX across carbon-carbon multiple bond proceeds in such a way that the proton adds to the less-substituted carbon atom, i.e. that already bearing the greater number of hydrogen atoms.
- We rationalized this in terms of the formation of the more favorable carbocation, which in the case of propene is the secondary carbocation rather than the alternative primary carbocation. Now, just the same sort of rationalization can be applied to the radical addition, in that the more favorable secondary radical is predominantly produced.
- This, in turn, leads to the addition of HBr in what is the anti-Markovnikov orientation. The apparent difference is because the electrophile in the ionic mechanism is a proton, and bromide then quenches the resultant cation. In the radical reaction, the attacking species is a bromine atom and a hydrogen atom is then used to quench the radical.
- This is effectively a reverse sequence for the addition process; nevertheless, the stability of the intermediate carbocation or radical is the defining feature. The terminologies Markovnikov or anti-Markovnikov orientation may be confusing and difficult to remember; consider the mechanism and it all makes sense.
- This radical anti-Markovnikov addition of HX to alkenes is restricted to HBr; both HI and HCl are added in a Markovnikov fashion by an ionic mechanism because the radical propagation steps are not favored.
- The C–I bond is relatively weak, so the addition of an iodine atom to the double bond is not favored. On the other hand, the H–Cl bond is relatively strong, and hydrogen abstraction using a radical is unfavorable.
- For many years, the addition of HBr to an alkene seemed quite mysterious and erratic, with Markovnikov or anti-Markovnikov orientation occurring apparently at random. Eventually, the problem was solved and traced to the purity of the compounds used.
- Impure reagents containing traces of peroxides led to the addition of anti-Markovnikov orientation, and we can now see that this is the consequence of a radical reaction. Reagents free from peroxides react via the ionic electrophilic addition mechanism, and we thus get predominantly Markovnikov orientation.
Radical Addition Of Hbr To Conjugated Dienes
The radical addition of HBr to an alkene depends upon the bromine atom added in the first step so that the more stable radical is formed. If we extend this principle to a conjugated diene, for example. but-1,3-diene, we can see that the preferred secondary radical will be produced if halogenation occurs on the terminal carbon atom. However, this new radical is also allylic, and an alternative resonance form may be written.
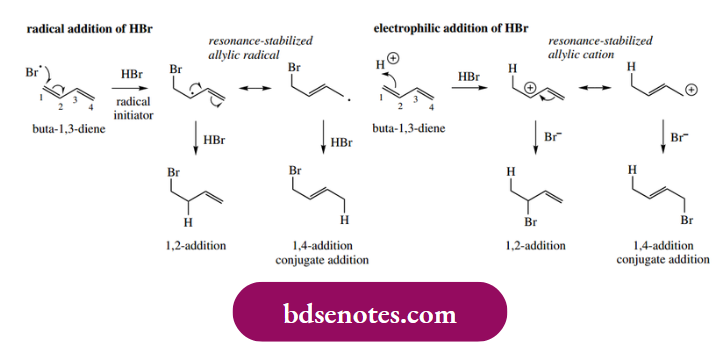
- A hydrogen atom is abstracted from HBr in the following step of the chain reaction to produce the addition product. Depending upon which resonance structure is involved, we shall get different products, the results of 1,2- and 1,4-addition. The 1,4-addition is termed conjugate addition.
- This is comparable to the electrophilic addition of HBr to butadiene (see Section 8.2), though the addition is in the reverse sense overall, in that Br adds before H in the radical reaction, whereas H adds before Br in the ionic mechanism. As with the electrophilic addition, we shall usually obtain a mixture of the two products.
Radical Polymerization Of Alkenes
The addition of a radical to an alkene generates a new radical, which potentially could add on to a further molecule of alkene, and so on, eventually giving a polymer. This becomes an obvious extension of the radical mechanisms we have already studied and is the basis for the production of many commercial polymers.
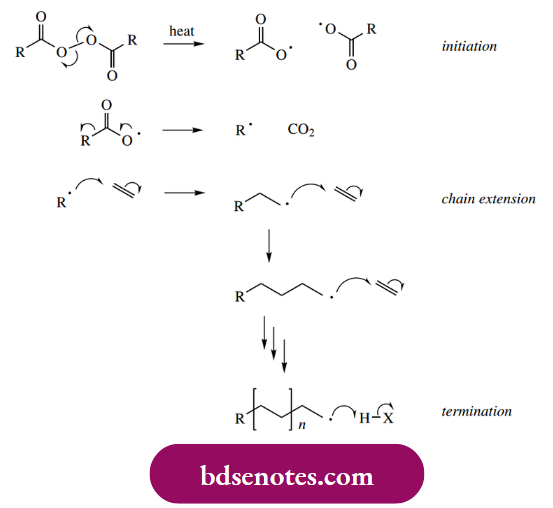
The radical initiator is usually a diacyl peroxide that dissociates to radicals that in turn add on to the alkene. This starts the chain reaction, which is terminated by hydrogen abstraction from some suitable substrate, for example. another polymeric radical that consequently becomes an alkene. In this general fashion, polymers such as polyethylene (polythene), polyvinyl chloride (PVC), polystyrene, and polytetrafluoroethylene (PTFE) may be manufactured.
