Introduction To Medicinal Chemistry
Medicinal chemistry is an interdisciplinary field study combining aspects of organic chemistry, physical chemistry, pharmacology, microbiology, biochemistry as well as computational chemistry.
It is concerned with the invention, discovery, design, identification, and preparation of biologically active compounds, the study of their metabolisms, the interpretation of their mode of action at the molecular level and the construction of “structure-activity relationships”.
Medicinal chemistry covers the following stages:
- New active substances or drugs are identified and prepared from natural sources, organic chemical reactions, or biotechnological processes. They are known as lead molecules.
- Optimization of lead structure to improve potency, and selectivity and to reduce toxicity.
- Development stage, which involves optimization of synthetic route for bulk production and modification of pharmacokinetic and pharmaceutical properties of active substance to render it clinically useful.
During the early stages of medicinal chemistry development, scientists were primarily concerned with the isolation of medicinal agents found in plants.
Today, scientists in this field are also equally concerned with the creation of new synthetic compounds as drugs. Medicinal chemistry is almost always geared toward drug discovery and development.
History And Development Of Medicinal Chemistry
The 19th century may be viewed as the birth period of modern medicinal chemistry with the introduction of the side-chain theory of drug action in 1885 by Berlin immunologist Ehrlich.
Later in 1891, he coined the term chemotherapy and defined it as “the chemical entities exhibiting selective toxicities against particular infectious agent.
- The modem drug receptor theory originated from this side-chain theory, which was supported during the same period (the mid-1890s) by Cambridge physiologist Langley who described it in his publications as “receptive substances”.
- Research on enzyme specificity (lock-and-key theory) by Fischer in 1894 and Henry’s hypothesis on enzyme-substrate complex formation in 1903 are recognized as key advancements in the principles of drug action and modern medicinal chemistry.
- Grimm’s and Erlenmeyer’s concepts of isosteric and boosterism also had a tremendous impact on the understanding of the structure-activity relationship (SAR) of drugs and the development of modern medicinal chemistry.
- Other notable advancements in the understanding of drug action and design that were made in the mid to late 20th century include intervention of charge transfer (Kosower, 1955); and induced-fit theony of drug action (Koshland, 1958).
Concepts of drug latentiation (Harper, 1959) and prodrug (Albert, 1960); application of mathematical methods to medicinal chemistry and transformation of SAR studies into quantitative SAR (QSAR) (Hansen and others, 1960s); and application of artificial intelligence to drug research (Chu, 1974).
- Diseases of protozoal and spirochetal origin responded to synthetic chemotherapeutic agents. Interest in synthetic chemicals that could inhibit.
- The rapid reproduction of pathogenic bacteria and the host organism to cope with invasive bacteria was dramatically increased.
- When Domagk reported that the red dyestuff 2,4-diaminoazobenzene-4′-sulfonamide (Prontosil) dramatically cured dangerous, systemic gram-positive bacterial infections in men and animals.
- The observation by Woods and Fildes in 1940 that the bacteriostatic action of sulfonamide-like drugs was antagonized by p-aminobenzoic acid, was one of the early examples in which a balance of stimulatory and inhibitory properties depended on the structural analogies of chemicals.
Together with the discovery of penicillin by Heming in 1929 and its subsequent examination by Florey and Chain in 1941 led to a water-soluble powder of much higher antibacterial potency and lower toxicity than those of previously known synthetic chemotherapeutic agents.
- With the discovery of a variety of highly potent anti-infective agents, a significant change was introduced into medical practice.
- Inventing and developing a new medicine is a long, complex, costly, and highly risky process that has few peers in the commercial world.
- Research and development (R and D) for most of the medicines available today has required 12-24 years for a single new medicine, from starting a project to the launch of a drug product.
- In addition, many expensive, long-term research projects completely fail to produce a marketable medicine.
- In the drug development phase, experience has shown that only approximately about 15-25 drug candidates survive the detailed safety and efficacy testing (in animals and humans) required for it to become a marketed product.
This is a high-stakes, long-term, and risky activity, but the potential benefits to the millions of patients with serious diseases provide a constant motivating force.
- The most striking differences from the longstanding practice of medicinal chemistry in the new millennium are:
- Data reduction of huge amounts of rapidly derived HTS biological results,
- Greater emphasis upon multi-technique chemical structure considerations, and most importantly,
The simultaneous attention given to all of the ADMET-related parameters along with efficacy and efficacy-related selectivity (E/S) during lead compound selection and further design or enhancement coupled with an expanding knowledge base that offers.
- The possibility for achieving synergistic benefits by taking advantage of various combinations of multi-agent, prodrug, soft drug, and or multivalent drug strategies.
- Medicinal chemists today live in exciting times. They are key participants in the effort to produce more selective, more effective, and safer medicines to treat the diseases of mankind.
- Their work can have a beneficial effect on millions of suffering patients – surely an important motivating factor for any scientist.
Physicochemical Properties Ih Relation To Biological Action
The ability of a chemical compound to elicit a pharmacological or therapeutic effect is related to the influence of various physical and chemical (physicochemical) properties of the chemical substance on the biomolecule that it interacts with.
Physical Properties: The physical property of a drug is responsible for its action.
Chemical Properties: The drug reacts extracellularly according to simple chemical reactions like neutralization, chelation, oxidation, etc.
Various Physico-Chemical Properties are:
- Solubility
- Partition Coefficient
- Ionization
- Hydrogen Bonding
- Chelation
- Surface activity
- Isosterism
1. Solubility
The solubility of a substance at a given temperature is defined as the concentration of the dissolved solute, which is in equilibrium with the solid solute. Sufficient solubility and membrane permeability an important factors for oral absorption.
The measurement of aqueous solubility depends upon the following facts.
- Buffer and Ionic strength
- Polymorphism and Purity of the sample
- pH
- Supersaturation
- The thermodynamic versus Kinetic solubility
The solubility of organic medicinal agents may be expressed in terms of its
affinity philicity repulsion or phobicity for either an aqueous (hydro) or lipid (lipo) solvent.
- hydrophilic …… water-loving.
- lipophobic …… lipid hating.
- lipophilic …… lipid-loving.
- hydrophobic …… water hating.
The most important intermolecular attractive forces (bonds) that are involved in the solubilization process are described as follows.
- Vander Waals attraction (induced dipole): They are the weakest intermolecular forces (0.5-1.0 kcal/mole) that occur between nonpolar groups (For Example. hydrocarbons). They are highly distance and temperature-dependent.
- Dipole-Dipole Bonding: These forces occur when electronegative elements are attached to carbon. They are stronger (1.0 to 10 kcal/mole) and occur electrostatically between electron-deficient and electron-rich atoms (dipoles).
- Hydrogen bonding is a specific example of this bonding and serves as a prime contributor to hydrophilicity.
- Ionic Bonding: Ionic bond is electrostatic attraction between cations and anions. These ionic attractions are common in inorganic compounds and salts of organic molecules and are relatively strong (5 kcal/mole).
- Probably the most important factor in the prediction of water solubility in ionic drugs is their ability to ionize. The degree of ionization of a drug is by far the best predictor of solubility for most compounds, which are acidic or basic.
- Ion-Dipole Bonding: This is the electrostatic force between a cation/anion and a dipole. It is relatively strong (1-5 kcal/mole) and is low temperature and distance dependent. Ion dipole bonding is an important attraction between organic medicinal agents and water.
Hence, the relative solubility of an organic medicinal agent is a function of the presence of both lipophilic and hydrophilic features within its structure, which serve to determine the extent of interaction of the organic medicinal agent with lipid or aqueous phases.
In ascending homologous senes, the physicochemical properties like boiling point viscosity. surface activity and partition coefficient increase then the aqueous solubility decreases.
The solubility characteristics of a drug can be increased or decreased by derivatization.
Example: Methylprednisolone acetate (water-insoluble) is changed to Methylprednisolone sodium succinate (water-soluble).
Example: Conversion of chloramphenicol (slightly soluble) to chloramphenicol palmitate (insoluble).
Methods to improve the solubility of drugs
- Structural modification
- Use of cosolvents
- Employing surfactants
- Complexation
Partition Co-efficient
The partition coefficient is one of the physicochemical parameters that influence drug transport and drug distribution, how the drug reaches the site of action from the site of application.
The partition coefficient is defined as the equilibrium constant of drug concentration for a molecule in two phases.
- P [Unionized molecule] = \(\frac{\text { [Drug] lipid }}{\text { [Drug] water }}\)
- P [Ionized molecule] = \(\frac{\text { [Drug] lipid }}{[1-\text { a }][\text { Drug] water }}\)
where, a = degree of ionization in aqueous solution.
Factors Affecting Partition Coefficient:
- pH
- Cosolvents
- Surfactant
- Complexation
Partition coefficients are difficult to measure in a living system. They are usually determined in vitro 1-octanol as a lipid phase and phosphate buffer of pH 7.4 as the aqueous phase.
The partition coefficient, P is dimensionless and its logarithm, log P is widely used as the measure of lipophilicity.
The log P is measured by the following methods:
- Shake flask method
- Chromatographic method
- Spectroscopy method
Phenobarbitone has a high lipid or water partition coefficient of 5.9. Thiopentone sodium has a chloroform or water partition coefficient of about 100, so it is highly soluble in lipids.
Surfactant
A surfactant is defined as a material that can reduce the surface tension of water at low concentrations. Surface active agents affect drug absorption which depends on:
- The chemical nature of surfactant.
- Its concentration.
- Its effect on biological membranes and the miscellaneous formation.
At lower concentrations, the surfactant enhances the absorption rate, the same in higher concentrations reduces the absorption rate.
Applications:
- The anthelmintic activity of hexylresorcinol.
- Bactericidal activity of cationic quaternary ammonium compounds.
- Bactericidal activity of aliphatic alcohols.
- Disinfectant action of phenol and cresol.
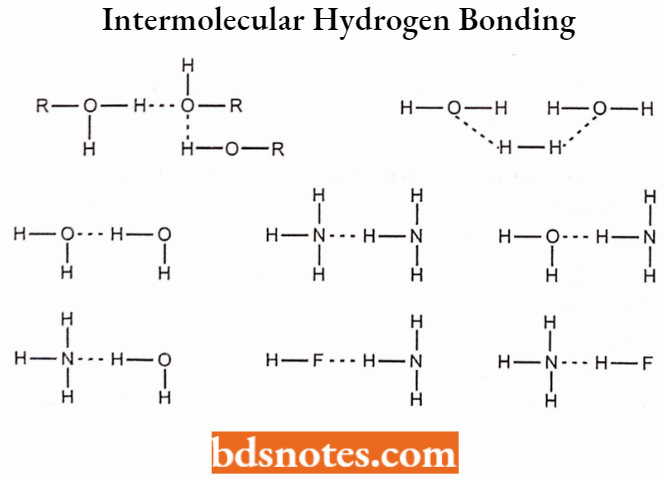
Hydrogen Bond
The hydrogen bond is a special dipole-dipole interaction between non-bonding electron pairs of hetero atoms like N, S, and O and electron-deficient hydrogen atoms in polar bonds such as OH, NH, F, etc.
