Hybridization And Bond Angles
From our discussions of bonding, we have learnt something about the arrangement of bonds around various atoms. These concepts are fundamental to our appreciation of the shape of molecules, i.e. stereochemistry.
Before we delve into these matters, let us recap a little on the disposition of bonds around carbon. Bonding at four-valent carbon is tetrahedral, with four sp3-hybridized orbitals mutually inclined at 109.5°.
Remember that the tetrahedral array is demonstrated by experimental measurements, and that hybridization is the mathematical model put forward to explain this observation.
We can conveniently represent the tetrahedral arrangement in two dimensions by using a wedge–dot convention. In this convention, single bonds written as normal lines are considered to be in the plane of the paper.
Bonds in front of this plane, i.e. coming out from the paper, are then drawn as a wedge, whilst bonds behind the plane, i.e. going into the paper, are drawn as a broken or dotted bond.
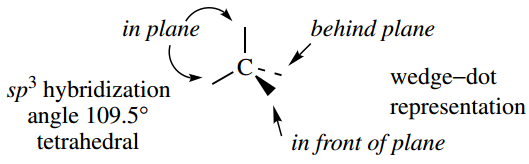
As we get more familiar with this representation, we may begin to abbreviate it by showing either the wedge or the dotted bond, rather than both.
Of course, it is important to remember that these abbreviated forms actually represent a tetrahedral array and not something with three bonds planar plus one other.
Drawing Stereostructures:
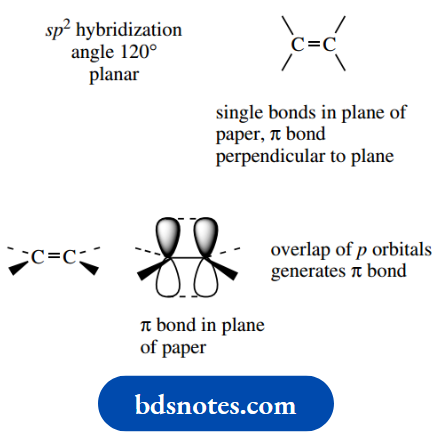
Bonding at three-valent carbon is trigonal planar with bond angles of 120°, an observation that we account for through sp2 hybridization plus the formation of a π bond by the overlap of p orbitals.
Thus, an alkene double bond involves electrons in sp2 hybrid orbitals making σ single bonds and the remaining electrons in p orbitals overlapping to produce the π-bond component of the double bond.
We can draw this as a planar representation, all single bonds in the plane of the paper, or show the π bonding in the plane of the paper, so that some bonds now require to be drawn in wedge form and others in dotted form.
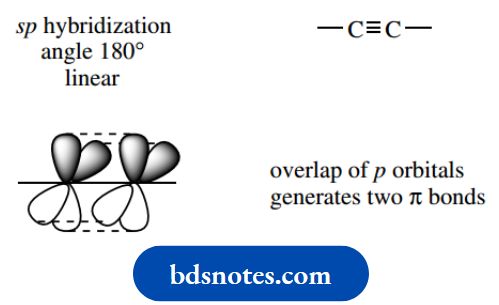
Bonding at two-valent carbon is linear, i.e. bond angles are 180°, and the triple bond comprises two π bonds and a σ single bond formed from sp hybrid orbitals.
The two π bonds are at right angles to each other. sp hybrid Although most of the atoms in the framework of an organic molecule tend to be carbon, other atoms, such as oxygen and nitrogen, are routinely encountered.
We can consider the arrangement of bonds around these atoms as approximately the same as the sp3– hybridized tetrahedral array seen with carbon. One (nitrogen) or two (oxygen) of the sp3 orbitals will be occupied by lone pair electrons.
The consequences of this include the fact that the two single bonds to oxygen are not linear, but are inclined at about 109°, and the three bonds to nitrogen are similarly not planar.
When oxygen or nitrogen are linked to another atom, For Example. carbon, by double bonds, the arrangement will be equivalent to the trivalent carbon, i.e. trigonal planar with a π bond perpendicular to the plane.
Lone pair electrons (one lone pair for nitrogen, two in the case of oxygen) will occupy nonbonding sp2 orbitals.
A triple bond to nitrogen, as bonding at nitrogen and oxygen approximates to that at carbon via lone pairs; in cyanide, will dictate a linear arrangement, with a nitrogen lone pair occupying a nonbonding sp orbital.
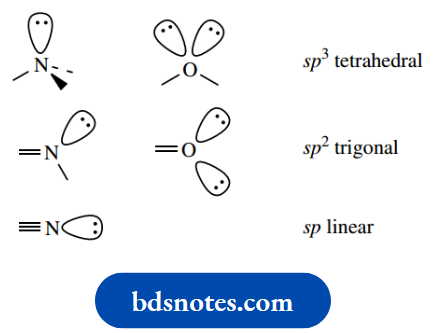
Bond angles depend upon the type of hybridization as just described, but in most molecules, they appear to be very similar.
There can often be a small degree of variation because of the nature of the precise atoms being bonded, and the presence of lone pair electrons but the level of consistency is very high
Similarly, bond lengths are also remarkably consistent, depending mainly on the nature of the atoms bonded and whether bonds are single, double, aromatic, or triple.
With bond lengths and bond angles being sufficiently consistent between molecules, it is possible to predict the shape and size of a molecule using simple molecular models or computer graphics.
