Post Translational Processing Of Proteins
The process of gene expression is not over when the genetic code has been used to create the sequence of amino acids that constitutes a protein.
- To be useful to the cell, this new polypeptide chain must fold up into its unique three-dimensional conformation, bind any small molecule cofactors required for its activity, be appropriately modified by protein kinases or other protein-modifying enzymes, and assemble correctly with the other protein subunits with which it functions.
- Thus, the polypeptide that emerges from the ribosome is inactive, and before taking on its functional role in the cell.
It Must Undergo At Least The First Of The Following Four Types Of Post Translational Processing:
- Protein folding
- Proteolytic cleavage
- Chemical modification
- Inteine splicing
Protein Folding
The proteome is the final product of genome expression and comprises all the proteins present in a cell at a particular time.
- A ‘typical’ mammalian cell, for example, a hepatocyte, is thought to contain 10,000-20,000 different proteins, about 8 × 109 individual molecules in all, representing approximately 0.5ng of protein or 18 – 20% of the total cell weight (airmen, 2002).
- A protein, like a DNA molecule, is a linear unbranched polymer. The monomeric subunits of a protein are called amino acids and the resulting polymers, or polypeptides, are rarely more than 2000 units in length.
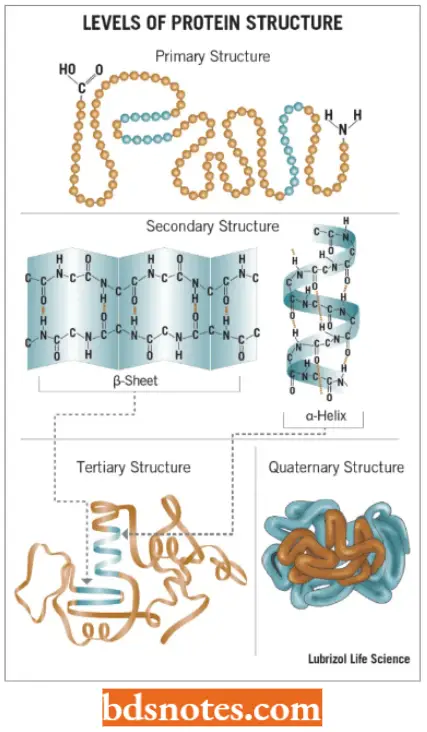
Four Levels Of Protein Structure: Proteins are found to have four distinct levels of structure. These levels are hierarchical, the protein being built up stage by stage, with each level of structure depending on the one below it.
“Understanding post-translational modifications through FAQs: Q&A explained”
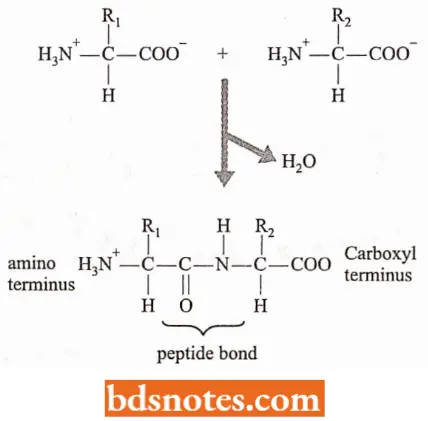
Primary structure: The primary structure of a protein is formed by joining amino acids into a polypeptide.
- The amino acids are linked by peptide bonds which are formed by a condensation reaction between the carboxyl group of one amino acid and the amino group of a second amino acid.
- The two ends of the polypeptide are chemically distinct: one has a free amino group and is called the amino, NH2, – or N – terminus; the other has a free carboxyl group and is called the carboxyl, COOH – or C – terminus.
- The direction of the polypeptide can therefore be expressed as either N → C (left to right) or C → N (right to left).
“Importance of studying post-translational modifications for biology students: Questions explained”
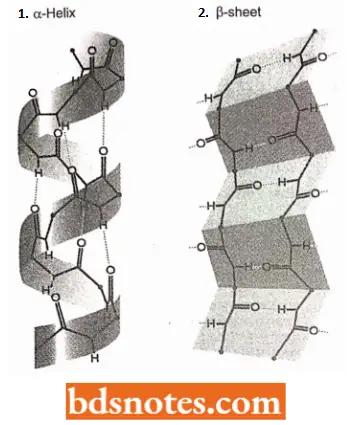
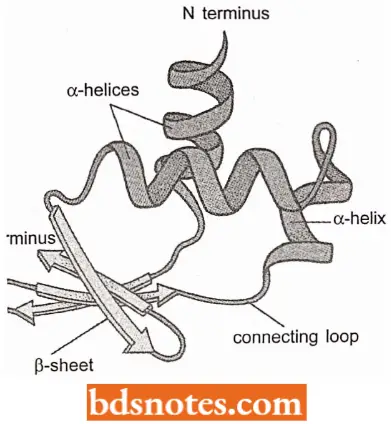
Secondary Structure: The secondary structure of protein refers to the different conformations that can be taken up by the polypeptide.
- The two types of secondary structures are the β-helix and α-sheet, both of which are stabilized by hydrogen bonds that form between different amino acids in the polypeptide.
- Most polypeptides are long enough to be folded into a series of secondary structures one after another along the molecule.