These are weak bonds and are denoted as dotted lines.
O-H…….O, HN-H O
The compounds that are capable of forming hydrogen bonding are only soluble in water. Hydrogen bonding is classified into two types.
Intermolecular Hydrogen Bonding:
In the Intermolecular Hydrogen Bonding type, hydrogen bonding occurs between two or more two molecules of the same compound and results in the formation of a polymeric aggregate.
- Intermolecular hydrogen bonding increases the boiling point of the compound and also its solubility in water.
- The molecules that can develop intermolecular hydrogen bonding improve their solubility by the formation of intermolecular hydrogen bonding with water.
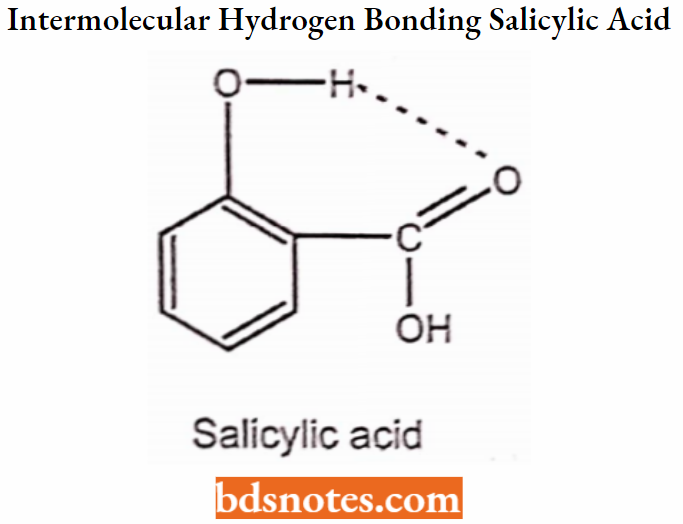
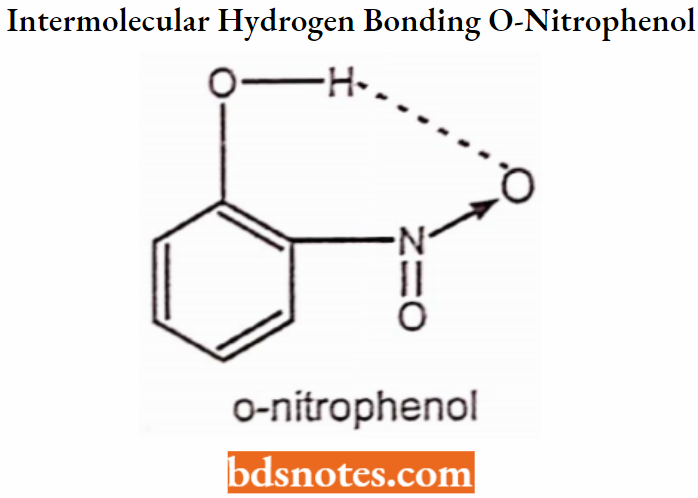
Intramolecular Hydrogen Bonding:
In Intramolecular Hydrogen Bonding, hydrogen bonding occurs within two atoms of the same molecule. This type of hydrogen bonding is commonly known as chelation and frequently occurs in organic compounds. Sometimes intramolecular hydrogen bonding develops a six or five-membered ring.
Hydrogen bonding and biological action
Example: Antipyrin i.e. 1-phenyl 2, 3-dimethyl 5-pyrazolone has analgesic activity.
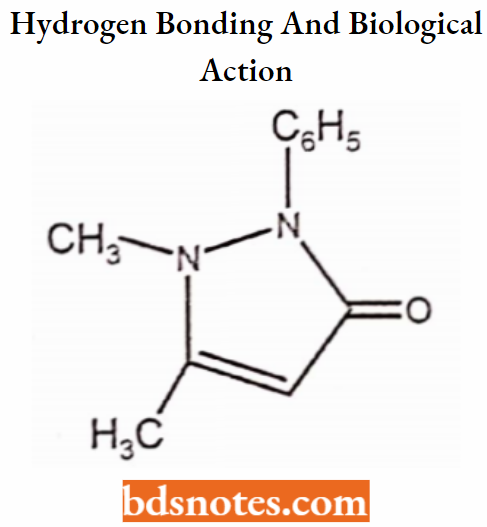
l-phenyl-3-methyl-5-pyrazolone is inactive
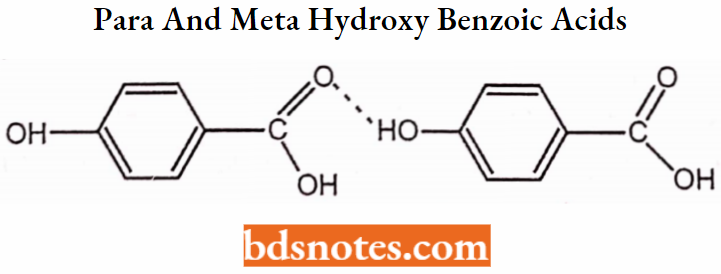
Salicylic acid (o-Hydroxy benzoic acid) has antibacterial activity
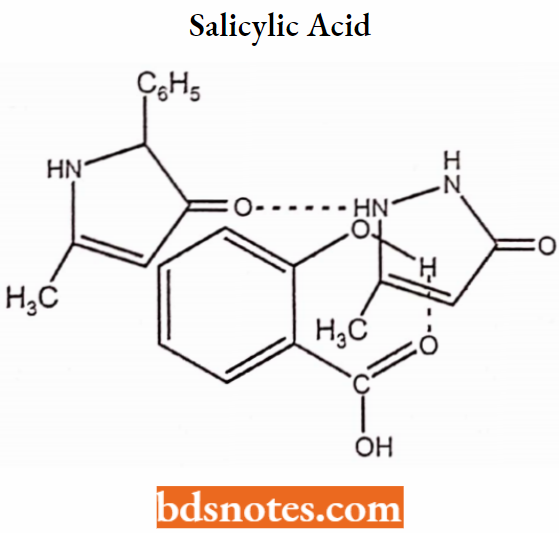
para and meta hydroxybenzoic acids are inactive.
Chelation
The compounds that are obtained by donating electrons to a metal ion with the formation of a ring structure are called chelates. The compounds capable of forming a ring structure with a metal are termed as ligands.
Importance of Chelates in Medicine:
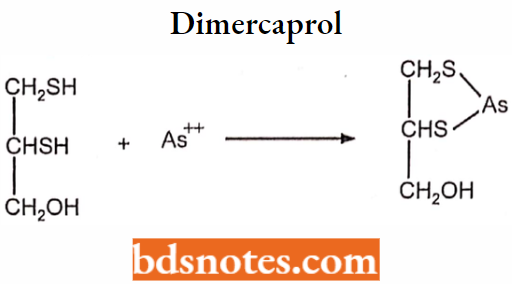
Antidote for metal poisoning.
Dimercaprol is a chelating agent.
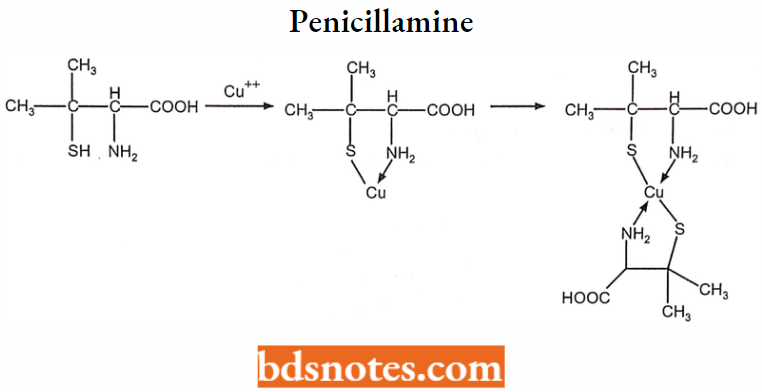
Penicillamine
8-Hydroxyquinoline and its analogs act as antibacterial and anti-fungal agents by complexing with iron or copper.
Undesirable side effects are caused by drugs, which chelate with metals.
A side effect of hydralazine antihypertensive agent is the formation of anemia and this is due to the chelation of the drug with iron.
Ionization and pKa
Most of the drugs are either weak acids or bases and can exist in either ionized or unionized states. The ionization of the drug depends on its pKa and pH.
- The rate of drug absorption is directly proportional to the concentration of the drug in the absorbable form but not the concentration of the drug at the absorption site.
- Example: Aspirin in the stomach will get readily absorbed because it is in the unionized form (99%).
- Example: Barbituric acid is inactive because it is a strong acid.
5, 5 disubstituted barbituric acid has CNS depressant action because it is a weak acid.
Acids are of two types:
- Unionized acid – HA
- Ionized acid – BH+
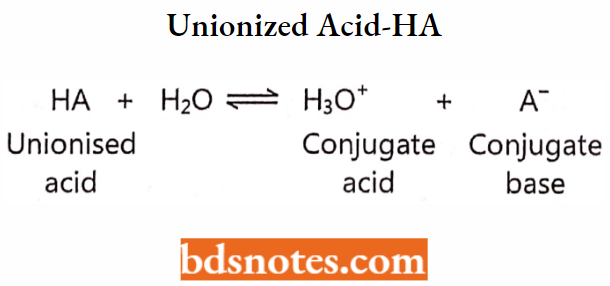
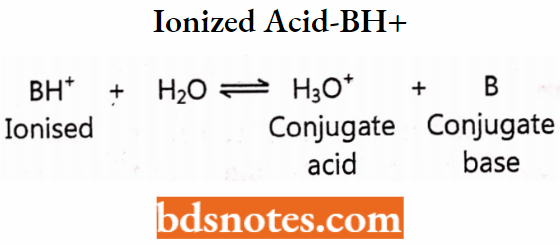
According to the Henderson-Hasselbalch equation,
⇒ \(\mathrm{pH}=\mathrm{pK}_{\mathrm{a}}+\log \frac{\text { [Unionised form] }}{\text { [Ionised form] }}\)
∴ % ionisation = \(\frac{100}{\left(1+10\left(\mathrm{pH}-\mathrm{pK}_{\mathrm{a}}\right)\right)}\)
By using the drug pKa, the formulation can be adjusted to pH to ensure maximum solubility in water or maximum solubility in a non-polar solvent. The pH of a substance can be adjusted to maintain water solubility and complete ionization.
Example: Phenytoin injection must be adjusted to pH 12 with sodium hydroxide to obtain 99.98% of the drug in ionized form.
Tropicamide eye drops, an anti-cholinergic drug have a pKa of 5.2 and the drug has to be buffered to pH 4 to obtain more than 90% ionization.