Stereoisomers
For a given molecular formula there is often more than one way of joining the atoms together, whilst still satisfying the rules of valency.
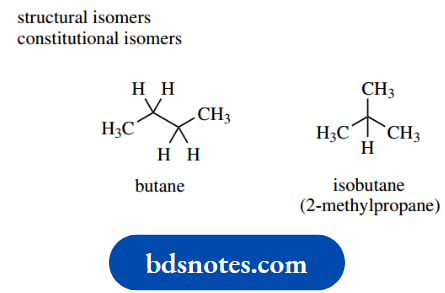
Such variants are called structural isomers or constitutional isomers compounds with the same molecular formula but with a different arrangement of atoms.
A simple example is provided by C4H10, which can be accommodated either by the straight-chained butane or by the branched-chain isobutane (2-methylpropane).
Stereoisomers, on the other hand, are compounds with the same molecular formula, and the same sequence of covalently bonded atoms, but with a different spatial orientation.
Two major classes of stereoisomers are recognized, conformational isomers and configurational isomers.
Conformational isomers, or conformers, interconvert easily by rotation about single bonds. Configurational isomers interconvert only with difficulty and, if they do, usually require bond breaking. We shall study these in turn
Conformational Isomers
Conformations Of Acyclic Compounds
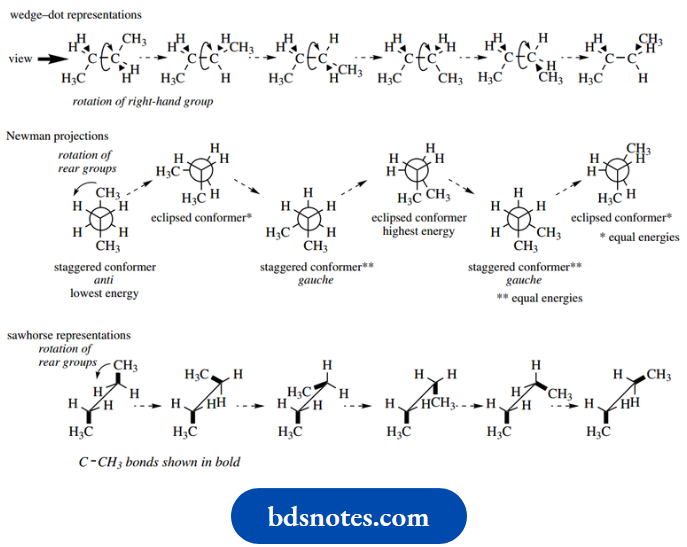
Let us consider first the simple alkane ethane. Since both carbons have a tetrahedral array of bonds, ethane may be drawn in the form of a wedge–dot representation.
Now let us consider the rotation of the right-hand methyl group about the C–C bond, and we eventually get to a different wedge–dot representation as shown.
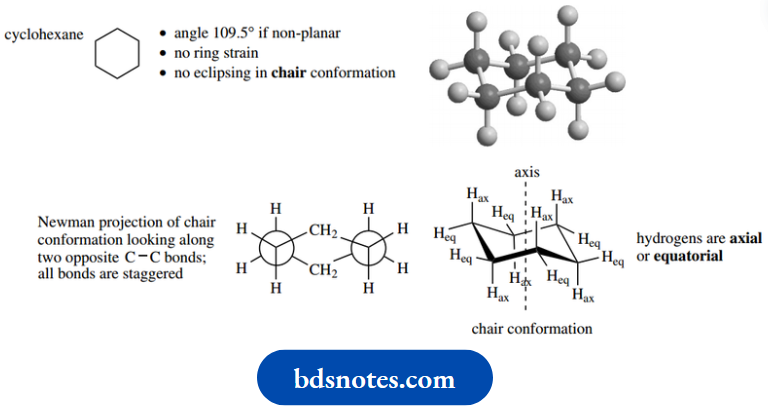
This is more easily visualized by looking at the molecule from one end down the C–C bond and this gives us what is termed a Newman projection.
The Newman projection shows the hydrogen atoms and their bonds, but the carbons are represented by a circle; since we are looking down the C–C bond, we cannot see the rear carbon.
A further feature is that the C–H bonds of the methyl closest to us are shown drawn to the centre of this circle, whilst those of the rear methyl are partially obscured and drawn only to the edge of this circle.
We can draw a similar Newman projection for the second wedge–dot representation, but the C–H bonds of the front and rear methyls will appear to be on top of each other.
We therefore draw a slightly modified version showing all bonds, but must remember that this really represents a system where the bonds at the rear are obscured by the bonds at the front.
In the sawhorse representation, the molecule is viewed from an oblique angle, and all bonds can be seen.
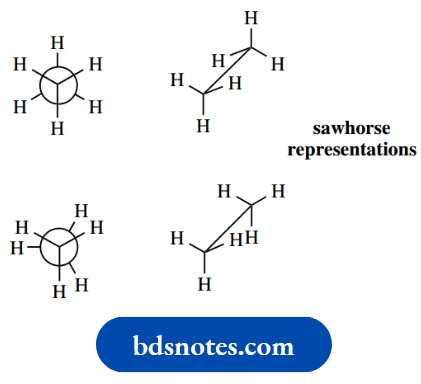
The two representations shown here are actually two different conformers of ethane; there will be an infinite number of such conformers, depending upon the amount of rotation about the C–C bond.