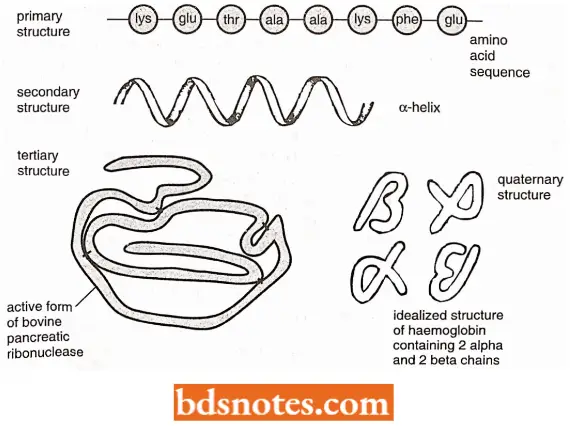
Tertiary Structure: The tertiary structure of protein results from folding the secondary structural components of the polypeptide into a three-dimensional configuration.
- The tertiary structure is stabilized by various chemical forces, notably hydrogen bonding between individual amino acids, and hydrophobic forces, which dictate that amino acids with non-polar (i.e., water-hating or hydrophobic) side groups must be shielded from water by embedding within the internal regions of the protein.
- There may also be covalent linkages called disulphide bridges between cysteine amino acids at various places in the polypeptide.
“Common challenges in understanding post-translational modifications effectively: FAQs provided”
Quaternary structure: The quaternary structure of proteins involves the association of two or more polypeptides, each folded into its tertiary structure, into a multi-subunit protein.
- Not all proteins form quaternary structures, but it is a feature of many proteins with complex functions, including several involved in genome expression.
- Some quaternary structures are held together by disulphide bridges between different polypeptides, but many proteins comprise looser associations of subunits stabilized by hydrogen bonding and hydrophobic effects.
- However, according to the functional requirements, quaternary proteins can revert to their component polypeptides, or change their subunit composition.
- All the information that a polypeptide needs to adopt its correct three-dimensional structure is contained within its amino acid sequence. This is one of the central principles of molecular biology.
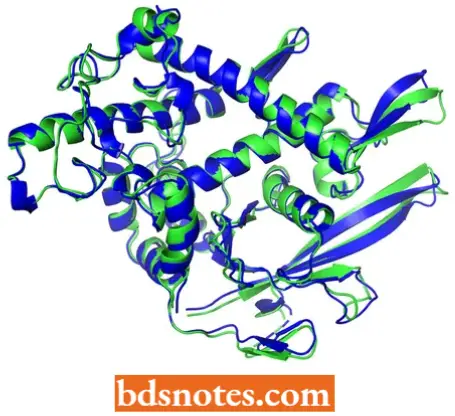
- The notion that the amino acid sequence contains all the information needed to fold the polypeptide into its correct tertiary structure derives from experiments carried out on ribonuclease in the 1960s (Anfinsen, 1973).
- The enzyme ribonuclease is a small protein, just 124 amino acids in length, containing four disulphide bridges and with a tertiary structure that is made up predominantly of α-sheet, with very little β-helix.
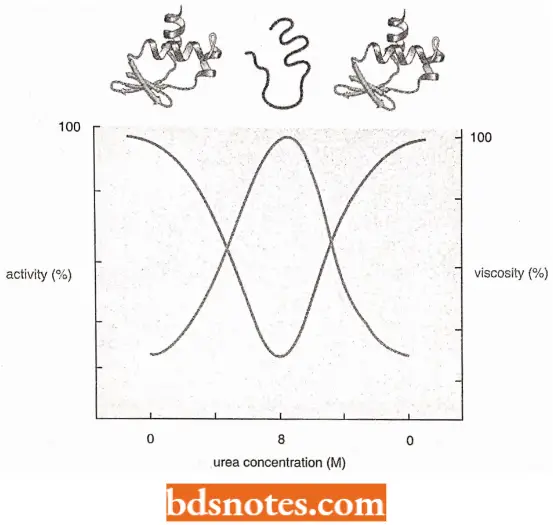
- Experiment showing spontaneous folding pathways for proteins: Studies of folding of proteins were carried out with ribonuclease that had been purified from cow pancreas and resuspended in butler.
- The addition of urea, a compound that disrupts hydrogen bonding, resulted in a decrease in the activity of the enzyme (measured by testing its ability to cut RNA) and an increase in the viscosity of the solution, indicating that the protein was being denatured by unfolding into an unstructured polypeptide chain.
- The critical observation was that when the urea was removed from the experimental solution, the viscosity decreased and the enzyme activity reappeared. The conclusion is that the protein refolds spontaneously when the denaturant (i.e., urea) is removed.
- In these initial experiments, the four disulphide bonds remained intact because they were not disrupted by urea but the same result occurred when the urea treatment was combined with the addition of a reducing agent to break the disulphide bonds: the activity was still regained on renaturation.
- This shows that the disulphide bonds are not critical to the protein’s ability to refold, they merely stabilise the tertiary structure once it has been adopted.
“Why is early learning of post-translational modifications critical for molecular biology? Answered”
A more detailed study of the spontaneous folding pathways for ribonuclease and other small proteins has led to the following two sets of the process:
- The secondary structural motifs along the polypeptide chain form within a few milliseconds
of the denaturant being removed. - During the next few seconds or minutes, the secondary structural motifs interact with one another and the tertiary structure gradually takes shape, often via a series of intermediate conformations.
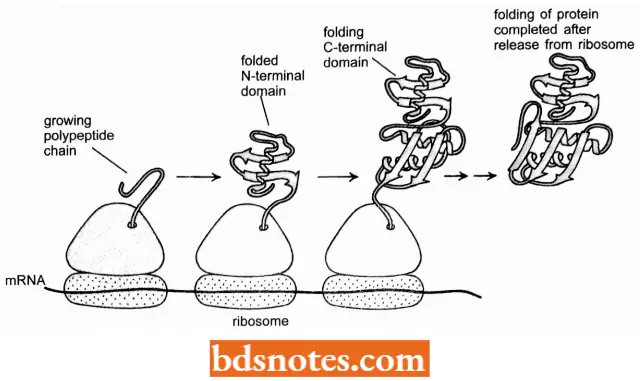
- In other words, the protein follows a folding pathway. However, there may be more than one possible pathway that a protein can follow to reach its correctly folded structure. Such in vitro folding pathways were found true for only the small proteins; large proteins fail to refold.