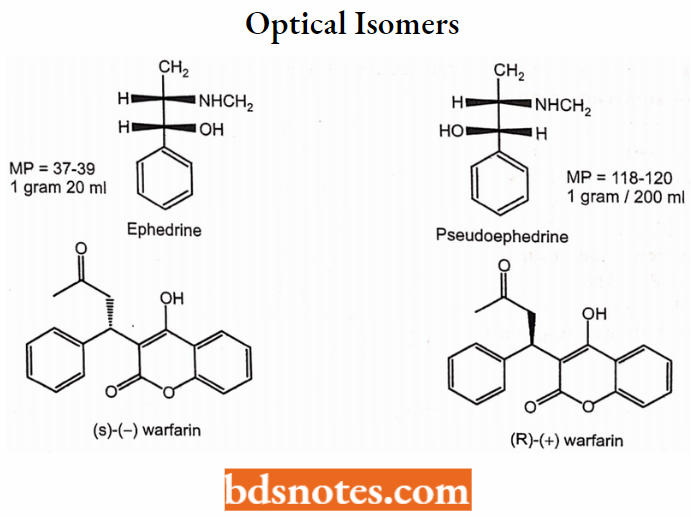
Optical Isomers
Stereochemistry, enantiomers, symmetry, and chirality are important concepts in the therapeutic and toxic effects of drugs. A chiral compound containing one asymmetric center has two enantiomers.
- Although each enantiomer has identical chemical and physical properties, it may have different physiological activities like interaction with receptors, metabolism, and protein binding.
- An optical isomer in biological action is due to one isomer being able to achieve a three-point attachment with its receptor molecule while its enantiomer would only be able to achieve a two-point attachment with the same molecule.
- Example: Ephedrine and Pseudoephedrine
The category of drugs where the two isomers have qualitatively similar pharmacological activity but have different quantitative potencies.
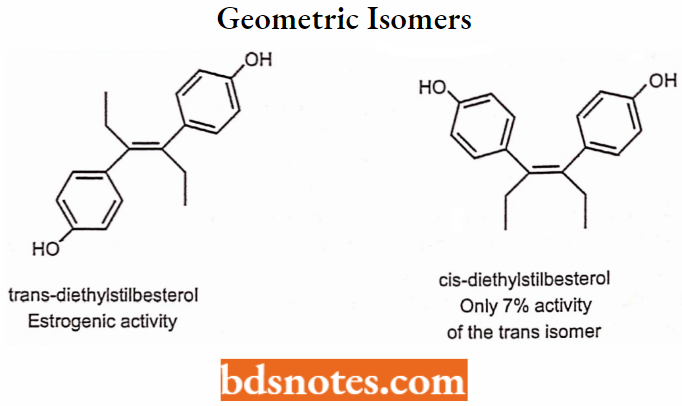
Geometric Isomerism
Geometric isomerism is represented by cis/trans isomerism resulting from restricted rotation due to carbon-carbon double bonds or in a rigid ring system.
Isosterism
Langmuir introduced the term isosterism in 1919, which postulated that two molecules or molecular fragments containing an identical number and arrangement of electrons should have similar properties and be termed isosteres. Isosteres should be isoelectric i.e. they should possess the same total charge.
Bioisosterism is defined as compounds or groups that possess near or equal molecular shapes and volumes, approximately the same distribution of electrons, and exhibit similar physical properties.
They are classified into two types:
- Classical bioisosteres
- Non-classical bioisosteres.
1. Classical Bioisosteres:
They have similarities in shape and electronic configuration of atoms, groups, and molecules which they replace. The classical bioisosteres may be,
Univalent atoms and groups:
- Cl, Br, I
- CH3NH2, -OH, -SH
Bivalent atoms and groups:
- R-OR, RNH-R, RSR, RSeR
- -CONHR, -COOR, -COSR
Trivalent atoms and groups:
- -CH=, -N=
- -P=, -As=
Tetravalent atoms and groups:
=C=, =N=, =P=
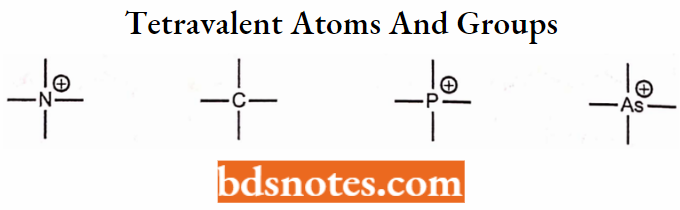
Application of Classical Bioisosteres in drug design:
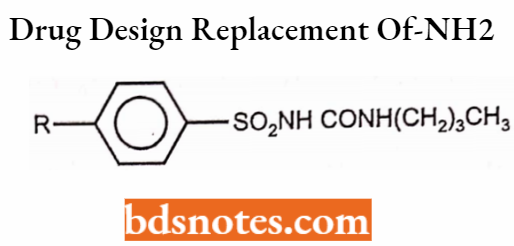
Replacement of – NH2 group by -CH3 group.
Carbutamide: R = NH2
Tolbutamide: R = CH3
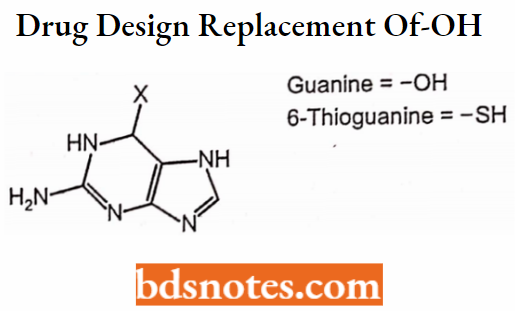
Replacement of -OH and -SH
2. Non-classical Bioisosteres:
They do not obey the stearic and electronic definition of classical isosteres. These isosteres retain activity by the retention of their properties such as pKg, and electrostatic potentials, which can alter selective enzyme processes.
Examples:
- Halogens: Cl, F, Br,CN
- Ether: -S-, -O-
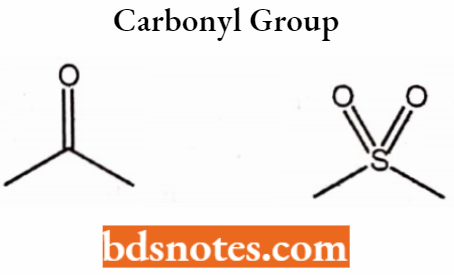
- Carbonyl group
- Hydroxyl group -OH, -NHSO2R, CH2OH
- Catechol
Protein Binding
The reversible binding of protein with non-specific and non-functional sites on the body protein without showing any biological effect is called as protein binding.
∴ \(\text { Protein }+ \text { Drug } \rightleftharpoons \text { Protein-drug complex }\)
Depending on whether the drug is a weak or strong acid, base or neutral, it can bind to single blood proteins to multiple proteins. The most significant protein involved in the binding of drugs is albumin, which comprises more than half of blood volume.
Metabolism
Metabolism is the body’s mechanism for processing, using, inactivating, and eventually eliminating foreign substances, including drugs. Drug exerts influence upon the body, it is gradually metabolized, or neutralized.
- The liver, the blood, the lymph fluid, or any body tissue that recognizes the drug as a foreign substance can break down or alter the chemical structure of drugs, making them less active, or inert.
- Drugs also can be neutralized by diverting them to body fat or proteins, which hold the substances to prevent them from acting on body organs.
- Once a drug is metabolized, it is the kidneys that normally filter the neutralized particles, called metabolites, as well as other waste and water, from the blood. Drugs can also be excreted out of the body by the lungs, in sweat, or feces.
- Metabolism is an essential pharmacokinetic process, which renders lipid soluble and non-polar compounds to water soluble and polar compounds so that they are excreted by various processes.
This is because only water-soluble substances undergo excretion, whereas lipid-soluble substances are passively reabsorbed from renal or extra-renal excretory sites into the blood under their lipophilicity.
Metabolism is a necessary biological process that limits the life of a substance in the body.
Site Of Drug Metabolism
The major site of drug metabolism is the liver (microsomal enzyme systems of hepatocytes). The primary site for the metabolism of almost all drugs is because it is relatively rich in a large variety of metabolizing enzymes.
- Secondary organs of biotransformation; Kidney (proximal tubule), Lungs (Type 2 cells), Testes (Sertoli cells), Skin (epithelial cells), plasma, nervous tissue (brain), intestines.
- Metabolism by organs other than the liver (called extrahepatic metabolism) is of lesser importance because a lower level of metabolizing enzymes is present in such tissues.
- Within a cell, drug-metabolizing activity is found in the smooth endoplasmic reticulum (microsomes) and the cytosol. Drug metabolism can also occur in mitochondria, nuclear envelope, and plasma membrane.
A few drugs are also metabolized by non-enzymatic means called as non-enzymatic metabolism, For Example. atracurium, a neuromuscular blocking drug, is inactivated in plasma by spontaneous non-enzymatic degradation (Hoffman elimination) in addition to that by pseudocholinesterase enzyme.
Metabolic Reaction Or Biotransformation Reactions
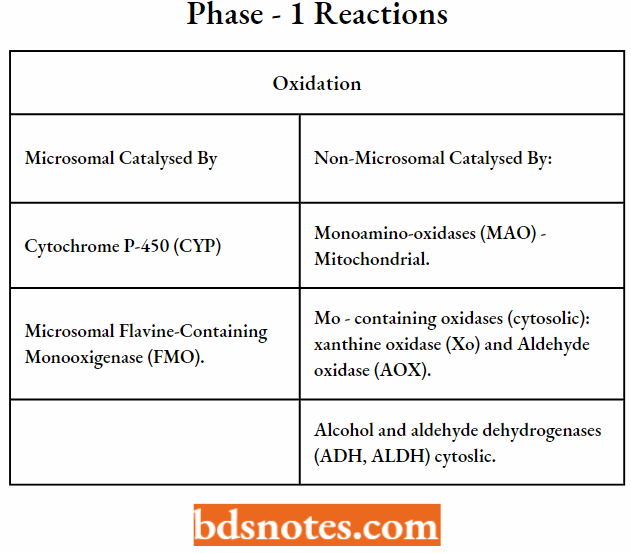
Metabolic conversions are classified as either Phase 1 (oxidation, reduction, or hydrolysis) or Phase 2 (conjugation). The enzymes involved in Phase-1 reactions are primarily located in the endoplasmic reticulum of the liver cell, they are called microsomal enzymes.
Phase-1 reactions are non-synthetic and generally produce more water-soluble and less active metabolites. The most common phase-I reactions are:
- Oxidative processes (aromatic hydroxylation; aliphatic hydroxylation; N-, O-, and S-dealkylation; N-hydroxylation; N-oxidation; sulfoxidation; deamination; and dehalogenation).
- Reductive process (azodye-reduction, nitroreduction) and
- Hydrolytic reactions.

Phase – 1 Reaction
Oxidation:
It is an important metabolic reaction. Energy is derived by the oxidative combustion of organic compounds containing carbon and hydrogen atoms.
- During the oxidation reaction, the hydrophilicity of drugs was increased by the addition of a functional group.
- Enzymes involved in the phase-I oxidative reaction are microsomal mono-oxygenases or mixed functional oxidases (MAO).