Although there is fairly free rotation about this bond, there does exist a small energy barrier to rotation of about 12 kJ mol-1 due to the repulsion of the electrons in the C–H bonds.
By inspecting the Newman projections, it can be predicted that this repulsion will be a minimum when the C–H bonds are positioned as far away from each other as possible.
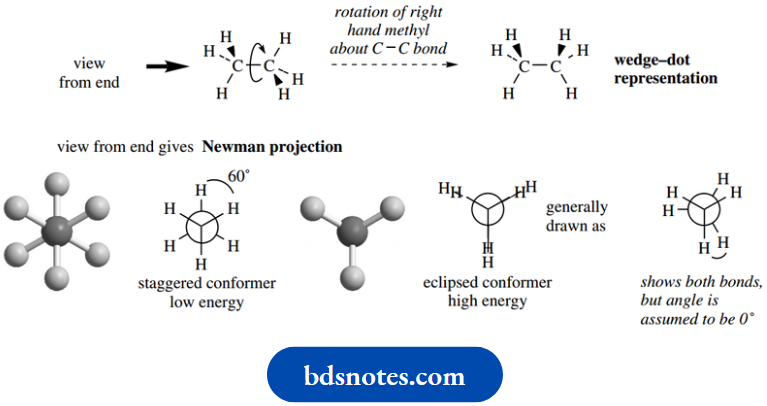
This is when the dihedral angle between the C–H bonds of the front and rear methyls is 60°, as exists in the left-hand conformer.
This conformation is termed the staggered conformation. On the other hand, electronic repulsion will be greatest when the C–H bonds are aligned, as in the right-hand conformer.
This conformation is termed the eclipsed conformation. In between these two extremes, there will be other conformers of varying energies, depending upon the degree of rotation.
Energies for these will be greater than that of the staggered conformer, but less than that of the eclipsed conformer.
Indeed, if one considers a gradual rotation about the C–C bond, the energy diagram will take the form of a sine wave, because rotations of either 120° or 240° will produce an indistinguishable conformer of identical energy. This is shown in.
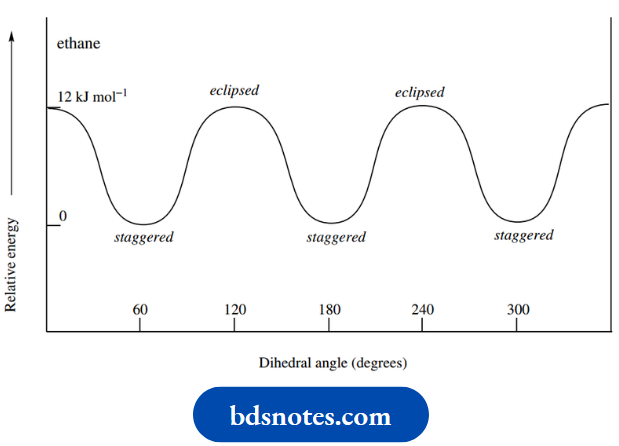
It follows that the preferred conformation of ethane is a staggered one; but, since the energy barrier to rotation is relatively small, at room temperature there will be free rotation about the C–C bond.
Let us now consider rotation about the central C–C bond in butane. Rotation about either of the two other C–C bonds will generate similar results as with ethane above.
Wedge–dot, Newman, and sawhorse representations are all shown; use the version that appears most logical to you.
As we rotate the groups, we shall get a series of staggered and eclipsed conformers. The energy barrier to rotation will be larger than the 12 kJ mol-1 seen with ethane.
This is because, in addition to the similar electronic repulsion in the bonds, there is now a spatial interaction involving the large methyl groups.
It follows that the repulsive energy associated with a methyl–methyl interaction will be larger than a methyl–hydrogen interaction, which in turn will be larger than that arising from hydrogen–hydrogen interactions.
Logically then, we predict that the energy of the eclipsed conformer in which the methyl groups are aligned will be higher than that in which there are methyl–hydrogen alignments and that there will be two equivalent versions of the latter.
Similarly, of the low-energy staggered conformers, there will be two equivalent ones where the carbon–methyl bonds are inclined at 60° to each other, and one in which the carbon–methyl bonds are inclined at 180°.
We can also predict that the latter conformer, which has the methyl groups as far away from each other as possible, will be of lower energy than the alternative staggered conformers, where there must be at least some spatial interaction between the methyl groups.
The staggered conformer with maximum separation of methyl groups is termed the anti-conformer (Greek: ant i = against), whilst the two other ones are termed gauche conformers (French: gauche = left).
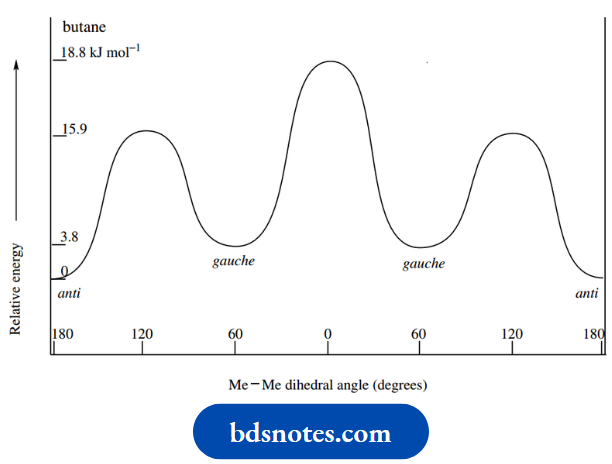
The energy diagram observed reflects these predictions, and the energy difference between the low-energy staggered anti-conformer and the highest energy eclipsed conformer is about 18.8 kJ mol-1.