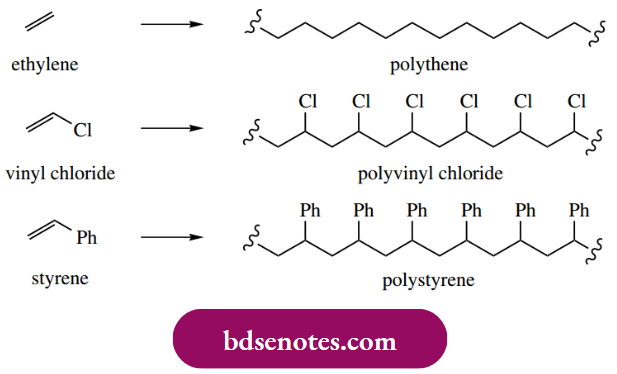
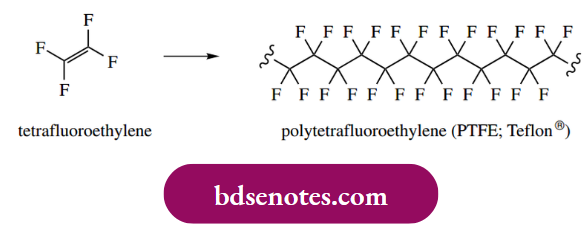
We met a rather similar process, cationic polymerization, under electrophilic reactions in Section 8.3. In practice, radical polymerization is more effective than cationic polymerization, and industrial polymers are usually produced by radical processes.
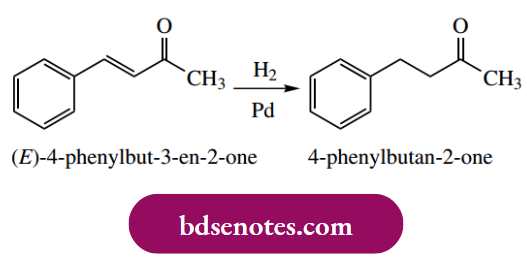
Addition Of Hydrogen To Alkenes And Alkynes: Catalytic Hydrogenation
The addition of hydrogen to carbon-carbon multiple bonds (reduction) may be achieved using gaseous hydrogen in the presence of a finely divided noble metal catalyst. This is termed catalytic hydrogenation.
- It is not a radical reaction as we have seen above, and does not feature initiation, propagation, and termination steps. However, since it appears to involve atomic hydrogen, it has much more in common with radical reactions than ionic ones, and we consider it here for convenience.
- The catalyst used is typically platinum, palladium, rhodium, or ruthenium, or sometimes an appropriate derivative. Precise details of the reaction remain vague, but we believe the catalyst surface binds to both the substrate, for example. an alkene, and hydrogen, weakening or breaking the π bond of the alkene and the σ bond of hydrogen. Sequential addition of hydrogen atoms to the alkene carbons then occurs and generates the alkane, which is then released from the surface.
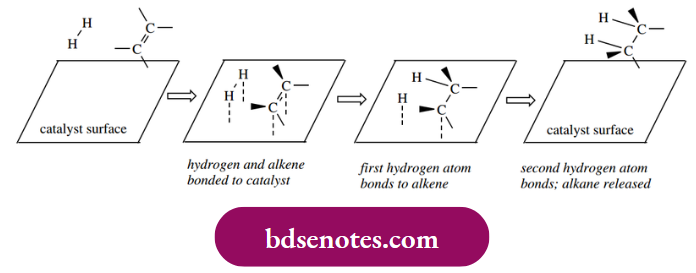
Catalytic hydrogenation delivers hydrogen to one face of the alkene; the consequence is the syn addition of hydrogen. This is a departure from our usual observations with ionic mechanisms, where the groups typically add to a double bond with anti-stereochemistry. The stereochemical consequences of this are illustrated in the following examples.
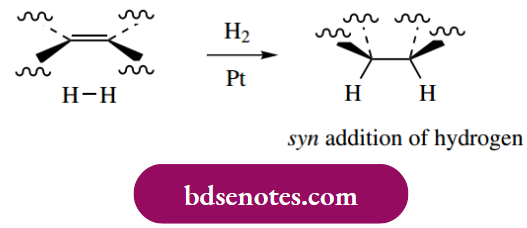
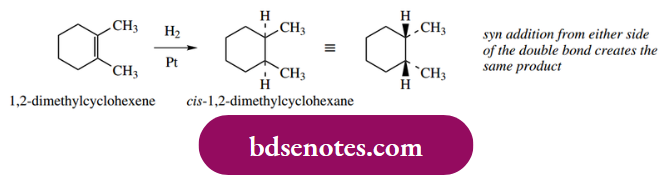
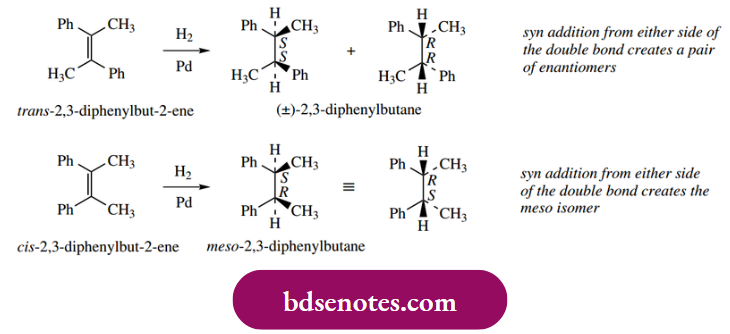
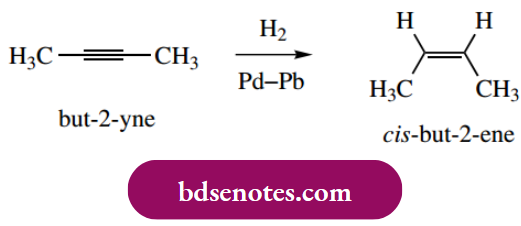
Alkynes may also be hydrogenated, initially to alkenes, and then further to alkanes. By suitable modification of the catalyst, it has proved possible to stop the reaction at the intermediate alkene. Typically, platinum or palladium catalysts partially deactivated (poisoned) with lead salts are found to be suitable for the reduction of alkynes to alkenes. Again, syn addition is observed.
Isolated double and triple bonds are reduced readily, whereas conjugated alkenes and aromatic systems are difficult to hydrogenate. Carbonyl double bonds react only very slowly, if at all, so it is possible to achieve selective reduction of C=C double bonds in the presence of aromatic and carbonyl functions.