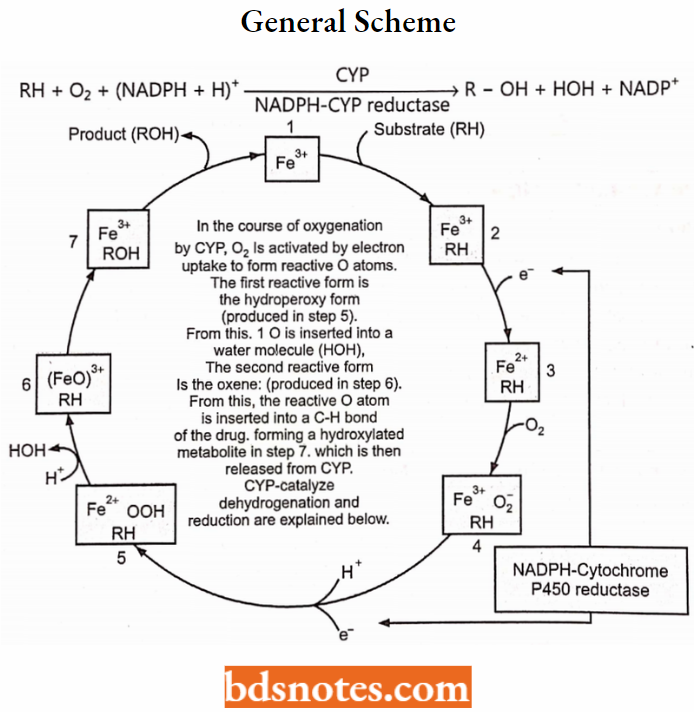
These reactions require molecular oxygen (O2) and reducing NADPH+H+ for the reaction which was located in the endoplasmic reticulum of the liver.
Oxidations Catalyzed by Cytochrome P-450 (CYP):
CYP is a heme-containing protein embedded in the membranes of the smooth endoplasmic reticulum (SER), the fragments of which in a tissue homogenate are sedimented after ultracentrifugation (at 100,000 g) in the microsomal fraction.
CYP constitutes a superfamily of enzymes that are classified into families (numbered) and subfamilies (marked with capital letters), the latter of which contain the individual enzymes (numbered), For Example., CYP1A2, CYP2C9, CYP2D6, CYP3A4.
CYP is an extremely versatile enzyme as it can catalyze numerous types of reactions, out of which three types are presented below.
Oxygenation:
Oxygenation involves the insertion of an O atom (from O2) into a C-H bond, forming a hydroxylated metabolite, or into a C=C double bond, forming an epoxide. A hydroxylated metabolite may be stable, or unstable.
From an unstable hydroxylated metabolite, a group may break off spontaneously: an alkyl group, ammonia, a halogen atom, or sulfur atom; such reactions are called oxidative dealkylation, oxidative deamination, oxidative dehalogenation, and oxidative desulfuration, respectively.
General scheme:
Insertion of it produces a Stable Metabolite (Oxygenation):
1. Hydroxylation:
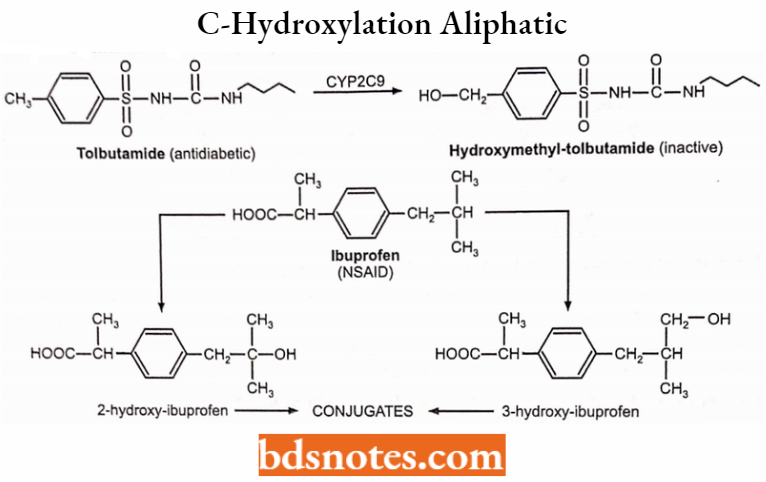
- C-hydroxylation (insertion of 0 into a C-H bond to form a hydroxyl group).
- Aliphatic hydroxylation: Tolbutamide (-), terfenadine (+), cyclophosphamide (+).
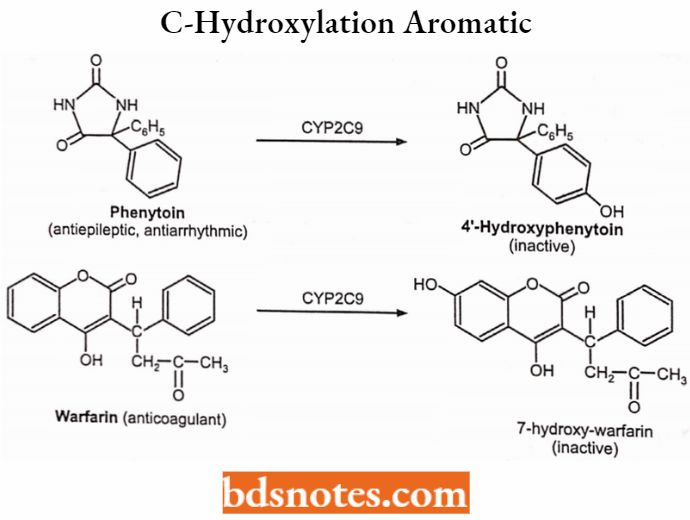
- Aromatic hydroxylation: Warfarin (-), phenytoin (-), propranolol (-).
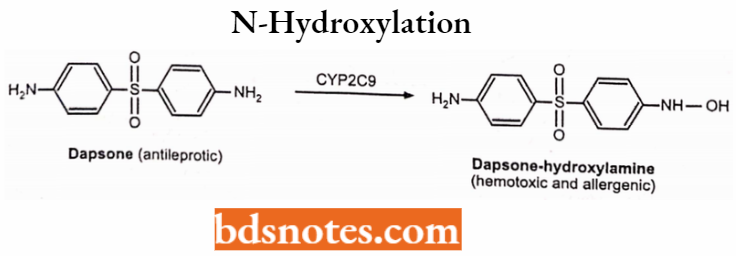
- N-hydroxylation (insertion of 0 into an N-H bond): dapsone.
C-hydroxylation: Aliphatic:
C-hydroxylation: Aromatic:
N-hydroxylation:
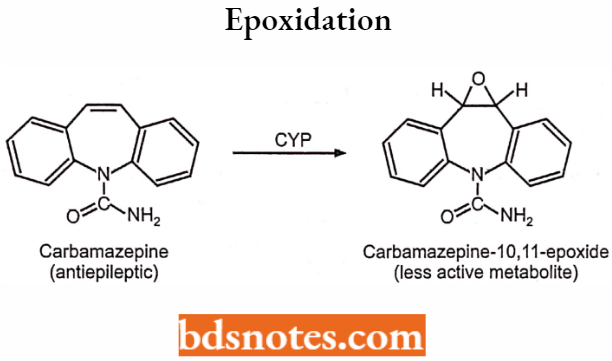
2. Epoxidation (insertion of O into a C=C bond to form an epoxide): Carbamazepine.
Insertion of it produces an Unstable Metabolite (Oxygenation):
First, the drug becomes hydroxylated at the C atom of the alkyl group that is linked to the N (or the O) atom. This hydroxylated metabolite is unstable.
It breaks spontaneously into two molecules: the dealkylated metabolite (For Example., an amine or alcohol or phenol), and an aldehyde (For Example., formaldehyde after demethylation, acetaldehyde after deethylation, etc.)
Oxidative dealkylation (dealkylated metabolite + an aldehyde):
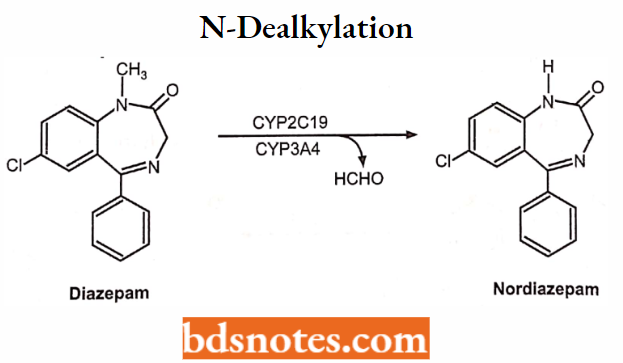
1. N-dealkylation: Diazepam → nordiazepam (+), Carisoprodol → meprobamate (+), Amitriptyline nortriptyline (+) desmethyl-nortriptyline (-) Lidocaine → monoethylglycylxylidine (+) → glycylxylidine (-) Caffeine → paraxanthine (-), or theophylline, or theobromine.