There will still be free rotation about C–C bonds in butane at room temperature, but the larger energy barrier compared with that for ethane means that the staggered conformers are preferred.
Calculations show that, at room temperature, about 70% of molecules will be in the anti-conformer and about 15% in each gauche conformed.
Conformations Of Cyclic Compounds
Cyclopropane, Cyclobutane, Cyclopentane, Cyclohexane
The practical consequences of conformational isomerism become much more significant when we consider cyclic compounds. The smallest ring system will contain three atoms; in the case of hydrocarbons, this will be cyclopropane.
Now, simple geometry tells us that the inside angle in cyclopropane must be 60°. This is considerably less than the 109.5° of tetrahedral carbon.
The consequences are that the amount of overlap of the sp3 orbitals in forming the C–C bonds must be considerably less than in an acyclic system like ethane. With poorer overlap, we get a potentially weaker bond that can be broken more easily.
We term this ring strain, and although three-membered rings exist and are quite stable, they are frequently subject to ring-opening reactions.

A further feature of three-membered rings is that they must be planar, and a consequence of this is that, in cyclopropane, all C–H bonds are in the high energy eclipsed state.
There can be no conformational mobility to overcome this. In cyclobutane, the internal angle is 90°. Consequently, there is high ring strain, but this is not as great as in cyclopropane.
If cyclobutane were planar, all C–H bonds would be in the high-energy eclipsed state. It transpires that cyclobutane is not planar, since it can adopt a more favourable conformation in which eclipsing is reduced, and the ring appears puckered.
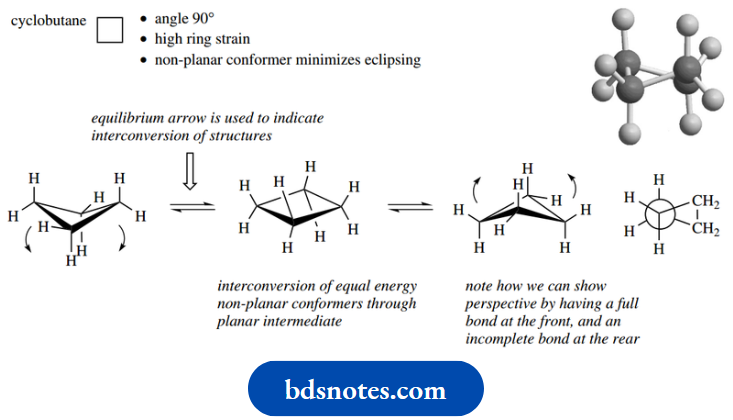
This appears to be achieved by pushing pairs of opposite carbons in different directions; but, in reality, it is only a combination of rotations about C–C bonds as we have seen with the simpler acyclic compounds.
It is not possible to achieve the ideal 60° staggered arrangement, but it does produce a lower energy conformed. Of course, there are two alternative ways of doing this, depending on whether pairs of carbons are ‘pushed’ or ‘pulled’.
Both conformers will be produced equally and can interconvert at room temperature because the energy barrier is fairly small at about 5.8 kJ mol-1.
The interconversion of the two forms is depicted by the equilibrium arrow, comprised of two half arrows. At equilibrium, both conformers coexist, and in this case, in equal amounts since they have the same energy.
The planar form of cyclobutane will be the energy maximum in the interconversion of conformers.
Compounds With Cyclopropane Or Cyclobutane Rings
A cyclopropane ring has the highest level of ring strain in the carbocycles. This means that they are rather susceptible to ring-opening reactions, but it does not mean that they are unstable and cannot exist.
Indeed, there are many examples of natural products that contain cyclopropane rings, and these are perfectly stable under normal conditions.
One group of natural cyclopropane derivatives of special importance is the pyrethrins, insecticidal components of pyrethrum flowers, and widely used in agriculture and in the home.
These compounds have very high toxicity towards insects without being harmful to animals and man and are rapidly biodegraded in the environment.
The pyrethrins are esters of two acids, chrysanthemum acid and pyrethrin acid, with three alcohols, pyrethrolone, cicerone, and jasmine, giving six major ester structures.
The acids contain the cyclopropane ring, and this appears essential for insecticidal activity.