Radical Addition Of Oxygen Autoxidation Reactions
The slow spontaneous oxidation of compounds in the presence of oxygen is termed autoxidation (autooxidation). This radical process is responsible for a variety of transformations, such as the drying of paints and varnishes, the development of rancidity in foodstuff fats and oils, the perishing of rubber, air oxidation of aldehydes to acids, and the formation of peroxides in ethers.
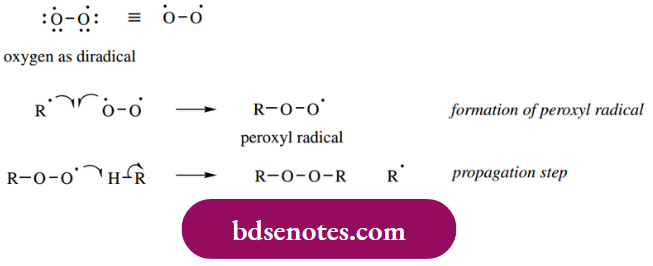
- Unsaturated hydrocarbons undergo autoxidation because allylic hydrogens are readily abstracted by radicals (see Section 9.2). Molecular oxygen in its low-energy arrangement is a diradical, with only one bond between the atoms, and consequently an unpaired electron on each atom.
- Thus, oxygen can abstract hydrogen atoms like other radicals, though it is not a particularly good hydrogen abstractor. Instead, sequences are initiated by light or by other promoters that generate radicals, and oxygen is involved in the propagation steps.
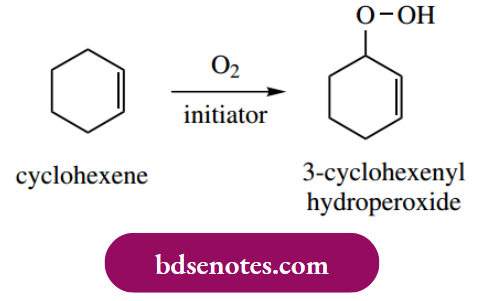
The processes that occur when cyclohexene reacts with oxygen in the presence of an initiator to give the allylic hydroperoxide exemplify this nicely.
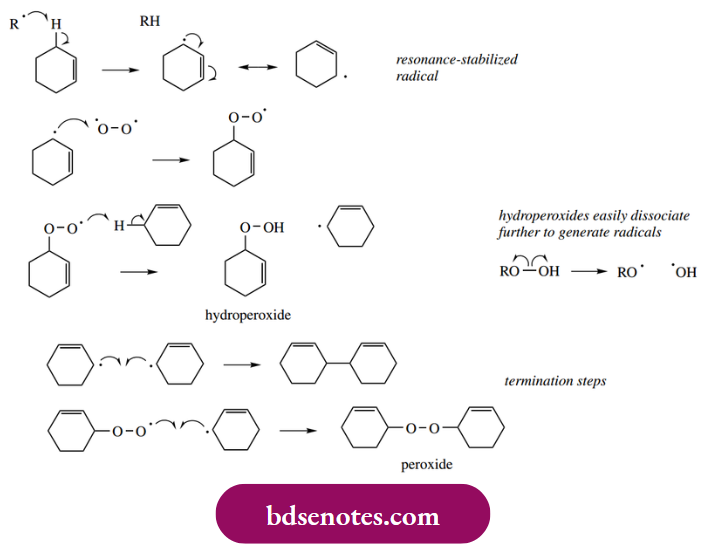
- Thus, the radical from the initiation reaction abstracts hydrogen from the allylic position of cyclohexene, as we have seen previously, to give the resonance-stabilized radical. In the propagation steps, this radical then reacts with oxygen, producing a peroxyl radical, which then abstracts hydrogen from a further molecule of the substrate.
- The product is thus the hydroperoxide, the reaction having occurred at the allylic position of the alkene. Two possible chain-termination steps might be the combination of two cyclohexyl radicals or the formation of peroxide, as shown. The hydroperoxide itself can easily dissociate to produce radicals that may then initiate other chain reactions.
- Peroxyl radicals are not particularly reactive and thus tend to be highly selective. They tend to abstract hydrogen atoms most readily from tertiary, allylic, and benzylic C–H bonds. These are systems with the weakest bonds and that have maximum stabilization in the radical produced.
Autoxidation In Fats And Oils The Origins Of Rancidity
Oxygen-mediated autoxidation can occur with unsaturated acid components of fats and oils, which are esters of fatty acids with glycerol (see Box 7.16). This leads initially to hydroperoxides that decompose further to produce low molecular weight carboxylic acids.
These are the causes of rancidity, unpleasant odor, and taste associated with badly stored fats. Linoleic acid is a typical unsaturated fatty acid component, and hydrogen abstraction will occur from the methylene between the two non-conjugated double bonds. The radical thus produced benefits from extensive delocalization, as shown by the resonance forms that can be drawn.
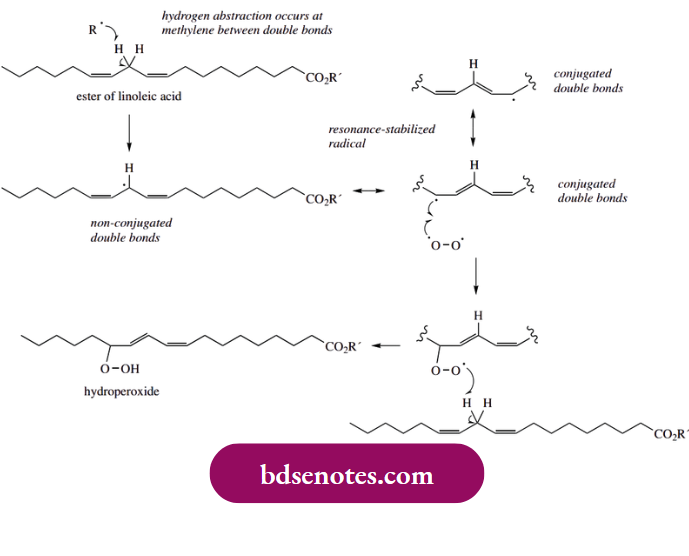
However, the resonance forms in which the double bonds are conjugated are inherently more stable than those with the unconjugated double bonds. Accordingly, the hydroperoxide subsequently formed upon reaction with oxygen will have conjugated double bonds. Abstraction of a hydrogen atom to form the hydroperoxide is part of the chain propagation process. Fragmentation of the hydroperoxide can then lead to chain shortening, as illustrated.