N-dealkylation: Example:
O-dealkylation: Example:
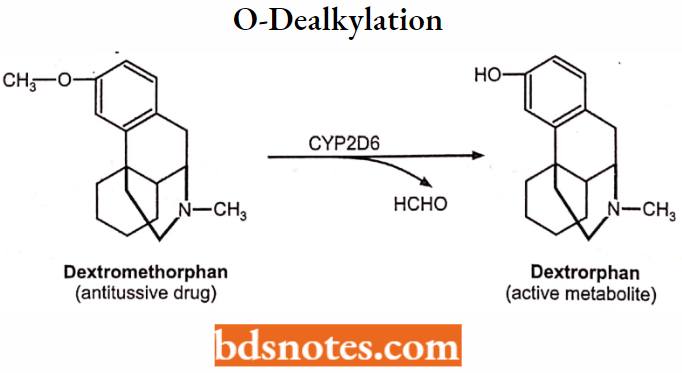
2. Oxidative deamination (→ O-containing metabolite + NH3):
Amphetamine → Phenylacetone (-)
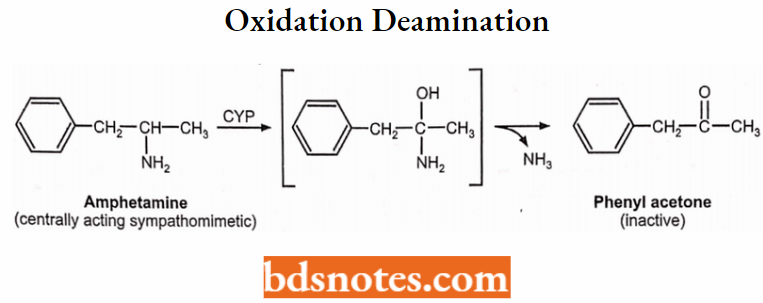
3. Oxidative dehalogenation (→ O-containing metabolite + HBr):
Halothane → trifluoroacetyl chloride (+)
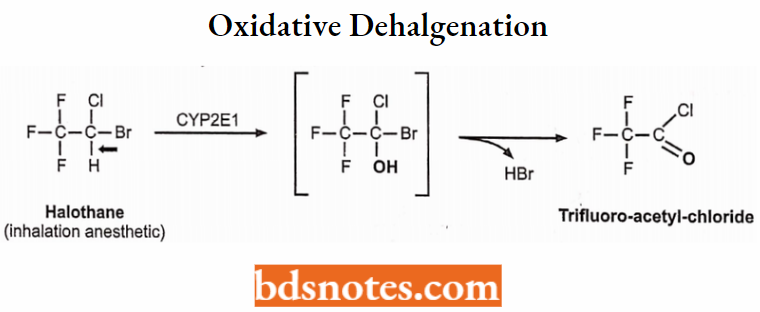
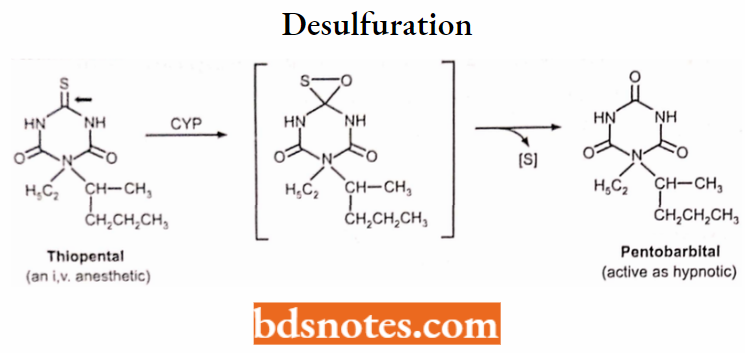
4. Oxidative desulfuration (→ O-containing metabolite + S):
Thiopental → Pentobarbital (+), Parathion → Paraoxon (+)
CYP-Catalyzed Dehydrogenation Reactions:
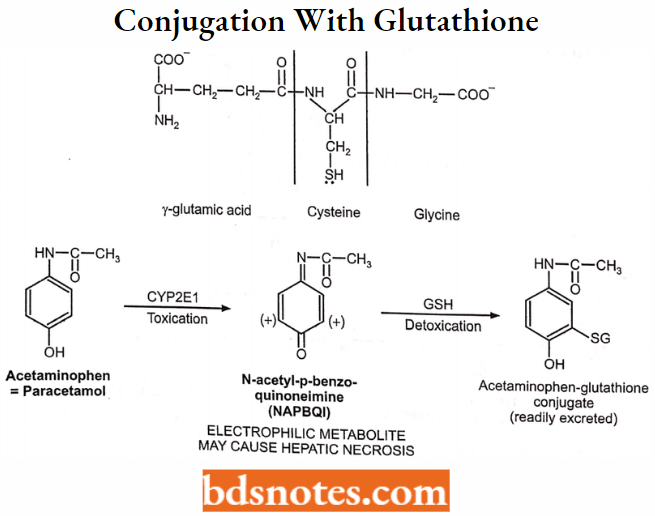
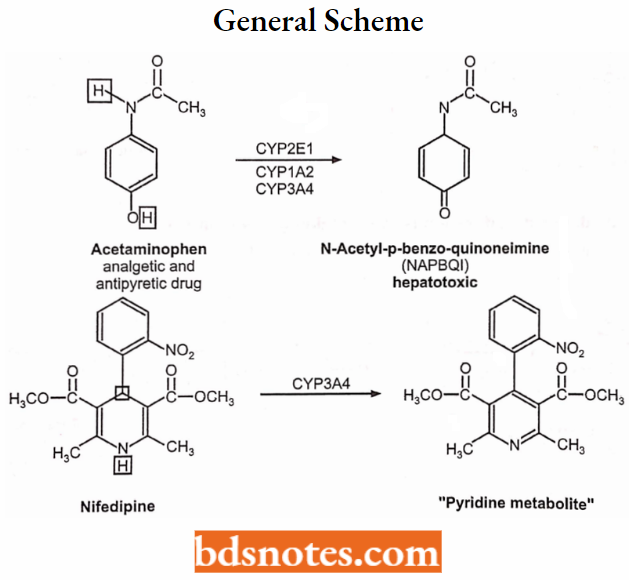
CYP also catalyzes dehydrogenation, i.e. removal of 2 H atoms from a drug molecule (this is how the reactive hepatotoxic paracetamol metabolite is formed).
General scheme:
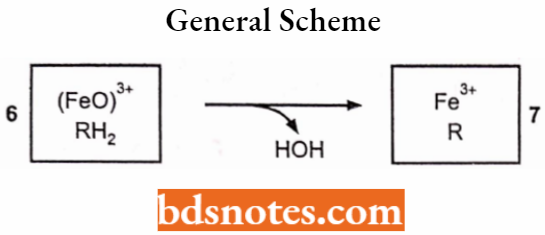
Note: While oxygenation produces 1 molecule of hydroxylated metabolite plus 1 molecule of HOH, dehydrogenation produces 2 molecules of HOH.
Examples:
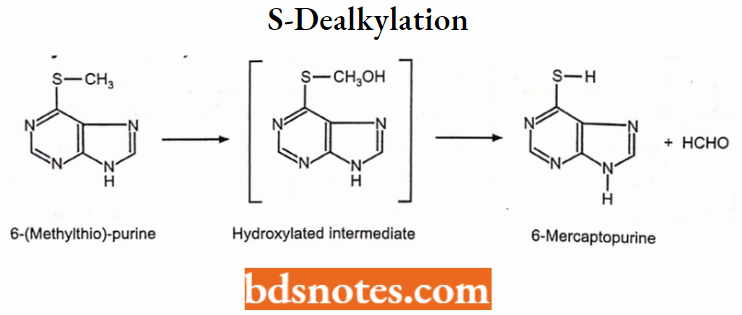
S-dealkylation: S-dealkylation involves oxidative cleavage of alkyl carbon-sulfur bonds.
Desulfuration:
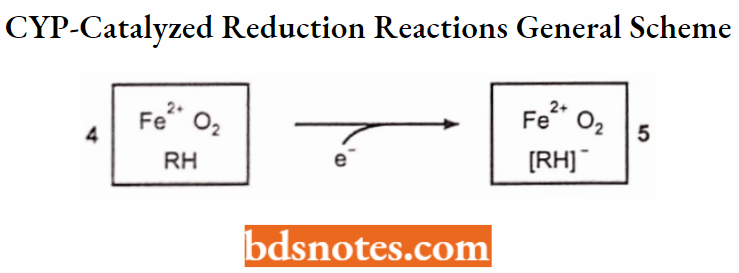
CYP-catalyzed Reduction Reactions:
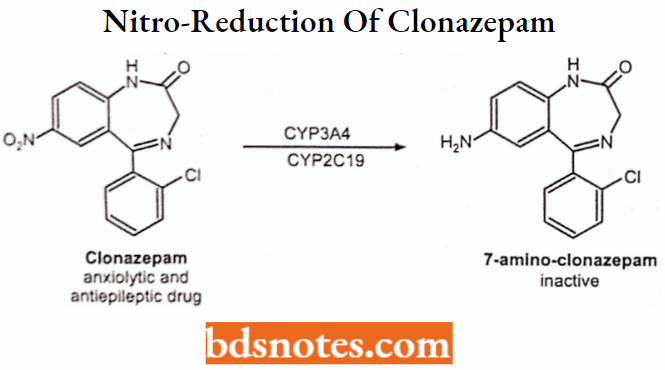
- CYP may also catalyze reduction, by transferring only 1 electron to a compound (For Example. in a reductive dehalogenation reaction) or as many as 6 electrons to a nitro group, thus converting it into an amino group (nitro reduction).
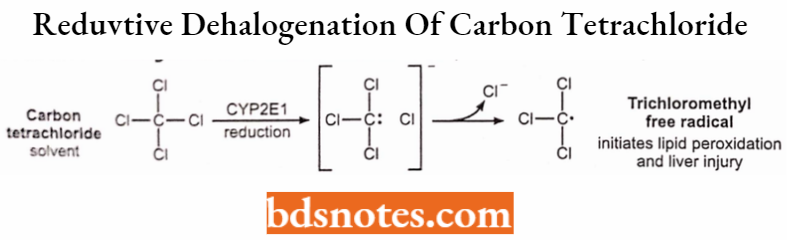
- In CYP-catalyzed reduction, the second electron is not transferred to the CYP-bound 02, but to the CYP-bound substrate, For Example. carbon tetrachloride or clonazepam (see also under nitro reduction).
- With carbon tetrachloride, its reduced intermediate then undergoes a homolytic cleavage to form a free radical and a chloride anion.
The reactive trichloromethyl free radical formed in the liver can initiate lipid peroxidation in the liver cell membranes and can thus induce hepatic necrosis.
General Scheme:
Reductive dehalogenation of carbon tetrachloride (1-electron reduction):
Nitro-reduction of clonazepam to 7-amino-clonazepam (6-electron reduction):
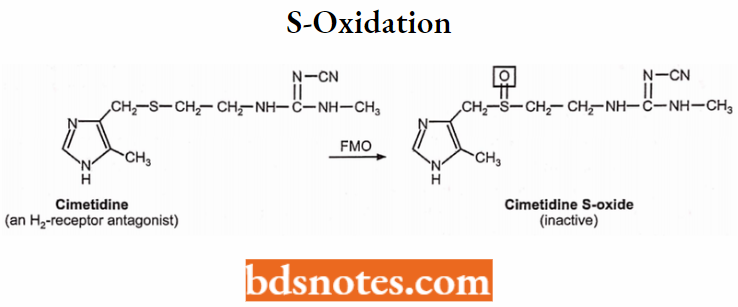
Oxidations catalyzed by Microsomal Flavin-containing Monooxygenase (FMO):
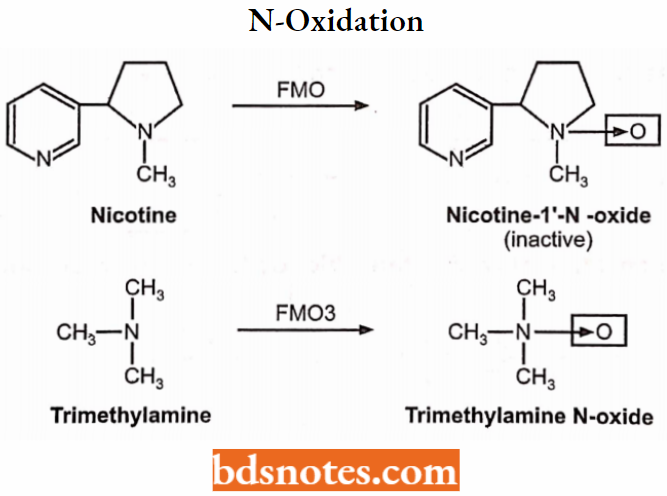
- Oxidation of S atom: Cimetidine → cimetidine-S-oxide.
- Oxidation of text. N atom: Nicotine → nicotine-l-N-oxide, Trimethylamine → trimethylamine N-oxide.