- Experiments have demonstrated that once a protein domain in a multidomain protein emerges from the ribosome, it forms a compact structure within a few seconds that contains most of the final secondary structure β-helices and α-sheets) aligned in roughly the right way.
- For many protein domains, this unusually open and flexible structure, which is called a molten globule, is the starting point for a relatively slow process in which many side chain adjustments occur that eventually form the correct tertiary structure.
- Nevertheless, because it takes several minutes to synthesize a protein of average size, a great deal of the folding process is completed by the time the ribosome releases the C-terminal end of a protein (Federov and Baldwin, 1997).
Role Of Molecular Chaperones In Folding Of Proteins: Most of our current understanding of protein folding in the cell is based on the discovery of proteins that help other proteins to fold. These are called molecular chaperones and have been studied most vividly in E. coli.
- Both eukaryotes and archaea possess equivalent proteins (chaperones), although some of the details of the way they work are different (Hartl, 1996; Slavotinck and Biesecker, 2001).
- Molecular chaperones in E.coli. The molecular chaperones in E.coli can be divided into two groups: Hsp 70 chaperones (Hsp = heat shock proteins) and chaperonins.
- The Hsp70 chaperones, include the proteins called HSP70 (coded by the dnak gene and sometimes called Dnak protein), Hsp40 (coded by dnaJ) and Grp E.
- The chaperonins, the main version of which in E.coli is the GroEL- GroES complex. Molecular chaperones do not specify the tertiary structure of a protein, they merely help the protein find that correct structure.
- The two types of chaperones do this in different ways. For example, the Hsp70 family (of chaperones) bind to hydrophobic regions of proteins, including proteins that are still being translated.

“Steps to explain types of post-translational modifications: Phosphorylation vs glycosylation: Q&A guide”
“Is post-translational modification-related risk reversible if addressed promptly? Answer provided”
- They prevent protein aggregation by holding the protein in an open conformation until it is completely synthesized and ready to fold.
- The Hsp70 chaperones are also involved in other processes that require shielding of hydrophobic regions in proteins, such as transport through membranes and disaggregation of proteins that have been damaged by heat stress.
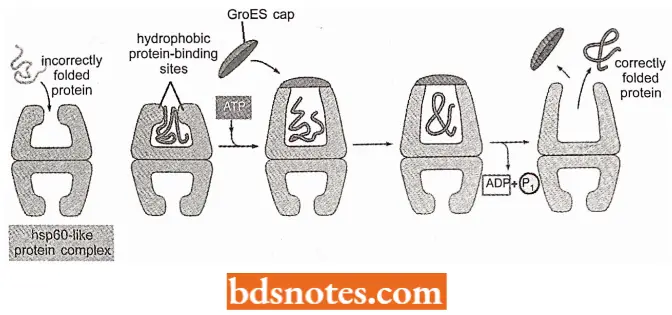
- The chaperonins work in quite a different way. GroEL and GroES form a multi-subunit structure that looks like a hollowed-out bullet with a central cavity Molecular chaperones facilitate correct folding of protein subunits.
- A single unfolded protein enters the cavity and emerges folded. The mechanism for this is not known but it is postulated that GroEL/GroES acts as a cage that prevents the unfolded protein from aggregating with other proteins and that the inside surface of the cavity changes from hydrophobic to hydrophilic in such a way as to promote the burial of hydrophobic amino acids within the protein.
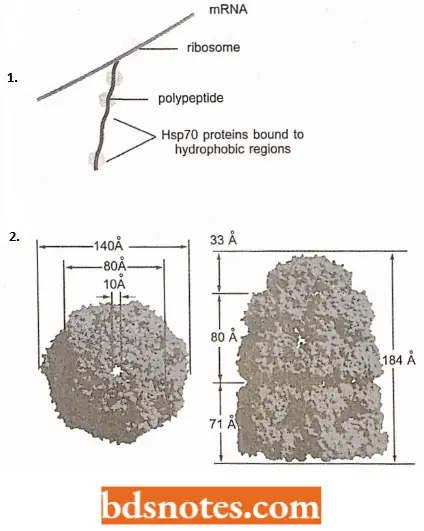
- It is later found that the cavity unfolds proteins that have folded incorrectly, passing these unfolded proteins back to the cytoplasm so that they can have a second attempt at adopting their correct tertiary structure Molecular chaperones and chaperons in eukaryotes.
- Eukaryotic cells have at least two families of molecular chaperones, known as the hsp60 and hsp70 proteins.
- Different family members function in different organelles. For example, mitochondria contain their own hsp60 and hsp70 molecules that are distinct from those that function in the cytosol (where hsp60 is called TCP-1 in vertebrate cells), and a special hsp70 (called BIP) helps to fold proteins in the endoplasmic reticulum.
- The hsp70 machinery acts early in the life of many proteins, binding to a string of about seven hydrophobic amino acids before protein leaves the ribosome.
- The hsp60-like proteins form a large barrel-shaped structure that acts later in the proteins’ life after it has been fully synthesized.
- This type of chaperone forms an “isolation chamber” into which misfolded proteins are fed, preventing their aggregation and providing them with a favourable environment in which they attempt to refold.