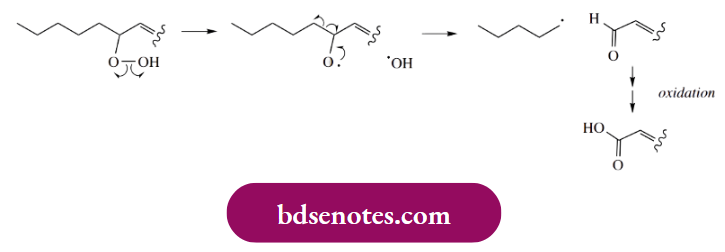
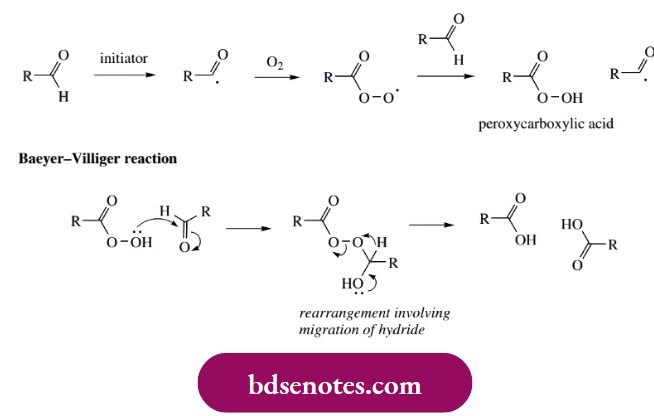
Acidic products result from further oxidation of aldehydes (or ketones), again by a radical process. Oxidation of an aldehyde to a carboxylic acid in the presence of air involves a peroxy acid. Finally, a reaction between the peroxy acid and a molecule of aldehyde yields two carboxylic acid molecules; this is not a radical reaction but is an example of a Baeyer–Villiger oxidation. Baeyer–Villiger reactions are valuable for converting a ketone into an ester, in which case we see a rearrangement involving the migration of an alkyl group.
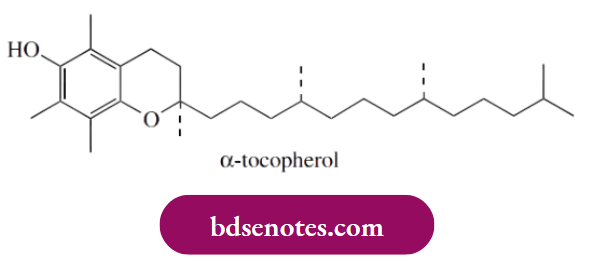
In Box 9.2 we shall see how vitamin E is used commercially to retard rancidity in fatty materials in food manufacturing; it reduces autoxidation by reacting with peroxyl radicals.
Antioxidants And Health
The human body is continually exposed to radicals, either from external sources such as pollutants or from endogenous sources because reactive oxygen species are involved in the natural processes used to detoxify chemicals and invading organisms.
- Although enzyme systems are present to protect radical production and damage, such systems cannot be completely efficient. There is growing evidence that several disease states can be linked to radical damage.
- Lipid membranes, proteins, and DNA are all susceptible to interaction with radicals, and natural molecules termed antioxidants provide an important defense against such damage.
- Antioxidants are compounds that inhibit autoxidation reactions by rapidly reacting with radical intermediates to form less-reactive radicals that are unable to continue the chain reaction. The chain reaction is effectively stopped since the damaging radical becomes bound to the antioxidant.
- Thus, vitamin E ( α-tocopherol) is used commercially to retard rancidity in fatty materials in food manufacturing. Its antioxidant effect is likely to arise by reaction with peroxyl radicals.
- These remove a hydrogen atom from the phenol group, generating a resonance-stabilized radical that does not propagate the radical reaction. Instead, it mops up further peroxyl radicals. In due course, the tocopheryl peroxide is hydrolyzed to α-tocopheryl quinone.
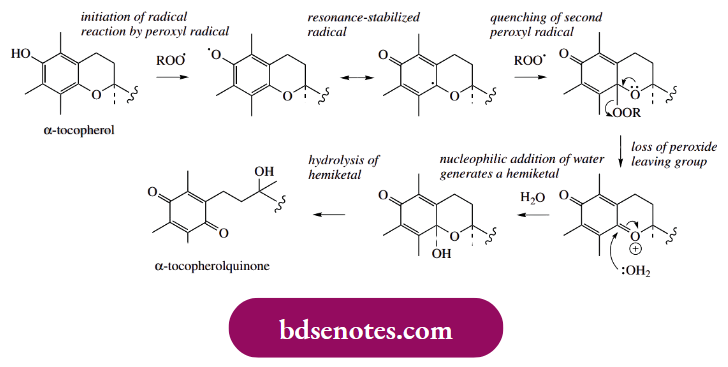
- Vitamin E in the diet is known to provide valuable antioxidant properties for humans, preventing the destruction of cellular materials, example. unsaturated fatty acids in biological membranes, and also helping to prevent heart disease.
- Other materials are similarly known to have beneficial antioxidant properties, and we are encouraged to incorporate sufficient levels of antioxidant-rich foods into our diets to minimize the risks of cardiovascular disease, cell degradation, and cancer.
- Carotenoids are plant chemicals that function along with chlorophylls in photosynthesis as accessory light-harvesting pigments, effectively extending the range of light absorbed by the photosynthetic apparatus.