S-oxidation:
N-oxidation:
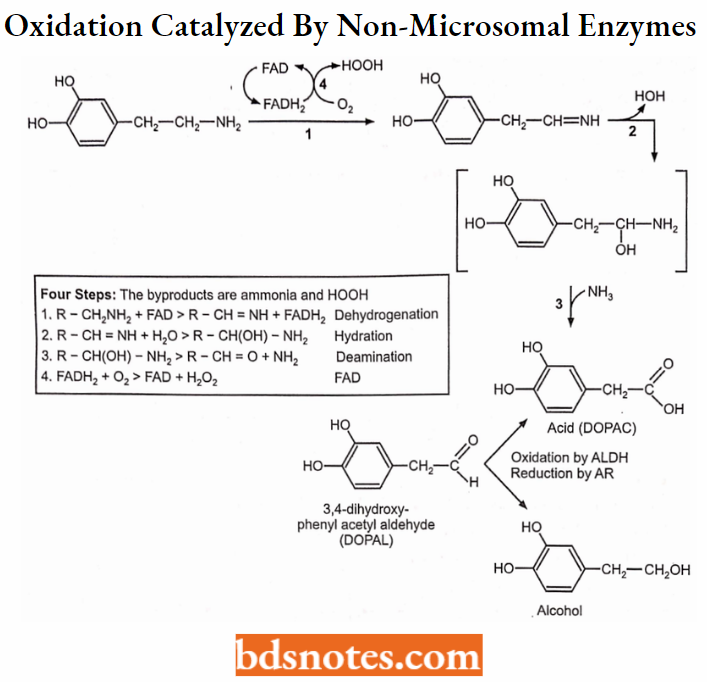
Oxidations catalyzed by non-microsomal enzymes:
The oxygen incorporated into the substrate is derived from HOH rather than O2
MAO catalyzes the oxidative deamination of amines:
Examples: Dopamine → 3,4-dihydroxy-phenylacetaldehyde.
Norepinephrine → 3,4-dihydroxy-mandelic aldehyde (-).
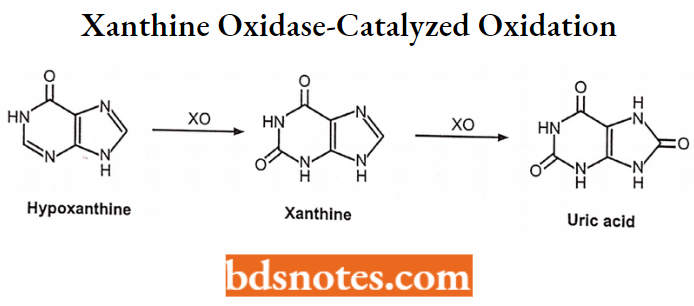
Xanthine oxidase-catalyzed oxidation:
Examples: Hypoxanthine → Xanthine → Uric acid
Allopurinol → Alloxanthine
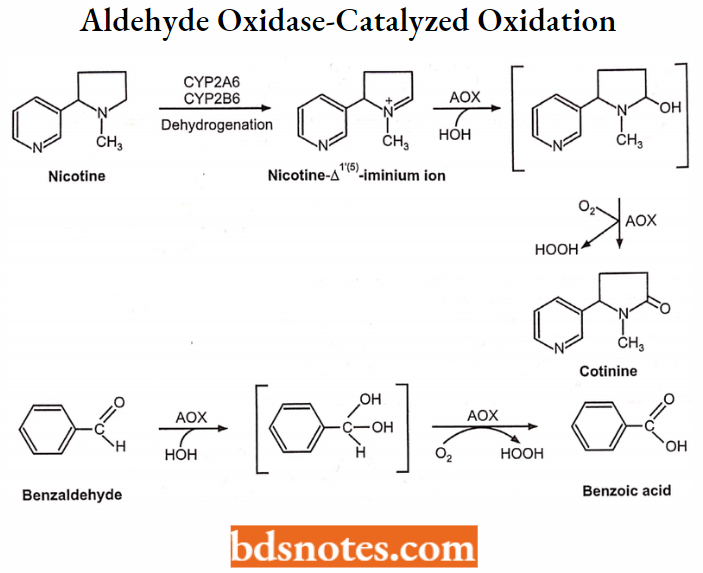
Aldehyde oxidase-catalyzed oxidation:
Examples: Nicotine → 1 (5 ) iminium ion → Cotinine – Benzaldehyde → Benzoic acid oxidation.
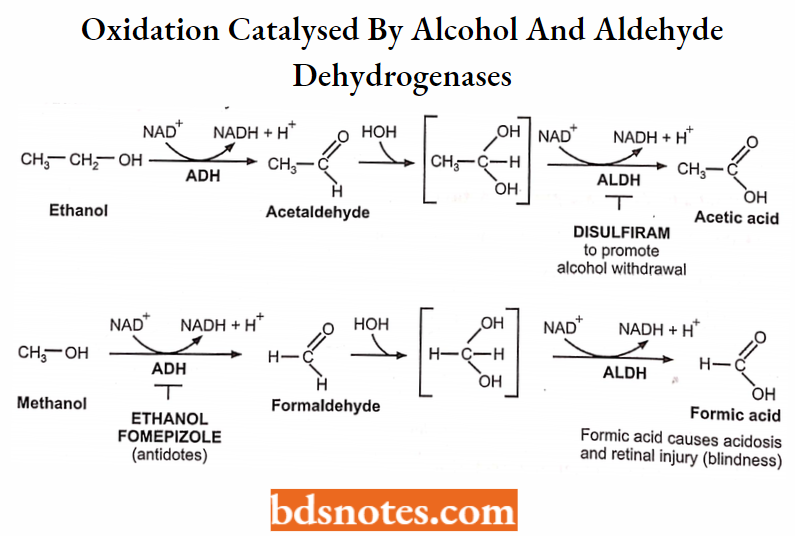
Oxidations catalyzed by alcohol and aldehyde dehydrogenases:
Examples: Methanol → Formaldehyde Formic acid
Ethanol → Acetaldehyde → Acetic acid.
Reduction:
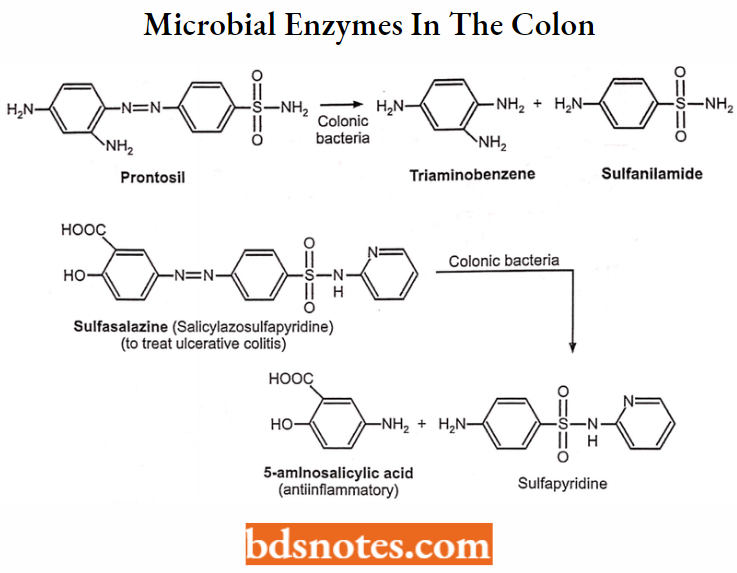
Azo-reduction is catalyzed by microbial enzymes in the colon.
Example: Prontosyl → Sulfanilamide + Triaminobenzene, Sulfasalazine (salicylazosulfapyridine) → 5-aminosalicylic acid sulfapyridine.
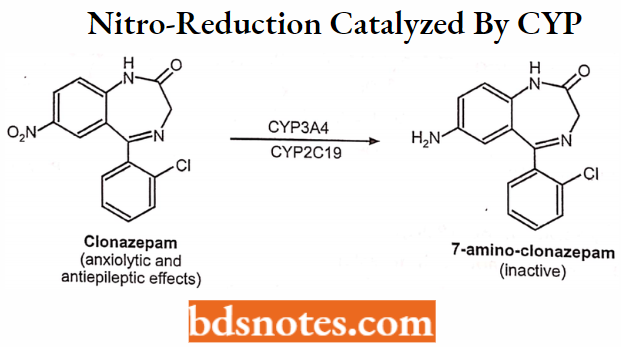
Nitro-reduction catalyzed by CYP
Example: Clonazepam → 7-amino-clonazepam.
Chloramphenicol → An arylamine metabolite.
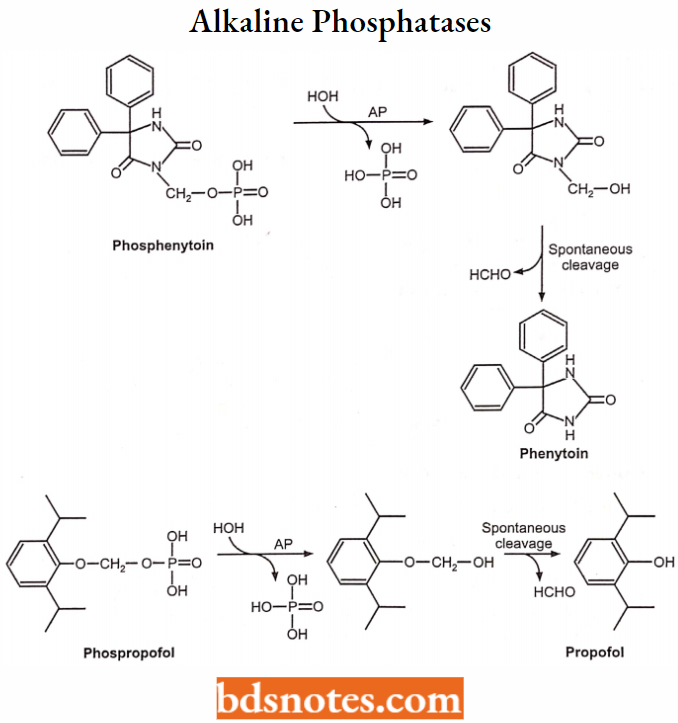
Hydrolysis:
Hydrolysis of esters: The active drugs are converted into inactive (or less active) metabolites. For Example. Succinylcholine, procaine, meperidine, acetylsalicylic acid.
By alkaline phosphatases: hydrolyzes phosphoric acid monoesters
Examples: (i.v. injectable prodrugs): Phosphenytoin (-) Phenytoin (-) Propofol (+) Clindamycin phosphate (-) Clindamycin (+).
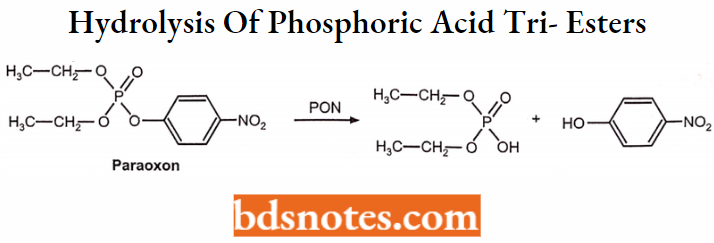
Hydrolysis by Paraoxonases (PON):
Hydrolysis of phosphoric acid tri-esters
Examples: Paraoxon and other organophosphate insecticides.