“Role of ubiquitination in protein degradation: Questions answered”
Proteolytic Cleavage
Proteolytic cleavage has the following two functions in the post-translational processing of proteins.
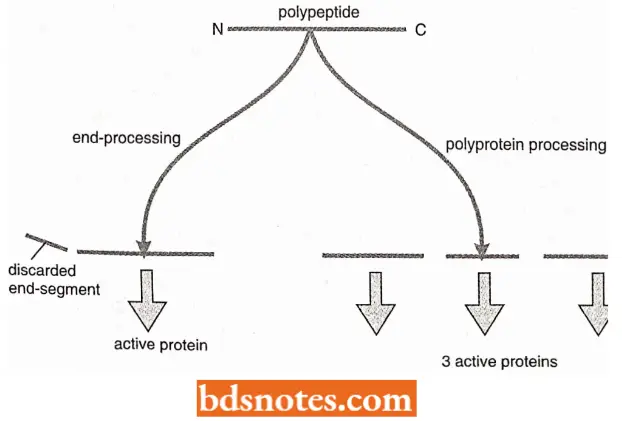
- It is used to remove short pieces from the N – and/or C – terminal regions of polypeptides, leaving a single shortened molecule that folds into the active protein.
- It is used to cut polyproteins into segments, all or some of which are active proteins. These events are relatively common in eukaryotes but less frequent in bacteria. Not all proteins undergo proteolytic cleavage.
Cleavage Of The Ends Of Polypeptides: Processing by proteolytic cleavage is common with secreted polypeptides whose biochemical activities might be injurious to the cell producing the protein.
- An example is provided by melittin, the most abundant protein in bee venom and the one responsible for causing cell lysis after injection of the bee sting into the person or animal being stung.
- Melttin (protein) lyses cells in bees as well as animals so have to be initially synthesized as inactive. precursor. This precursor, called pronielittin, has 22 amino acids at its N-terminus.
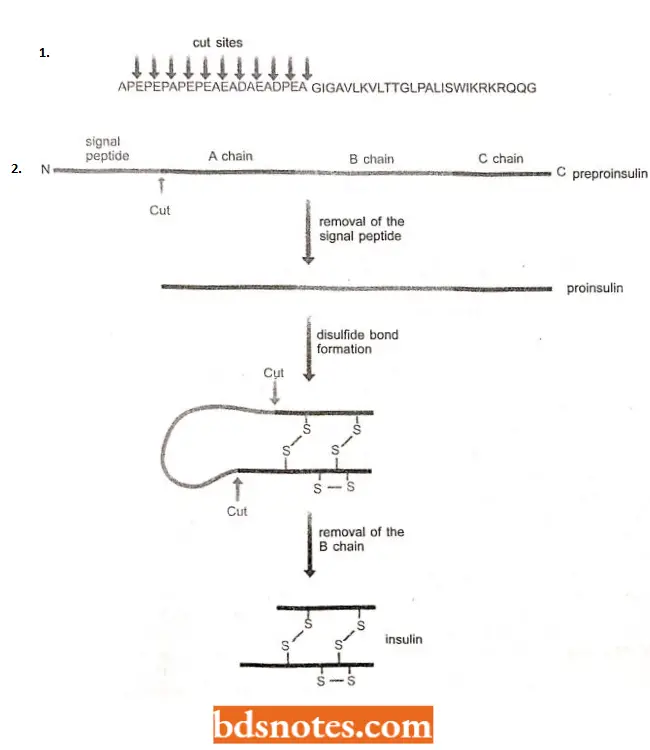
- The pre-sequence is removed by an extracellular protease that cuts it at all eleven positions, releasing the active venom protein.
- The protease does not cleave within the active sequence because its mode of action is to release dipeptides with the sequence X – Y, where X is alanine, aspartic acid or glutamic acid, and Y is alanine or proline, these motifs are not found in the active sequence.
“How does acetylation regulate gene expression? FAQ explained”
Amino Acid Abbreviation:
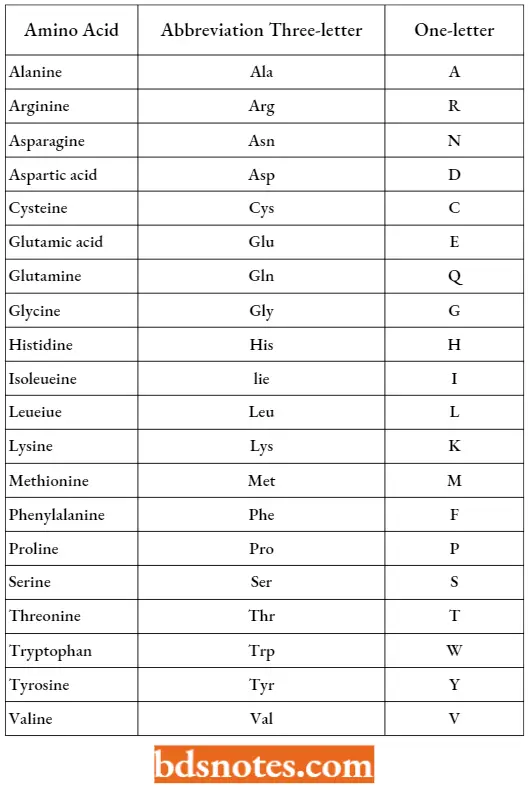
“Early warning signs of gaps in understanding post-translational modification basics: Common questions”
- Insulin also undergoes processing similar to melittin. It is a chromonal protein made in the islets of Langerhans in the vertebrate pancreas and responsible for controlling blood sugar levels. Insulin is synthesised as proproinsulin, which is 105 amino acids in length.