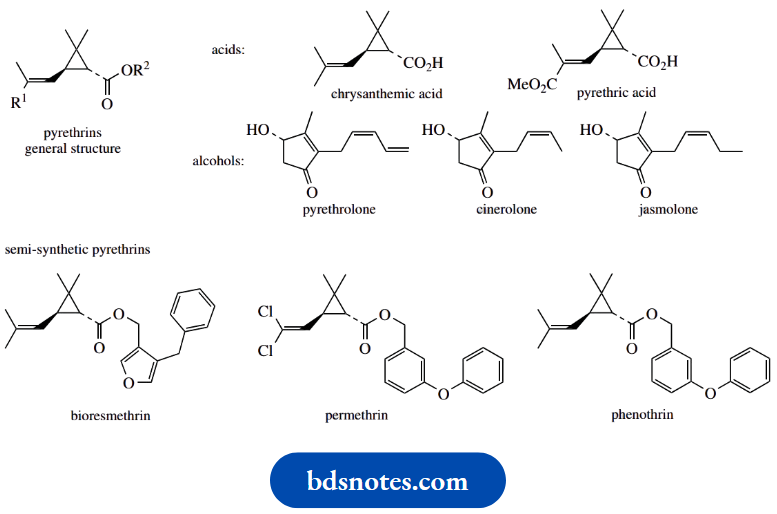
Many semi-synthetic esters, For Example. bioresmethrin, permethrin, and phenothrin have been produced and these have increased toxicity towards insects and also extended lifetimes.
All such esters retain a high proportion of the natural chrysanthemum acid or pyrethrin acid structure. The drugs naltrexone and nalbuphine are semi-synthetic analogues of the analgesic morphine.
Morphine is a good painkiller, but has some unpleasant side effects, the most serious of which is the likelihood of becoming addicted.
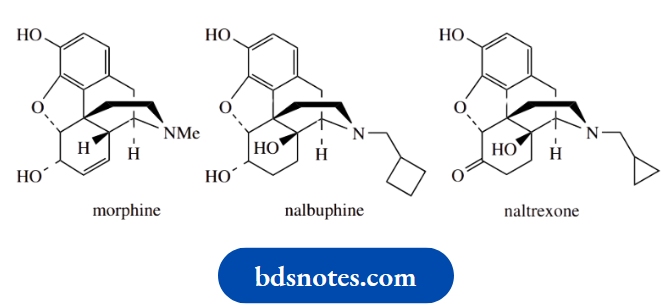
Nalbuphine is a modified structure containing a cyclobutane ring as part of the tertiary amine function.
Extending the size of the nitrogen substituent makes the drug larger and allows it to exploit extra binding sites on the receptor that morphine cannot interact with.
Nalbuphine is found to be a good analgesic with fewer side effects than morphine. Naltrexone incorporates a cyclopropane ring in the nitrogen substituent.
This, together with the other structural modifications, produces a drug that has hardly any analgesic effects but is a morphine antagonist.
Accordingly, it can be used to assist in the detoxification of morphine and heroin addicts.
Let us move on to cyclopentane, where geometry tells us the internal angle is 108°.
This is so close to the tetrahedral angle of 109.5° that cyclopentane can be considered essentially free of ring strain. However, planar cyclopentane would have all its C–H bonds eclipsed, which is obviously not desirable.
Accordingly, it adopts a lower energy conformation in which one of the carbon atoms is out of planarity. ‘Pushing’ this carbon out of the plane is achieved by rotation about C–C bonds, and it reduces eclipsing along all but one of the C–C bonds.
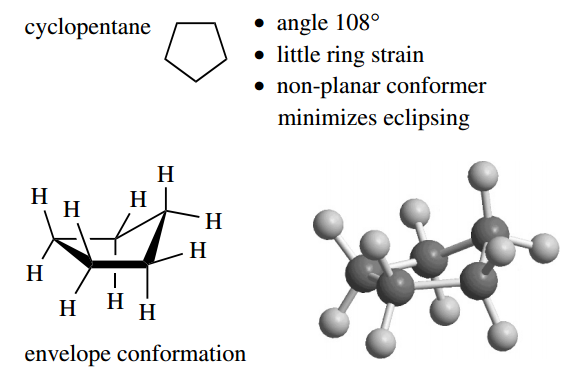
The energy barrier to this conformational change is about 22 kJ mol-1. There is no reason why any one particular carbon should be out of the plane, and at room temperature there is rapid interconversion of all possible variants.
Again, a planar form would feature as the energy maximum in the interconversions. The conformation with four carbons in the plane and one out of a plane is termed an envelope conformation.
This terminology comes from the similarity to an envelope with the flap open. For cyclohexane, the calculated internal angle is 120° if the molecule were to be planar, but the tetrahedral angle of 109.5° turns out to be perfect if the molecule is non-planar.
It is possible to construct a cyclohexane ring from tetrahedral carbons without introducing any strain whatsoever. The ring shape formed in this way is termed a chair conformation.
There is a considerable resemblance to a folding chair having a backrest and legrest, though the open seat might be regarded as a distinct disadvantage.
Not only is the bond angle perfect, but it also turns out that all C–H bonds are in a staggered relationship with adjacent ones. The chair conformation cannot be improved upon.
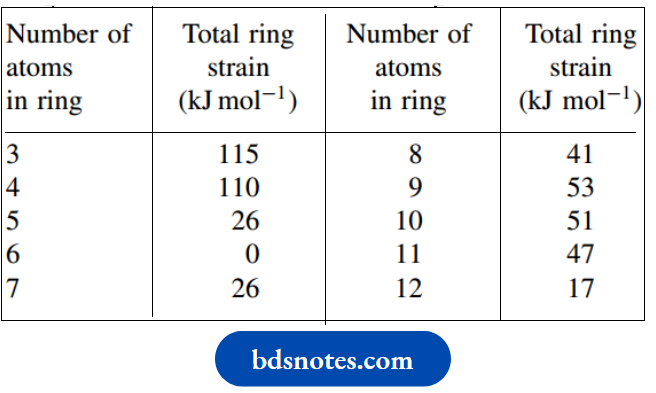
The total ring strain in various cycloalkanes compared with their strain-free acrylic counterparts has been estimated, as shown in Table.