- They also serve as important protectants for plants and algae against photo-oxidative damage, by quenching toxic oxygen species. Recent research also suggests that carotenoids are important antioxidant molecules in humans, quenching peroxyl radicals, minimizing cell damage, and affording protection against some forms of cancer.
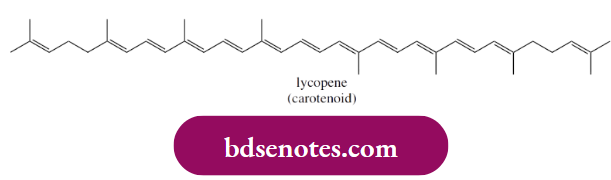
- The most significant dietary carotenoid in this respect is lycopene; tomatoes and processed tomato products feature as the predominant source. The extended conjugated system allows radical addition reactions and hydrogen abstraction from positions allylic to the double bond system.
Considerable quantities of natural polyphenolic compounds are consumed daily in our vegetable diet, and there is growing belief that some flavonoids are particularly beneficial, acting as antioxidants and giving protection against cardiovascular disease, certain forms of cancer, and, it is claimed, age-related degeneration of cell components. Their polyphenolic nature enables them to scavenge injurious radicals by hydrogen abstraction from phenol groups, as in α-tocopherol above.
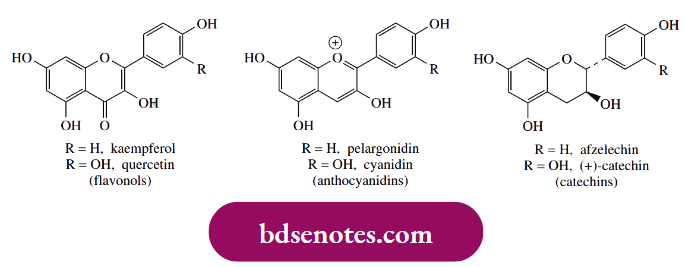
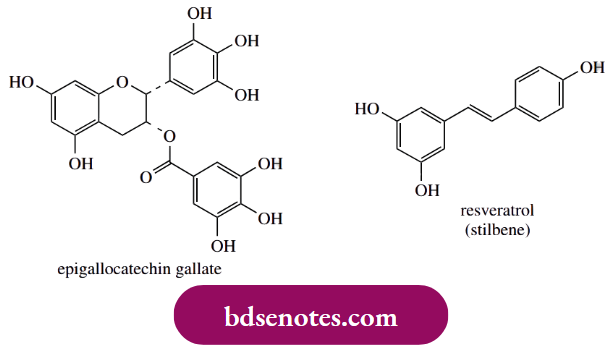
- Quercetin, in particular, is almost always present in substantial amounts in plant tissues and is a powerful antioxidant, chelating metals, scavenging radicals, and preventing oxidation of low-density lipoprotein. Flavonoids in red wine (quercetin, kaempferol, and anthocyanidins) and tea (catechins and catechin gallate esters) are also demonstrated to be effective antioxidants.
- A particularly efficient agent in green tea is epigallocatechin gallate. Resveratrol is another type of polyphenol, a stilbene derivative, that has assumed greater relevance in recent years as a constituent of grapes and wine, as well as other food products, with antioxidant, anti-inflammatory, anti-platelet, and cancer-preventative properties.
- Coupled with the cardiovascular benefits of moderate amounts of alcohol, and the beneficial antioxidant effects of flavonoids, red wine has now emerged as an unlikely but most acceptable medicinal agent. Vitamin C (ascorbic acid) is also a well-known antioxidant. It can readily lose a hydrogen atom from one of its enolic hydroxyls, leading to a resonance-stabilized radical.
- Vitamin C is acidic (hence ascorbic acid) because the loss of a proton from the same hydroxyl leads to a resonance-stabilized anion (see Box 12.8). However, it appears that vitamin C does not act as an antioxidant in quite the same way as the other compounds mentioned above.
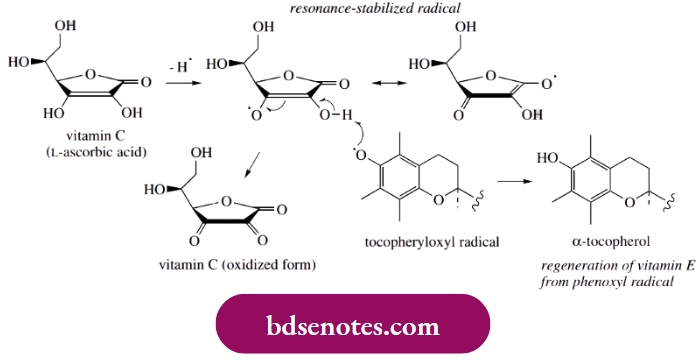
The main function of vitamin C as a radical producer is to provide a regenerating system for tocopherol (see above). Thus, tocopheryloxyl radicals can remove hydrogen atoms from vitamin C to regenerate functioning molecules of tocopherol.
A tocopheryloxyl radical may well be the agent that removes the first hydrogen atom from vitamin C. A second such radical can then abstract a further hydrogen atom and produce the oxidized tricarbonyl form of vitamin C.
Radical Oxidations In Prostaglandin Biosynthesis Cyclo Oxygenase
We saw that the widely used analgesic aspirin exerted its action by acetylating the enzyme cyclooxygenase (COX) which is involved in the production of prostaglandins. Prostaglandins are modified C20 fatty acids synthesized in animal tissues and they affect a wide variety of physiological processes, such as
- Blood pressure, gastric secretion, smooth muscle contraction, and platelet aggregation. Inflammation is a condition that occurs as a direct result of increased prostaglandin synthesis, and many of the non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin and ibuprofen, exert their beneficial effects by reducing prostaglandin formation.