- The processing pathway involves the removal of the first 24 amino acids to give proinsulin, followed by two additional cuts which excise the central segment leaving two active parts of the protein, the A and B chains, which link together by the formation of two disulphide bonds to the form mature insulin.
- The first segment to be removed, the 24 amino acids from the N-terminus, is a signal peptide, a highly hydrophobic stretch of amino acids that attaches the precursor protein to a membrane before transport across that membrane and out of the cell.
Proteolytic Cleavage Of Polyprotein: In the examples, proteolytic processing results in a single mature protein. This does not happen every time.
- Some proteins are initially synthesized as polyproteins, long polypeptides that contain a series of mature proteins linked together in a head-to-tail fashion. Cleavage of the polyprotein releases the individual proteins, which may have very different functions from one another.
- Polyproteins are very common in eukaryotes.
- Several types of viruses that infect eukaryotic cells use them as a way of reducing the sizes of their genomes, a single polyprotein gene with one promoter and one terminator taking up less space than a series of individual genes.
- Polyproteins are also involved in the synthesis of peptide hormones in vertebrates.
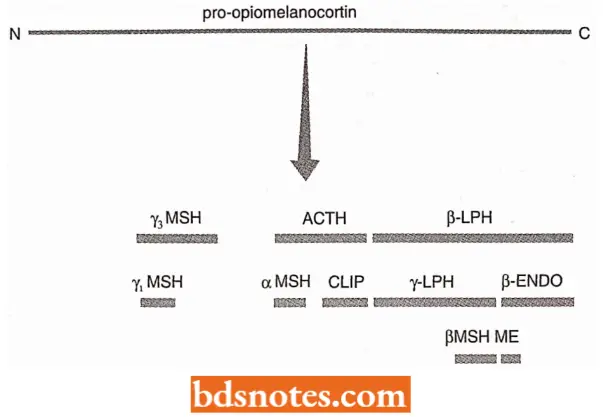
- For example, the polyprotein called pro-opiomelanocortin, made in the pituitary gland, contains at least ten different peptide hormones.
- These are released by proteolytic cleavage of the polyprotein, but not all can be produced at once because of overlaps between individual peptide sequences. Instead, the exact cleavage pattern is different in different cells.
“Asymptomatic vs symptomatic effects of ignoring post-translational modification principles: Q&A”
Chemical Modification
The genome can code for 21 different amino acids: the 20 specified by the standard genetic code, and selenocysteine, which is inserted into polypeptides by the context-dependent reading of a 5′ – UGA – 3′ codon.
- This range is increased dramatically by posttranslational chemical modification of proteins, which results in an immense supply of different types of amino acids.
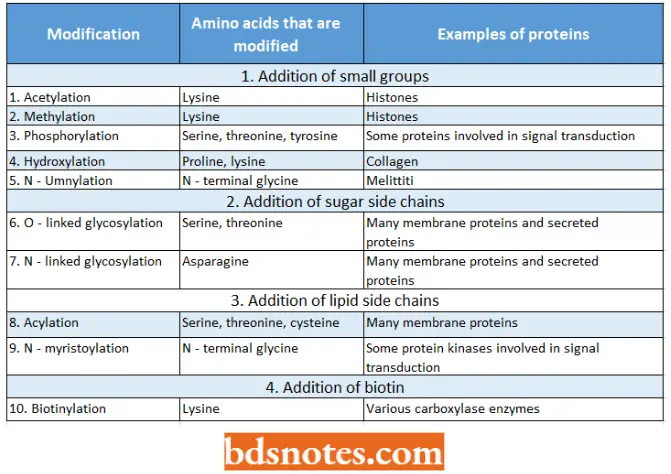
- The most simple sort of chemical modifications involve the addition of a small chemical group (for example., an acetyl, methyl or phosphate group;) to an amino acid side chain, or the amino or carboxyl groups of the terminal amino acids in a polypeptide (Bradshaw et ah, 1998).
- More than 150 different modified amino acids have been reported in different proteins; each modification is found to be carried out in a highly specific manner, i.e., each amino acid being modified in a highly specific manner and the same amino acids being modified in the same way in every copy of the proteins, for example., histone H3.

Examples Of Post-Translational Chemical Modifications:
“Can targeted interventions improve outcomes using post-translational modification knowledge? FAQs provided”
- We have observed how acetylation and methylation of histone H3 and other histones have an important influence on chromatin structure and hence on genome expression.
- Other types of chemical modification have important regulatory roles, for example., phosphorylation is used in transduction.
- Glycosylation is a complex type of modification of amino acids. It involves the attachment of large carbohydrate side chains to polypeptides (Drickamer and Taylor, 1998).
There Are Following Two General Types Of Glycosylation:
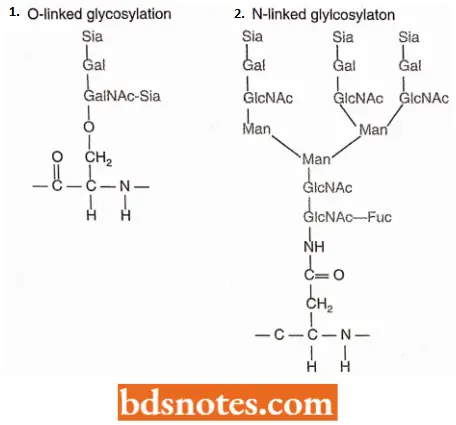
- O-linked glycosylation. It is the attachment of a sugar side chain via the hydroxyl group of a serine or threonine amino acid.