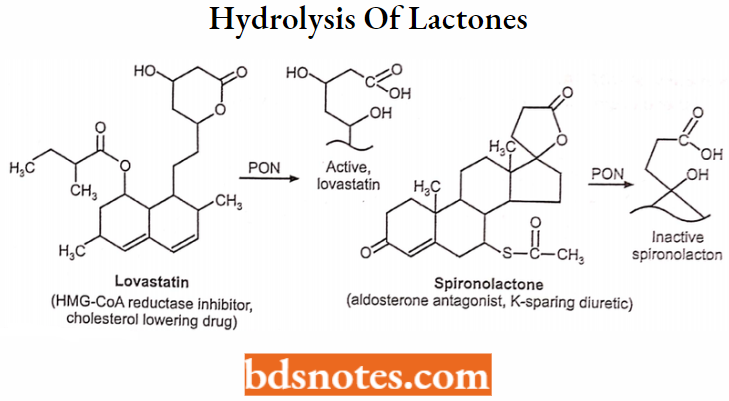
Hydrolysis of lactones:
Examples: Statins, For Example. lovastatin, or simvastatin (lactone) Hydroxy-acid (+), Spironolactone → Hydroxy-acid (-).
Phase-2 Reactions (Conjugations)
Phase-2 reactions were also conjugation reactions. Metabolic transformation of drugs in phase – 1 generally produced less water soluble or pharmacologically inactive metabolites.
Some exhibit more or less different activity from the parent drug. Phase-2 reactions or conjugation as glucuronic acid, sulfate, an acetyl group, or other amino acids in the presence of enzyme transferases and then excreted out.
Phase – 2 reactions (conjugations) involved the following reactions:
- Glucuronidation.
- Sulfation.
- Conjugation with glycine (Gly), glutamine, and other amino acids.
- Conjugation with glutathione (GSH) or mercaptopuric acid.
- Acetylation.
- Methylation.
Glucuronidation:
Glucuronidation involves the reaction of drug metabolite with glucuronic acid in the presence of the enzyme UDP-glucuronyl transferase. Drug substrate attached to glucuronic acid of uridine diphosphate glucuronic acid (UDPGA).
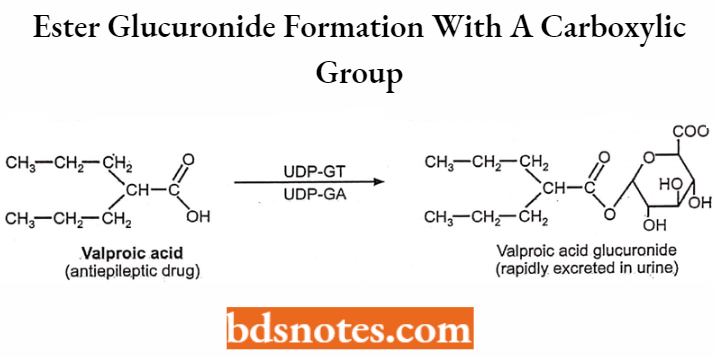
Glucuronides thus formed are inactive and excreted into the urine and bile. Molecules with phenolic hydroxyl, alcoholic hydroxyl, and carboxylic acid groups undergo glucuronidation reaction readily.
Glu-1-P → UDPG → UDPGA → Substrate drug molecule → Glucuronide metabolites.
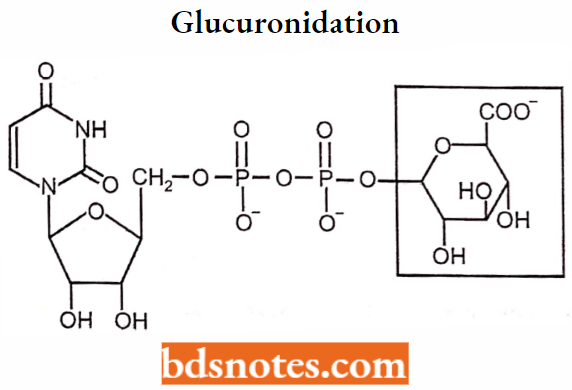
Structure of UDPGA:
Ether glucuronide formation (from compounds with a hydroxyl group).
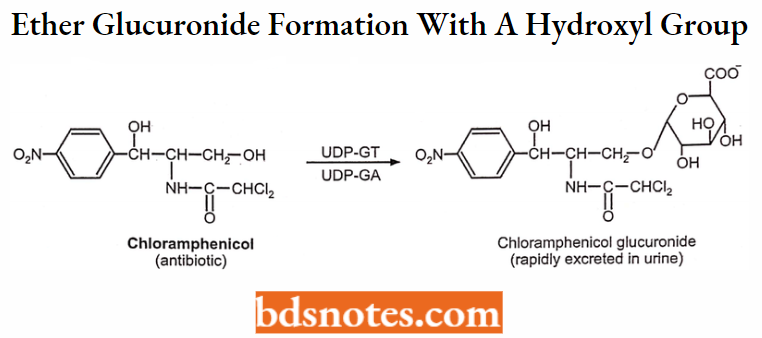
Ester glucuronide formation (from compounds with a carboxylic group).
Sulfation:
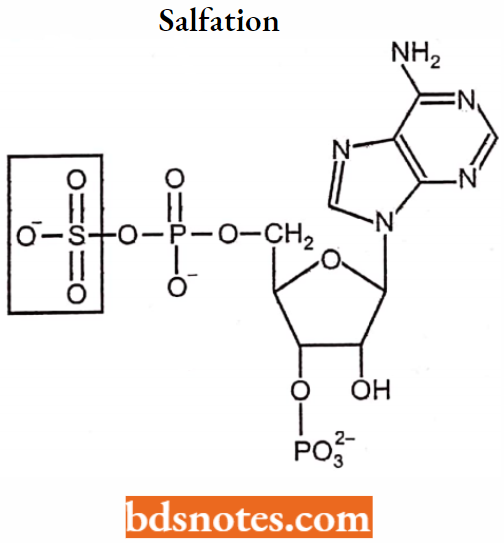
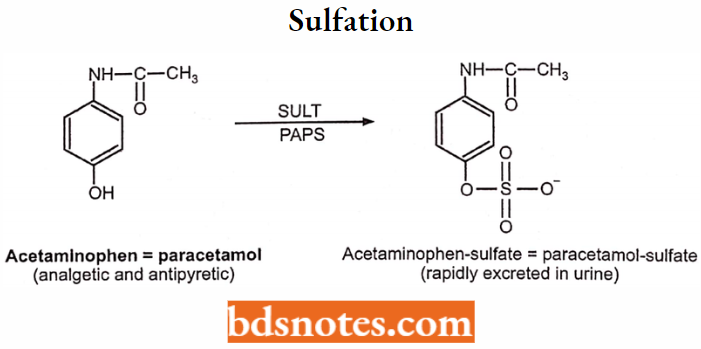
Sulfation sulfate conjugates are formed by the conjugation of sulfate moiety with the drug substrate. Compounds containing alcoholic hydroxyl, phenolic hydroxyl, and other compounds containing amino groups undergo sulfate conjugation.
The sulfate moiety from cofactor 3′-phosphoadenosine-5′-phosphosulphate (PAPS). The transfer of sulfate takes place in the presence of the enzyme sulphotransferase.
Example:
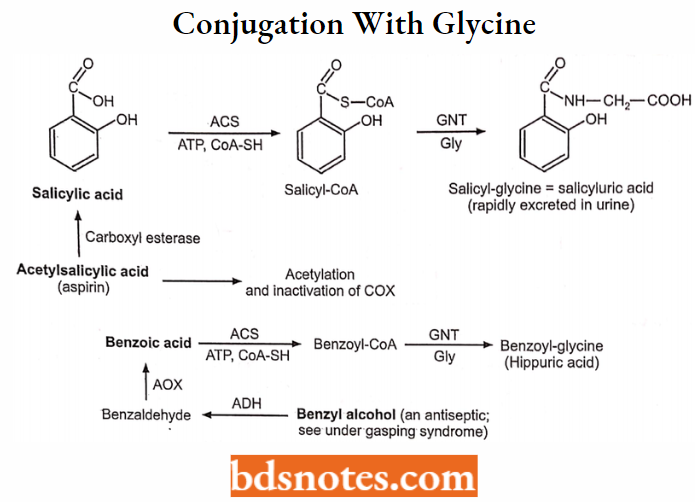
Conjugation with Glycine (GLY)
Examples:
Conjugation with Glutathione (GSH) or Mercaptopuric Acid:
Enzymes: Glutathione S-transferases (GST).
Cosubstrate: Glutathione (GSH).
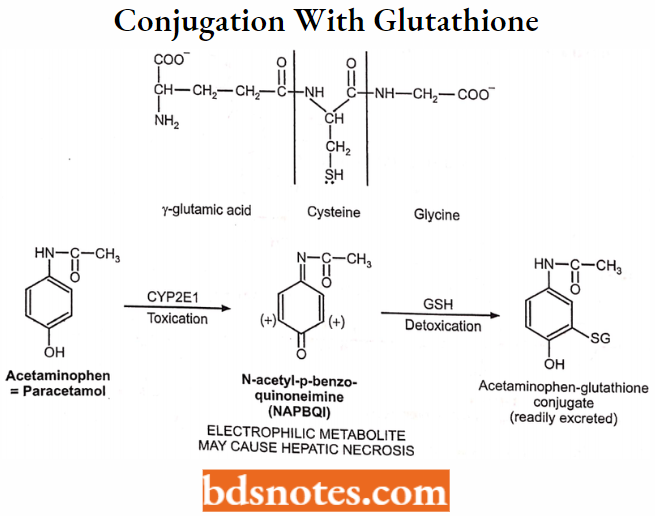
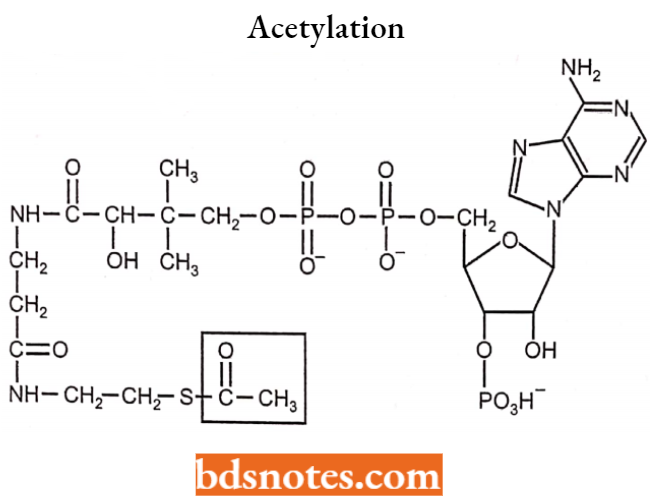
Acetylation:
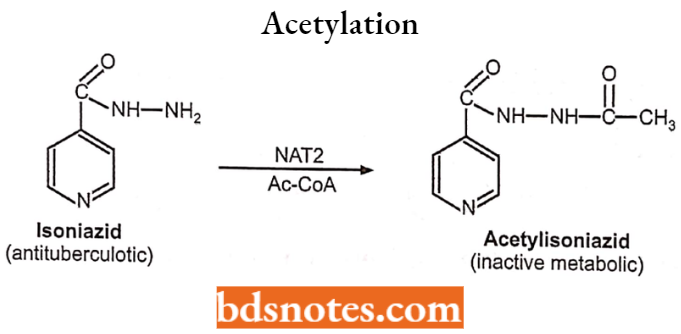
Enzymes: N-acetyltransferases (NAT).