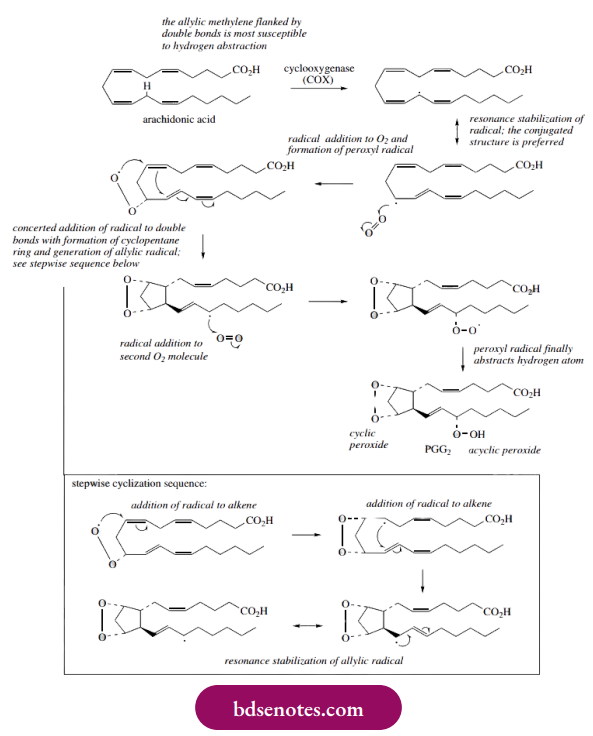
- Prostaglandin biosynthesis from the unsaturated fatty acid arachidonic acid looks very complicated. Breaking the process down into separate steps should reassure us that we have met these reactions already.
- In the reaction catalyzed by COX, arachidonic acid is converted into prostaglandin G2 (PGG2) by incorporating two molecules of oxygen and producing a compound with both cyclic and acyclic peroxide functions. This may be rationalized by radical reactions essentially identical to those we have seen above.
- The major difference is that the initiation reaction giving a radical is achieved by the enzyme, rather than by typical chemical processes. In arachidonic acid, the allylic methylene group flanked by two double bonds is most susceptible to hydrogen abstraction, because of the resonance stabilization conferred.
- There are two such positions in arachidonic acid, but the enzyme is selective. Reaction with oxygen occurs so that a conjugated diene results (see Box 9.1). This leads to a peroxyl radical.
- Formation of PGG2 is then depicted as a concerted cyclization reaction, initiated by the peroxyl radical, through addition to the various double bonds, the enzyme holding the substrate in the required manner to achieve ring formation. It is easier to consider this cyclization via the stepwise sequence shown.
- The resultant radical then reacts with a second oxygen molecule, which abstracts hydrogen from a suitable substrate and generates a hydroperoxide, giving the structure PGG2. The hydrogen atom donor is likely another molecule of arachidonic acid, thus continuing the chain reaction.
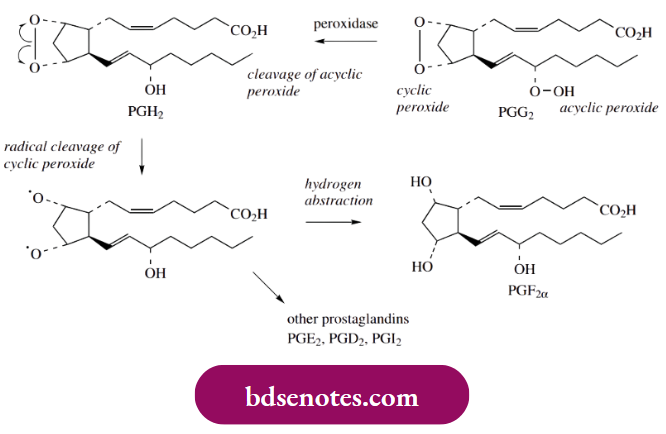
The acyclic peroxide group in PGG2 is then cleaved by a peroxidase enzyme and hydrogen abstraction yields prostaglandin H2 (PGH2), which occupies a central role and can be modified in several different ways.
These further modifications can be rationally accommodated by the initial cleavage of the cyclic peroxide to a diradical. For example, simple quenching of the radicals by abstraction of hydrogen atoms gives rise to prostaglandin F2 α (PGF2 α).
Phenolic Oxidative Coupling
Many natural products are produced by the coupling of two or more phenolic systems, in a process readily rationalized using radical reactions. The reactions can be brought about by oxidase enzymes, including peroxidase and laccase systems, known to be radical generators.
- Other enzymes catalyzing phenolic oxidative coupling have been characterized as cytochrome P-450-dependent proteins, requiring NADPH and O2 cofactors, though no oxygen is incorporated into the substrate.
- Hydrogen abstraction from a phenol (one-electron oxidation) gives the radical, and the unpaired electron can then be delocalized via resonance forms in which the free electron is dispersed to positions ortho or para to the original oxygen function. We have already seen this property in the antioxidant effect of α-tocopherol and other phenolics.
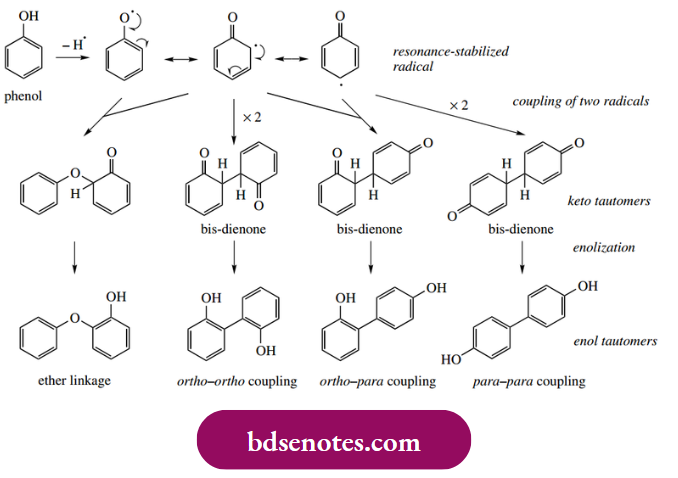
- In phenolic oxidative coupling reactions, these phenol-derived radicals do not propagate a radical chain reaction; instead, they are quenched by coupling with other radicals. Thus, the coupling of two of these resonance structures in various combinations gives a range of dimeric systems, as shown.