- N-linked glycosylation. It involves attachment through the amino group on the side chain of asparagine.
Glycosylation results in attachment to the proteins of grand structures comprising branched networks of 10 to 20 sugar units of various types.
- These side chains help to target proteins to particular sites in the cells and determine the stability of proteins circulating in the bloodstream.
- Another category of large-scale modification is acylation which involves the attachment of long-chain lipids, often to serine or cysteine amino acids. Acylation occurs with many proteins that become associated with membranes.
- A less common modification is biotinylation, in which a molecule of biotin is attached to a small number of enzymes that catalyze the carboxylation of organic acids such as acetate and propionate (Chapman, Smith and Cronan, 1999).
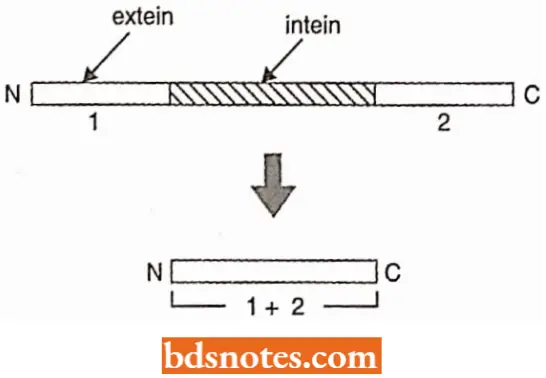
Inteine Splicing
The final type of post-translational processing is inteine splicing. It is a protein version of the more extensive intron splicing that occurs in the context of pre-RNAs.
- Inteins are internal segments of proteins that are removed soon after translation, the two external segments or exteins becoming linked together.
- The first intein was discovered in 1990 in S. cerevisiae and more than 100 inteins have been reported till today.
- Most interns are known in bacteria, archaea and lower eukaryotes. In a few cases, there is more than one intein in a single protein.
“Differential applications of reversible vs irreversible modifications: Questions answered”
- Most inteins are approximately 150 amino acids in length and like pre-mRNA introns, the sequences at the splice junctions of inteins have some similarity in most of the known examples.
- The first amino acid of the downstream extein is cysteine, serine or threonine.
- A few other amino acids within the intein sequence are also conserved. These conserved amino acids are involved in the splicing process, which is catalyzed by the intein itself.
Characteristics Of Inteins: Self-catalysis. The first interesting feature of inteins was discovered when the structure of two inteins was determined by X-ray crystallography (Duan et al., 1997; Klabunde et al.,
1998).
- These structures are similar in some respects to that of a Drosophila protein called Hedgehog, which is involved in the development of the segmentation pattern of the embryo of fruit fly. Hedgehog is an autoprocessing protein that cuts itself into two.
- The structural similarity with inteins lies in the part of the Hedgehog protein that catalyzes its self-cleavage.
- Molecular biologists believe that possibly the same protein structure has evolved twice, or possibly inteins and Hedgehog protein shared a common link at some stage in the evolutionary past.
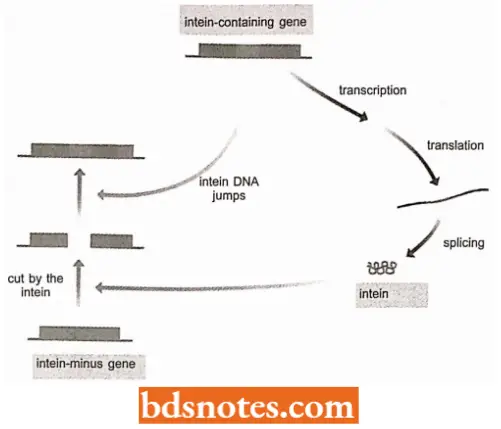
Intein Homing: The second interesting feature of intein is that with some inteins the excised segment is a sequence-specific endonuclease.
- The intein cuts DNA at the sequence corresponding to its insertion site in a gene coding for an intein-free version of the protein from which it is derived.
- If the cell also contains a gene coding for the intein-containing protein, then the DNA sequence for the intein can jump into the cut site converting the intein-minus gene into an intein-plus version, a process called intein homing (Pietrokovski, 2001).
Monitoring And Control Of Protein Quality
A protein that has a sizable exposed patch of hydrophobic amino acids on its surface is usually abnormal: it has either failed to fold correctly after leaving the ribosome, suffered an accident that partly unfolded it at a later time, or failed to find its normal partner subunit in a larger protein complex.
- Such a protein is useless to the cell. Further, such protein complexes can be dangerous. Many proteins with an abnormally exposed hydrophobic region can form large aggregates, precipitating out of solution.
- We shall see that in rare cases, such aggregates form and cause severe human diseases. But in the vast majority of cells, powerful protein quality control mechanisms prevent such disasters.
- Cells have evolved elaborate mechanisms that recognize and remove the hydrophobic patch proteins. Two of the mechanisms depend on the molecular chaperones just discussed, which bind to the patch and attempt to repair the defective protein by giving it another chance to fold.

“Difference between phosphorylation and glycosylation in proteins: Q&A explained”
- At the time, by covering the hydrophobic patches, these chaperones transiently prevent protein aggregation. Proteins that very rapidly fold correctly on their own do not display such patches and are therefore bypassed by chaperones.
- All of the options of quality control that a cell makes for a difficult-to-fold newly synthesized protein have been outlined.
- As indicated, when attempts to refold a protein fail, a third option is called into play that destroys the protein by proteolysis.
- The proteolytic pathway begins with the recognition of an abnormal hydrophobic patch on the surface of a protein and it ends with the delivery of the entire protein to a protein destruction machine, a complex protease called proteosome.