Thus, small rings like cyclopropane and cyclobutane have considerable ring strain, and cyclohexane is effectively strain-free.
Larger rings (8–11 atoms) have more ring strain than might be predicted, certainly much more than cyclohexane, but any puckering that reduces ring strain actually creates eclipsing. We shall meet rings containing more than six carbons only infrequently.
How To Draw Chair Conformations Of Cyclohexane
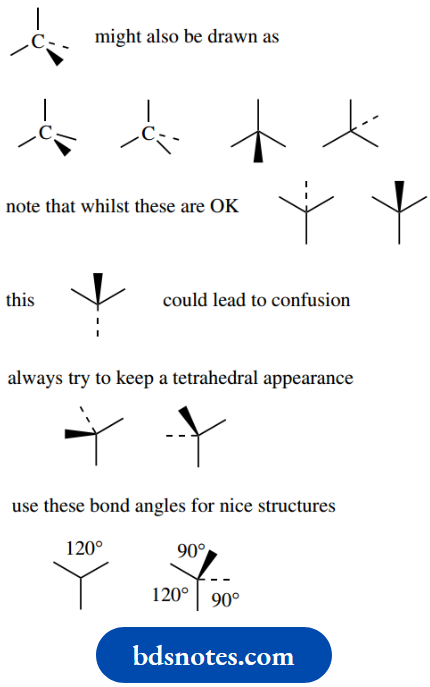
You can only appreciate stereochemical features if you can draw a representation that correctly pictures the molecule.
One of the most challenging is the chair conformation of cyclohexane. Practice makes perfect; so this is how it is done.
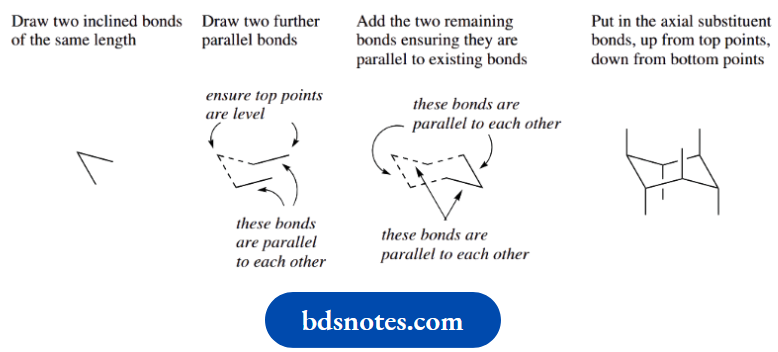
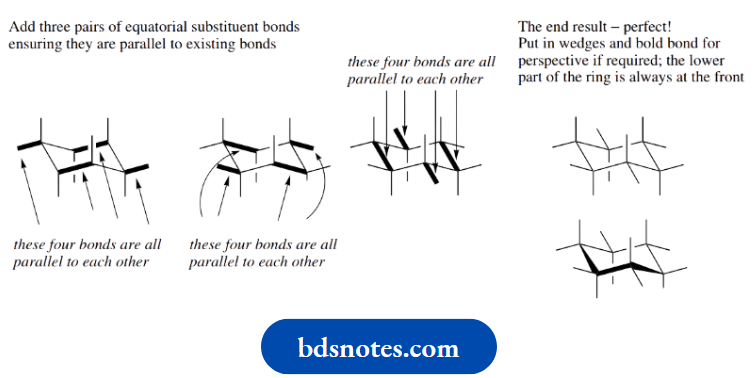
Note that the wedges and bold bonds help to show how we are looking at the cyclohexane chair.
In practice, particularly to speed up the drawing of structures, we tend to omit these. Then, by convention, the lower bonds represent the nearest part of the ring.

When one looks at the hydrogens in the chair conformation of cyclohexane, one can see that they are of two types.
Six of them are parallel to the central rotational axis of the molecule, so are termed axial. The other six are positioned around the outside of the molecule and are termed equatorial.
One might imagine, therefore, that these two types of hydrogen would have some different characteristics, and be detectable by an appropriate spectral technique.
Such a technique is NMR spectroscopy; but, at room temperature, only one type of proton is detectable.
At room temperature, all hydrogens of cyclohexane can be considered equivalent; this is a consequence of conformational mobility and the interconversion of two-chair conformation.
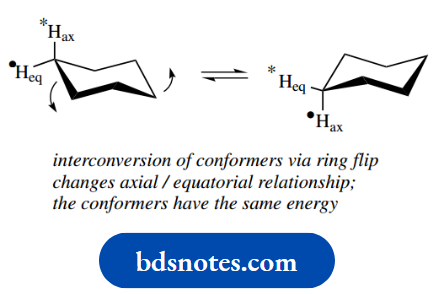
This interconversion may be considered as the simultaneous pushing down/pulling up of carbons on opposite sides of the ring, as indicated in the lefthand structure.
As a result, the ring ‘flips’ into an alternative conformation, also a chair, as in the righthand structure. This ring flip is actually achieved by rotation about several of the C–C bonds at the same time.