- The final products indicated are then derived by enolization, which restores aromaticity to the rings. We shall discuss the concept of enolization in some detail in Section 10.1; for the moment, a simple acid-catalyzed mechanism is shown below.
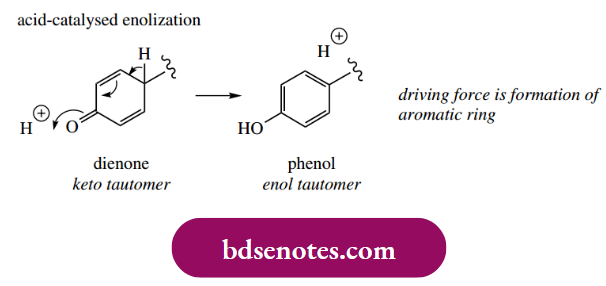
- Accordingly, carbon-carbon bonds involving positions ortho or para to the original phenols, or ether linkages may be formed. The reactive dienone systems formed as intermediates may, in some cases, be attacked by other nucleophilic groupings (see Section 10.10), extending the range of structures ultimately derived from this basic reaction sequence.
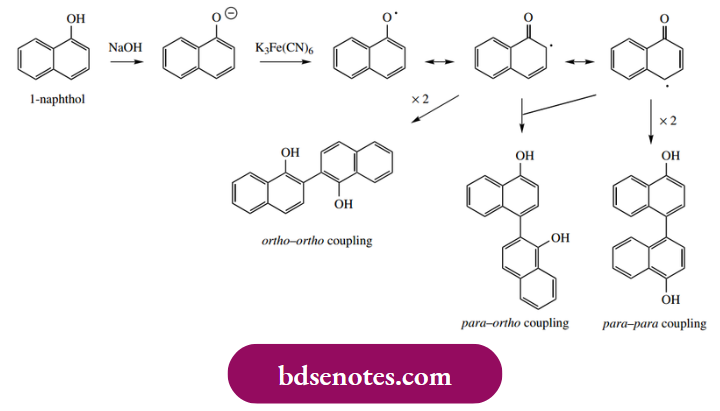
- The phenolic oxidative coupling process can also be demonstrated in laboratory experiments. Thus, treatment of 1-naphthol with alkaline potassium ferricyanide yields a mixture of products, including those shown overleaf. As an oxidizing agent, potassium ferricyanide, K3Fe(CN6), undergoes a change in oxidation state from Fe3+ to Fe2+, i.e. a one-electron change.
- This makes it capable of initiating radical reactions by removal of one electron from the phenolate anion (hence the requirement for alkaline conditions). Thus, the formation of 1-naphthol dimers having ortho–ortho, ortho–para, and para–para coupling modes are easily accommodated.
Phenolic Oxidative Coupling: The Biosynthesis Of Tubocurarine And Morphine
Tubocurarine is the principal component of some varieties of curare, the arrow poison of the South American Indians. Curare is prepared by extracting the bark of several different plants, and then concentrating the extract to a brown glutinous mass. Curare kills by paralyzing muscles, particularly those associated with breathing.
It achieves this by competing with acetylcholine at nicotinic receptor sites, thus blocking nerve impulses at the neuromuscular junction. Curare, and then tubocurarine, have found considerable use as muscle relaxants in surgery, but synthetic analogs have improved characteristics and are now preferred over natural products.
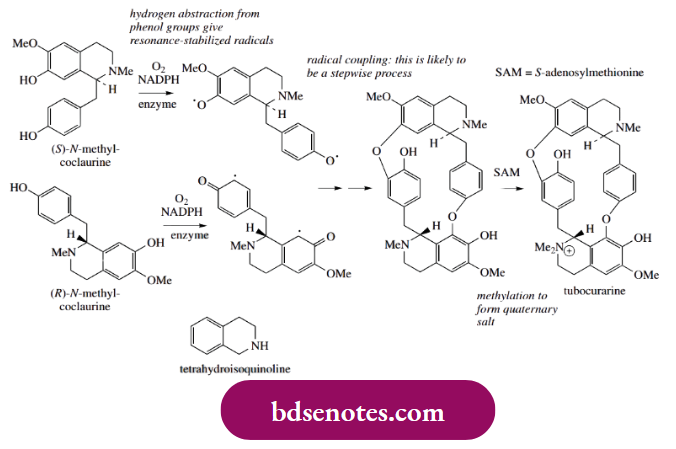
- The structure of tubocurarine has two benzyltetrahydroisoquinoline alkaloid units linked together, and this linking is achieved through phenolic oxidative coupling. We shall meet tetrahydroisoquinoline alkaloids as a product of biochemical Mannich-like reactions.
- Tubocurarine is formed in nature from two molecules of N-methylcoclaurine, one of each configuration. The coupling enzyme is a cytochrome P-450- dependent mono-oxygenase. Radical coupling is readily rationalized.
- The two diradicals, formed by hydrogen abstraction from the phenol group in each ring, couple to give ether bridges. This would be a consequence of the free electrons being localized on carbon in one system and on oxygen in the other.
- It is not proven, but more likely, that the radical coupling is a stepwise process involving simple monoradicals rather than the diradicals shown in the scheme. Tubocurarine is finally elaborated by enzymic methylation of one nitrogen atom to form the quaternary ammonium salt.
- This involves the participation of SAM as the methyl donor. In natural alkaloids, the coupling of two benzyltetrahydroisoquinoline molecules by ether bridges, as in tubocurarine above, is rather less frequent than that involving carbon-carbon bonding between aromatic rings.