- As described next, this process depends on an elaborate protein marking system that also carries out other central functions in the cell by destroying selected normal proteins.
Protein Degradation: The protein synthesis and processing events that we have studied so far result in new proteins that take up their place in the cell’s proteome. These proteins either replace existing ones that have reached the end of their working lives or provide new protein functions in response to the changing requirements of the cell.
- The concept that the proteome of a cell can change over time requires not only de novo protein synthesis but also the removal of proteins whose functions are no longer required.
- This removal must be highly selective so that only the correct proteins are degraded, and must also be rapid to account for the abrupt changes that occur under certain conditions, for example during key transitions in the cell cycle (Hunt, 1997).
- In eukaryotes, most breakdown involves a single system, involving ubiquitin and the proteasome. A link between ubiquitin and protein degradation was first established in 1975 when it was shown that this abundant 76-amino acid protein is involved in energy-dependent proteolysis reactions in rabbit cells (Varshavsky, 1997).
- Subsequent research identified a series of three enzymes that attach ubiquitin molecules, singly or in chain, to lysine amino acids in proteins that are targeted for breakdown.
- Whether or not a protein becomes ubiquitinated depends on the presence or absence within it of amino acid motifs that act as degradation-susceptible signals.
These signals have not been completely characterized but yeast is known to contain about 10 different such signals. These signals include the following two categories.
- The N-degron, a sequence element present at the N-terminus of a protein;
- PEST sequence, internal sequences that are rich in proline (P), glutamic acid (E), serine (S) and threonine (T).
These sequences are permanent features of the proteins that contain them and so cannot be straightforward ‘degradation signal’-ifthey were so then these proteins would be broken down as soon as they are synthesized.
- Instead, they must determine susceptibility to degradation and hence the general stability of a protein in the cell.
- How this might be linked to the controlled breakdown of selected proteins at specific times, for instance during the cell cycle, is little understood.
“Most common complications of poorly understood post-translational modifications: FAQs”
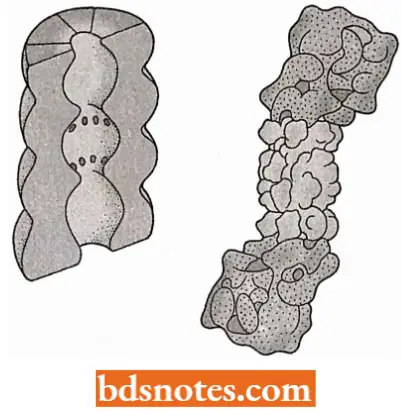
- The second component of the ubiquitin-dependent degradation pathway is the proteosome the structure within which ubiquitinated proteins are broken down.
- In eukaryotes, the proteasome is a large, multi-subunit structure with a sedimentation coefficient of26S, comprising a hollow cylinder of 20S and two “caps” of 19S (Groll et al., 1997; Ferrell et al., 2000).
- Archaea also have proteasomes of about the same size but these are less complex, being composed of multiple copies of just two proteins; eukaryotic proteasomes contain 14 different types of protein subunits.
- The entrance into the cavity within the proteasome is narrow, and a protein must be unfolded before it can enter. This unfolding probably occurs through an energy-dependent process and may involve a structure similar to chaperonins.
- After unfolding, the protein can enter the proteasome within which it is cleaved into short peptides 4-10 amino acids in length. These are released back into the cytoplasm where they are broken down into individual amino acids which can be reutilized in protein synthesis.
Abnormal Protein Folding Causes Destructive Human Diseases: When all of a cell’s protein quality controls fail, large protein aggregates tend to accumulate in the affected cell.
- Some of these aggregates, by adsorbing critical macromolecules to them, can severely damage cells and even cause cell death.
- The protein aggregates released from dead cells tend to accumulate in the extracellular matrix that surrounds the cells in a tissue, and in extreme cases, they can also damage tissues.
- Since the brain is composed of a highly organised collection of nerve cells, it is especially vulnerable. These protein aggregates primarily tend to cause diseases of neurodegeneration.
- Prominent among these are Huntington’s disease and Alzheimer’s disease – the latter causing age-related dementia in more than 20 million people in today’s world (Alberts et al., 2002).
- A protein aggregate, which has to survive, grow and damage an organism, must be highly resistant to proteolysis both inside and outside the cell.
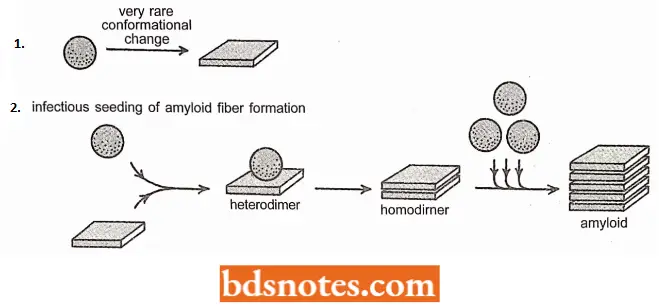
- Many of the protein aggregates that cause problems from fibrils built from a series of polypeptide chains that are layered one over the other as a continuous stack of p-sheets.
- These are so-called cross beta filaments, which tend to be highly resistant to proteolysis. This resistance presumably explains why this structure is observed in so many of the neurological disorders caused by protein staining deposits, called amyloid.
“Why are post-translational modification mechanisms often misunderstood in practice? Questions answered”
- One particular variety of these neurological diseases is quite notorious. These are prion diseases. Unlike Huntington’s or Alzheimer’s disease, a prion disease can spread from one organism to another, providing that the second organism eats a tissue containing the protein aggregate.