Cosubstrate: Acetyl coenzyme A (Ac-CoA).
Example:
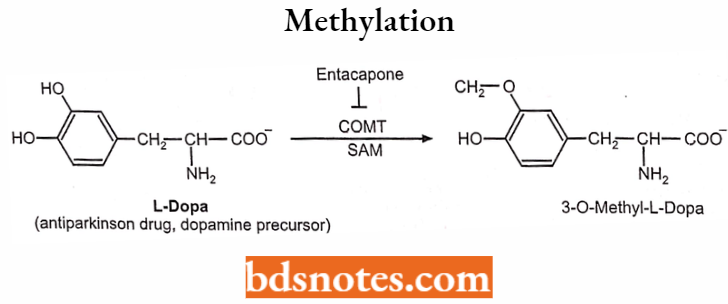
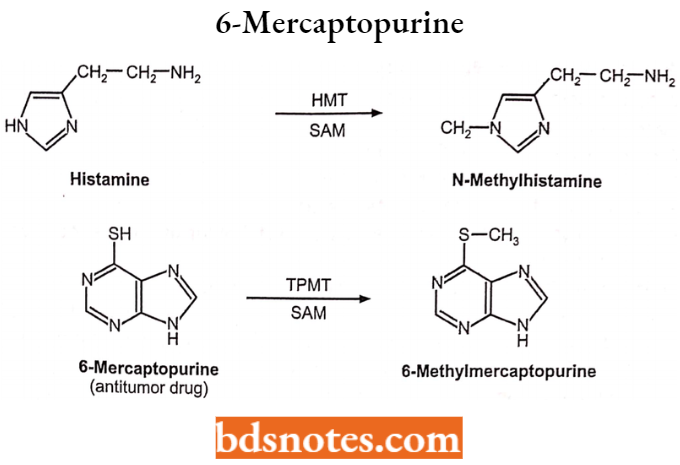
Methylation:
Enzymes: Methyltransferases (MT).
Cosubstrate: S-aldenocylmethionine (SAM)
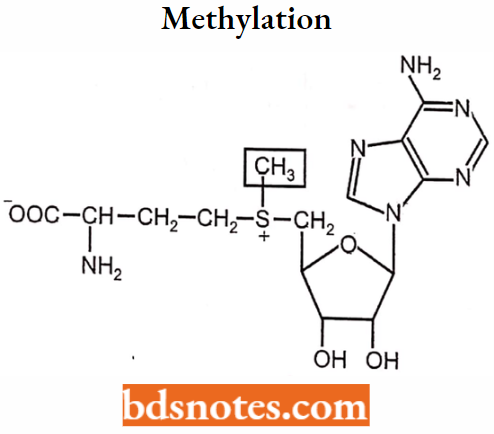
Example
Factors affecting drug metabolism including stereochemical aspects: The therapeutic efficacy, toxicity, and biological half-life of a drug greatly depend on the metabolism of the drug and several factors affect the metabolism of the drug.
Hence various factors affecting drug metabolism must be considered during administration and also in proper dosing of any drug to the patients.
Factors Affecting Metabolism: Several factors may influence the metabolic rate of a drug. Some of them are:
- Chemical factors include:
- Enzyme induction
- Enzyme inhibition
- Environmental chemicals
- Biological factors include:
- Age
- Diet
- Sex difference
- Species difference
- Strain difference
- Altered physiological factors
- Physicochemical properties of the drug
Chemical Factors:
1. Enzyme induction: The phenomenon of increased drug metabolizing ability of enzymes by several drugs and chemicals is called enzyme induction and the agents that bring about such an effect are called enzyme inducers.
Mechanisms of enzyme induction:
- Increase in both liver size and liver blood flow.
- Increase in both total and microsomal protein content.
- Increased stability of enzymes.
- Increased stability of cytochrome P-450.
- Decreased degradation of cytochrome P-450.
- Proliferation of smooth endoplasmic reticulum.
Consequences of enzyme induction include:
- Decrease in pharmacological activity of drugs.
- Increased activity where the metabolites are active.
- Altered physiological status due to enhanced metabolism of endogenous compounds such as sex hormones.
2. Enzyme inhibition: A decrease in the drug-metabolizing ability of an enzyme is called enzyme inhibition. The process of inhibition may be direct or indirect.
- Direct inhibition: It may result from interaction at the enzymic site bringing out the change in enzyme activity. Direct enzyme inhibition can occur by one of the following mechanisms:
- Competitive inhibition: It occurs when structurally similar compounds compete for the same site on an enzyme. Example: Methacholine and acetylcholine compete for the enzyme cholinesterase which causes the inhibition of acetylcholine metabolism.
- Non-competitive inhibition: It occurs when a structurally unrelated agent interacts with the enzyme and prevents the metabolism of drugs. Example: Inhibition of phenytoin by isoniazid.
- Product inhibition: It occurs when the metabolic product competes with the substrate for the same enzyme.
- Indirect inhibition: It is caused by one of the following mechanisms:
- Repression: It may be due to a fall in the rate of enzyme synthesis or a rise in the rate of enzyme degradation. Example: Due to the destruction of some enzymes by CCI4, CS2, etc.
- Altered physiology: It may be due to nutritional deficiency or hormonal imbalance.
3. Environmental chemicals: Several environmental agents influence the drug-metabolizing ability of enzymes.
Example: Halogenated pesticides such as DDT and polycyclic aromatic hydrocarbons contained in cigarette smoke have an enzyme induction effect.
Organophosphate insecticides and heavy metals such as mercury, nickel, cobalt, and arsenic inhibit the drug-metabolizing ability of enzymes. Other environmental factors that may influence drug metabolism are temperature, altitude, pressure, atmosphere, etc.
Biological Factors:
1. Age: The drug metabolic rate in the different age groups differs mainly due to variations in the enzyme content, enzyme activity, and hemodynamics.
- In neonates (up to 2 months) and infants (2 months to 1 year), the microsomal enzyme system is not fully developed. So, many drugs are metabolized slowly.
- For example, caffeine has a half-life of 4 days in neonates in comparison to 4 hrs in adults.
- Children (between 1 year and 12 years) metabolize several drugs much more rapidly than adults as the rate of metabolism reaches a maximum somewhere between 6 months and 12 years. As a result, they require a large mg/kg dose in comparison to adults.
- In elderly persons, the liver size is reduced, the microsomal enzyme activity is decreased and hepatic blood flow also declines as a result of reduced cardiac output, all of which contribute to decreased metabolism of drugs.
For example, chlomethiazole shows a high bioavailability among the elderly, therefore they require a lower dose.
2. Diet: The enzyme content and activity is altered by several dietary components. Generally, a low protein diet decreases and a high protein diet increases the drug-metabolizing ability as enzyme synthesis is promoted by a protein diet and also raises the level of amino acids for conjugation with drugs.
- A fat-free diet depresses cytochrome P-450 levels since phospholipids, which are important components of microsomes become deficient.
- Grapefruit inhibits the metabolism of many drugs and improves their oral bioavailability.
- Dietary deficiency of vitamins like vitamins A, B2, B3, C, and E) and minerals such as Fe, Ca, Mg, and Zn retard the metabolic activity of enzymes.
Starvation results in a decreased amount of glucuronides formed than under normal conditions.
3. Sex difference: Variations between males and females are observed following puberty. There is a related difference in the rate of metabolism that may be due to sex hormones.
Such sex differences were studied in rats which shows that male rats have greater drug metabolizing capacity than females.
Example: In humans, women metabolize benzodiazepines more slowly than men. Several studies have shown that women on contraceptive pills metabolize many drugs at a slow rate.
4. Species difference: Species differences have been observed in both Phase-1 and Phase-2 reactions. In Phase-1 reactions, both qualitative and quantitative variations in the enzyme and their activity have been observed.
- Qualitative differences among species generally result from the presence or absence of specific enzymes in those species.
- Quantitative differences result from variations in the amount and localization of enzymes, the amount of natural inhibitors, and the competition of enzymes for specific substrates.
- The human liver contains less cytochrome P-450 per gram of tissue than the livers of other species.
For example, rat liver contains approximately 30 to 50 nmol/g of Cytochrome P450, whereas human liver contains 10 to 20 nmol/g.
- Furthermore, the human liver is 2 percent of body weight, whereas the rat liver is approximately 4 percent. Similarly, in men, amphetamine and ephedrine are predominantly metabolized by oxidative deamination.
- Whereas in rats aromatic oxidation is the major route in Phase-2 reactions. Similarly in pigs, the phenol is excreted mainly as glucuronide whereas its sulphate conjugate dominates in cats.
5. Strain difference: The difference in drug-metabolizing ability between different species is related to genetics, the differences are observed between strains of the same species also. It may be studied under two headings:
- Pharmacogenetics: A study of inter-subject variability in drug response is called pharmacogenetics. The inter-subject variations in metabolism may either be monogenetically or polygenetically controlled.
- A polygenetic control is observed in twins. In identical twins (monozygotic), very little or no difference in the metabolism of halothane, phenylbutazone, dicoumarol, and antipyrine was detected but large variations were observed in fraternal twins (dizygotic).
- Ethnic variations: Differences observed in the metabolism of drugs among different are called ethnic variations. Such variations may be monomorphic or polymorphic.
Example: Approximately equal percent of slow and rapid acetylators are found among whites and blacks whereas the slow acetylators dominate the Japanese and Eskimo populations.
Physiological Factors:
- Pregnancy: Pregnancy is known to affect hepatic drug metabolism. Physiological changes during pregnancy are probably responsible for the reported alteration in drug metabolism. These 6 include elevated concentrations of various hormones such as estrogen, progesterone, placental growth hormones, and prolactin. For Example. in women, the metabolism of promazine and pethidine is reduced during pregnancy.
- Disease states: Many disease states affect the metabolism of drugs. Some of them are cirrhosis of the liver, alcoholic liver disease, cholestatic jaundice, diabetes mellitus, acromegaly, malaria, various bacterial and viral infections, etc. It can be seen that major effects are seen in the disease affecting the liver as the liver is quantitatively the important site for metabolism.
- Hormonal imbalance: The higher level of one hormone may inhibit the activity of a few enzymes while inducing that of others. Example: Effect observed in the pituitary growth hormone and stress-related changes in ACTH levels.
Physicochemical Properties of the Drug:
Molecular size and shape, pKa, acidity or basicity, lipophilicity, and steric and electronic characteristics of a drug influence in interaction with the active sites of enzyme and the metabolism to which it is subjected. However, such an interrelationship is not clearly understood.
Leave a Reply