- The principal opium alkaloids morphine, codeine, and thebaine are derived by this type of process, though the subsequent reduction of one aromatic ring to some extent disguises their benzyltetrahydroisoquinoline origins. (R)-Reticuline is firmly established as the precursor of morphine-like alkaloids.
- Both morphine and codeine are valuable analgesics. Morphine is extracted from opium, the dried latex of the opium poppy, and codeine is usually obtained from morphine by semi-synthesis, since the amounts in opium are rather small.
- Thebaine is a valuable raw material for the semi-synthesis of a wide range of morphine-like drugs. (R)-Reticuline, turned over and rewritten as in the scheme, is the substrate for hydrogen abstractions via the phenol group in each ring, giving the diradical.
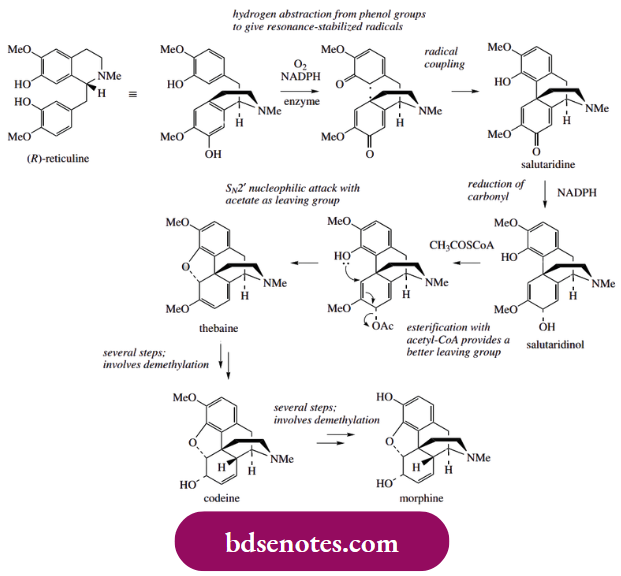
- Coupling ortho to the phenol group in the tetrahydroisoquinoline and para to the phenol in the benzyl substituent then yields the dienone salutaridine, found as a minor alkaloid constituent in the opium poppy.
- Only the original benzyl aromatic ring can be restored to aromaticity since the tetrahydroisoquinoline fragment becomes coupled para to the phenol function, a position that is already substituted.
- The alkaloid thebaine is obtained by way of salutaridinol, formed from salutaridine by stereospecific reduction of the carbonyl group involving NADPH as a reducing agent. Ring closure to form the ether linkage in thebaine would be the result of a nucleophilic attack of the phenol group onto the dienol system and subsequent displacement of the hydroxyl (termed an SN2 reaction).
- This cyclization step can be demonstrated chemically by treatment of salutaridinol with acid. In vivo, however, an additional reaction is used to improve the nature of the leaving group, and this is achieved by acetylation with acetyl-CoA.
- The cyclization then occurs readily and without any enzyme participation. Subsequent reactions involve the conversion of thebaine into morphine by way of codeine, a process that most significantly removes two O-methyl groups.
- The involvement of these O-demethylation reactions is rather unusual; metabolic pathways tend to increase the complexity of the product by adding methyls rather than removing them.
- In this pathway, it is convenient to view the methyl groups in reticuline as protecting groups, which reduce the possible coupling modes available during the oxidative coupling process; these groups are then removed towards the end of the synthetic sequence.
Phenolic Oxidative Coupling The Biosynthesis Of Thyroxine
The thyroid hormone thyroxine is necessary for the development and function of cells throughout the body. It increases protein synthesis and oxygen consumption in almost all types of body tissue. Excess thyroxine causes hyperthyroidism, with increased heart rate, blood pressure, overactivity, muscular weakness, and loss of weight.
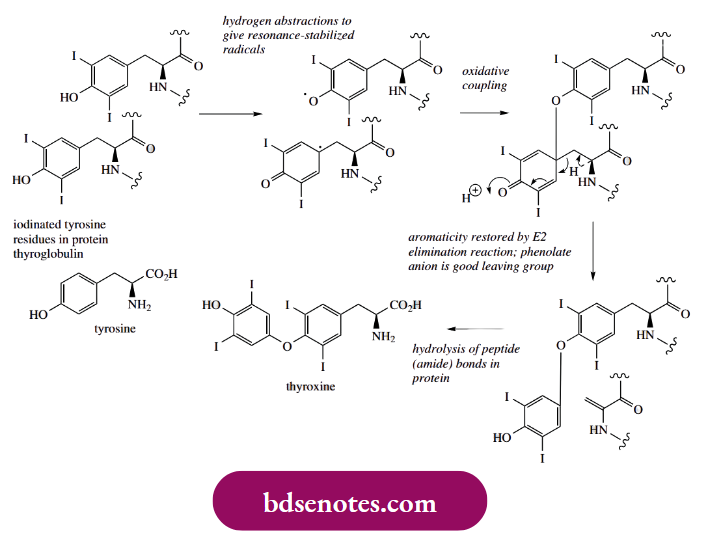
- Too little thyroxine may lead to cretinism in children, with poor growth and mental deficiency, or myxoedema in adults, resulting in a slowing down of all body processes. Thyroxine is a simple derivative of the aromatic amino acid tyrosine but is believed to be derived by the degradation of a larger protein molecule containing tyrosine residues.
- One hypothesis for their formation invokes suitably placed tyrosine residues in the protein thyroglobulin being iodinated to di-iodotyrosine. These residues then react together by phenolic oxidative coupling.
- Coupling allows the formation of an ether linkage, but since the position para to the original phenol is already substituted, it does not allow rearomatization through simple keto-enol tautomerization.
- Instead, rearomatization is achieved by an E2 elimination reaction in the side-chain of one residue, resulting in cleavage of the ring from the side-chain. This is feasible since the phenolate anion is a good leaving group. Thyroxine is then released from the protein by hydrolytic cleavage of peptide.
Leave a Reply