- A set of diseases- called scrapie in sheep, Creutzfeldt- Jacob disease (CJD) in humans, and bovine spongiform encephalopathy (BSE) in cattle-are caused by a misfolded, aggregated form of a protein called PrP (for prion protein).
- The PrP is normally located on the outer surface of the plasma membrane, most prominently in neurones.
- Its normal function is not known. However, PrP has the unfortunate property of being convertible to a very special abnormal conformation.
- This conformation not only forms protease-resistant, cross-beta filaments; it also is “infectious” because it converts normally folded molecules of PrP to the same form.
- This property creates a positive feedback loop that produces the abnormal form of PrP, called PrP and thereby allows PrP to spread rapidly from cell to cell in the brain, causing death of both animals and humans.
- It is dangerous to eat the tissues of animals that contain PrP*, as witnessed most recently by the spread of BSE (commonly referred to as the “mad cow disease”) from cattle to humans in Great Britain.
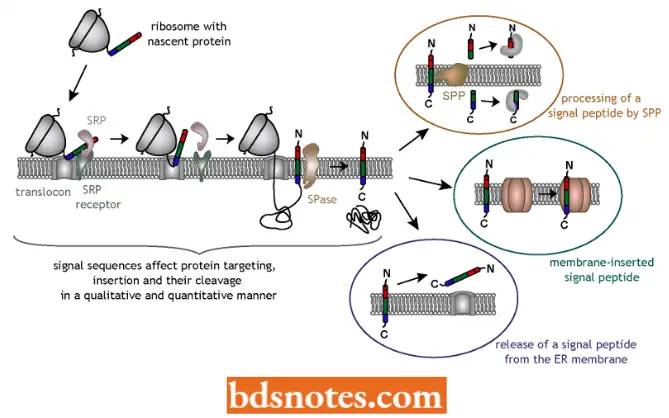
Signal Hypothesis (Protein Targeting)
Ribosomes are either free in the cytoplasm or associated with membranes, depending on the type of protein being synthesised. Membrane-bound ribosomes, which are indistinguishable from free ribosomes, synthesise proteins that enter membranes.
- These proteins either become a part of the membrane or in eukaryotes, either pass into membrane-bound organelles (for example., the Golgi apparatus, mitochondria, chloroplasts, vacuoles) or transported outside the plasma membrane.
- The signal hypothesis of Gunter Blobcl, a 1999 Nobel laureate, and his colleagues, explains the mechanism for membrane attachment. The mechanism applies to both prokaryotes and eukaryotes. Here, the signal hypothesis has been described for mammals).
“Success rate of interventions using modern post-translational modification techniques: FAQ”
- The signal for membrane insertion is in the form of a polypeptide. It is coded into the first one to three dozen amino acids of membrane-bound proteins. This signal peptide takes part in a chain of events that leads the ribosome to attach to the membrane and insertion of the protein. The first step occurs when the signal peptide becomes exposed on the outside of the ribosome.
- A ribonucleoprotein particle, called signal recognition particle (SRP), which consists of six different proteins and a 7S RNA (about 300 nucleotide long), recognizes the signal peptide.
- In the second step, the complex of signal recognition particle, ribosome, and signal peptide then passes to a membrane, where the SRP binds to a receptor called a docking protein (DP) or signal recognition particle receptor.
- During this time, protein synthesis or translation halts. The ribosome is brought into direct contact with the membrane, and other proteins which help anchor the ribosome.
- In the third step, the protein synthesis resumes, with the nascent protein usually passing directly into a translocation channel (called translocon). Once through the membrane, the signal peptide is cleaved from the protein by an enzyme called signal peptidase.
- Evidence for the signal hypothesis came about through recombinant DNA techniques.
- A signal sequence was placed in front of the a-globin gene, whose protein product is normally not transported through a membrane. When this gene was translated, the ribosome became membrane-bound, and the protein passed through the membrane.
Since different proteins enter different membrane-bound compartments (for example the Golgi apparatus), some mechanism must direct a nascent protein to its proper membrane. This specificity seems to depend on the exact signal sequence and membrane-bound glycoproteins called signal sequence receptors.
- After the ribosome binds to the docking protein, the signal peptide interacts with a signal sequence receptor, which presumably determines whether that protein is specific for that membrane. If it is, the remaining processes continue.
- If not, the ribosome may be released from the membrane. The signal peptide does not seem to have a consensus sequence similar to transcription or translation recognition boxes.
- Rather similarities (at least for the endoplasmic reticulum and bacterial membrane-bound proteins) include a positively charged (basic) amino acid (commonly lysine or arginine) near the beginning (N- terminal end), followed by about a dozen hydrophobic (nonpolar) amino acids, commonly alanine, isoleucine, leucine, phenylalanine and valine.
“Cost of ignoring post-translational modification principles vs benefits of systematic approaches: Q&A”
The Signal Peptide Of The Bovine Prolactin Protein:
NH2– Met Asp Ser Lys Gly Ser Ser Gin Lys Gly Ser Arg Leu Leu Leu Leu Leu Val Ser Asn Leu Leu Leu Cys Gin Gly Val Val Ser/Thr Pro Val .Asn Asn Cys – COOH
Post Translational Processing Multiple Choice Questions And Answers
Question 1. Peptide linkage is
- —CO—NH—
- —CO—NH2
- —COOH—NH2
- —CH—N
Answer: 1. —CO—NH—
Question 2. The tertiary structure of proteins having amino acids cysteine is achieved through
- Ionic bonds
- Covalent bonds
- Disulphide bonds
- Hydrogen bonds
Answer: 3. Disulphide bonds
Leave a Reply