The ring flip can be demonstrated with suitable molecular models, and it is possible to feel the resistance in the model to this rotation, which represents the energy barrier to the change.
Both conformers have the same energy, but the energy barrier is about 42 kJ mol-1. The energy barrier looks high compared with those in ethane or butane, but this is because the interconversion involves rotations about several C–C bonds at the same time.
Look at the hydrogen atoms shown labelled in the left-hand structure. Note particularly that, after the ring flip, the axial hydrogen becomes equatorial, whilst the equatorial hydrogen becomes axial. Similar changes occur at all other positions.
With rapidly interconverting conformers, the hydrogens cannot be distinguished by NMR spectroscopy and they all merge to give a single signal. However, as one cools.
In the sample, the energy available to overcome the interconversion energy barrier diminishes, until at a sufficiently low temperature, the interconversion stops, and two types of hydrogen are detectable in the NMR spectrum.
This temperature is −89°C. Measurement of this temperature allows the energy barrier to be calculated.
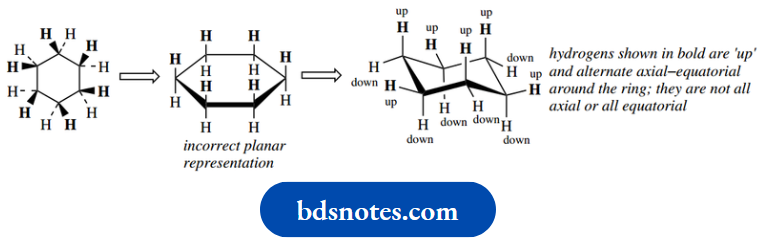
If we look at the two-dimensional hexagon representation for cyclohexane, we could put in the bonds to hydrogens as wedges (up bonds) or dotted lines (down bonds).
We now know the cyclohexane ring is not planar, but has a chair conformation. We shall frequently want to use the hexagon representation, and it will be necessary to assign hydrogens or other substituents onto the chair representation with the correct stereochemistry.
At this stage, it is salutary to look at both the two-dimensional hexagon and the chair representations of cyclohexane. Note particularly that we must not confuse ‘up’ with axial, and ‘down’ with equatorial.
As the structures show, ‘up’ hydrogens or substituents will alternate axial and equatorial as we go around the ring positions.
The chair is not the only conformation that cyclohexane might adopt. An alternative boat conformation is attained if the ring flip-type process is confined to just one carbon.
The name boat comes from the similarity to boats formed by paper folding; sea-worthiness is rather questionable.
Again, there is no ring strain in this conformation, but it turns out that some of the C–H bonds are eclipsed, as seen in the accompanying Newman projection.
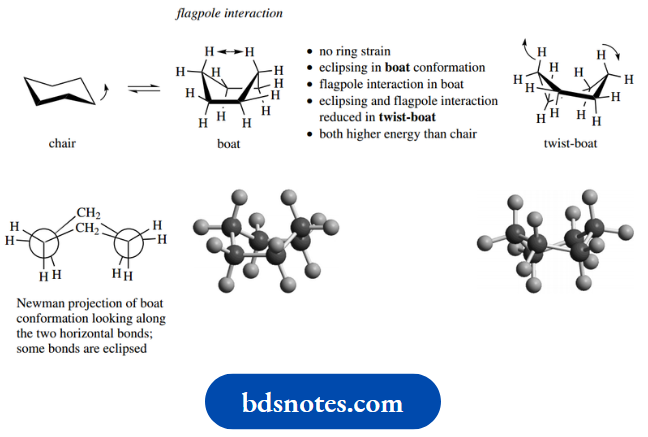
In addition, the hydrogens at the top of the structure are getting rather close to each other, and there is some interaction, termed a flagpole interaction, again from the nautical analogy.
Both the eclipsing and the flagpole interactions can be minimized when the boat conformation undergoes further subtle changes by rotation about C–C bonds to form the twist-boat.
This is a result of twisting the flagpole hydrogens apart. Making a molecular model of the boat conformation immediately shows how easy it is to modify it to the twist-boat variant; the boat conformation is quite floppy compared with the chair, which is very rigid.
An energy diagram linking the chair, boat and twist-boat conformations is shown. The boat conformation is represented by an energy maximum.
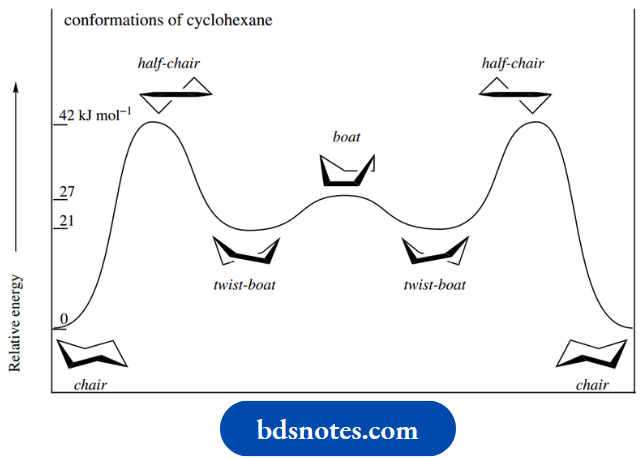
In practice, only the chair conformation is important for cyclohexane, since the energy differences between it and the other conformations make them much less favourable.
However, there are plenty of structures where cyclohexane rings are forced into the boat or twist-boat conformation because of other limiting factors.
For example, bornane is a terpene hydrocarbon where opposite carbons in a cyclohexane ring are bridged by a methylene group. This is stereochemically impossible to achieve with a chair – the carbons are too far apart.
However, it is possible with a boat confirmation. In such a structure, there are no further possibilities for conformational mobility – the conformation is now fused in and no further changes are possible, even though there may be unfavourable eclipsing interactions.
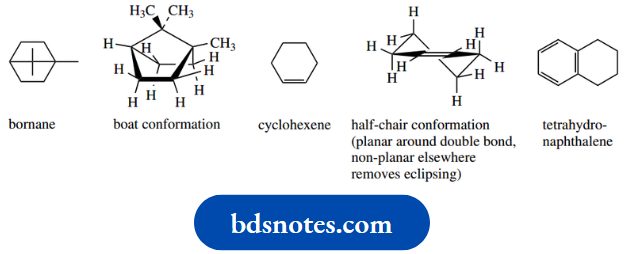
In cyclohexene, the double bond and adjacent carbons must all be planar. The remainder of the molecule avoids unfavourable eclipsing interactions by adopting what is termed a half-chair conformation.
This would also be found in a cyclohexane ring fused onto an aromatic ring (tetrahydronaphthalene) or fused to a three-membered ring.
The half-chair conformation in cyclohexane (without the double bond) is thought to be equivalent to the energy maximum in Figure that must be overcome in the chair–twist-boat interconversion.
Substituted cyclohexanes
The ring-flipping conformational mobility in the unsubstituted compound cyclohexane has little practical significance; but, when the ring is substituted, we have to take ring flip into account, because one particular conformation is usually favoured over the other
Let us look at a simple example, namely methylcyclohexane. Ring flip in the case of methylcyclohexane achieves interconversion of one conformer where the methyl group is equatorial into a conformer where this group is axial (compare the hydrogens in cyclohexane).
It turns out that the conformer with the equatorial methyl group is favoured over the conformer where the methyl group is axial. The energy difference of these two conformers is estimated to be about 7.1 kJ mol-1; this is the energy difference, not the barrier to interconversion.
Because of this energy difference, the equilibrium mixture at room temperature has about 95% conformers with the equatorial methyl and only 5% where the methyl is axial.
We can account for the difference in energy between the two conformers quite easily using the reasoning we applied earlier for the acyclic hydrocarbon butane.
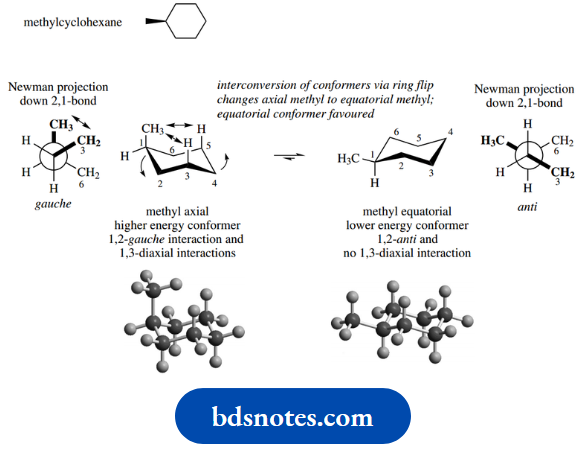
We need to consider a Newman projection looking down the 2,1 bond. When the methyl is axial, it can be seen that there will be a gauche interaction between this methyl and the ring methylene (C-3); a second, similar interaction will be seen if we look down the 6,1 bond.
Now, in the conformer where the methyl is equatorial, the Newman projection shows the most favourable anti-arrangement for the methyl and methylene(s); there will be a similar anti-interaction if we look down the 6,1 bond.
On this basis alone, we can predict that the equatorial conformer is of lower energy and, thus, more favoured.
However, there is a further feature that destabilizes the axial conformer, and that is the spatial interaction between the axial methyl and the axial hydrogens at positions 3 and 5, termed a 1,3-diaxial interaction.
Together, they account for the equilibrium mixture consisting mainly of the equatorial conformer. We can indicate this by using arrows of unequal size in the equilibrium equation.
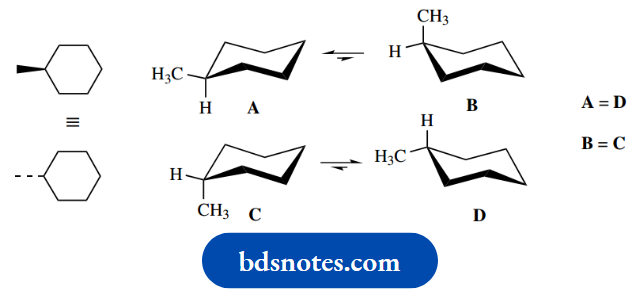
Note that it is not necessary to consider both forms of cyclohexane, where the methyl is either wedged (up) or dotted (down).
If the cyclohexane ring were planar, the two structures would be the same, since one merely has to turn the structure over to get the other.
Although the cyclohexane ring is not planar, it turns out that the two structures are still identical, because of the ring flip process
This is shown below. One set of conformers is simply the upside-down version of the other.
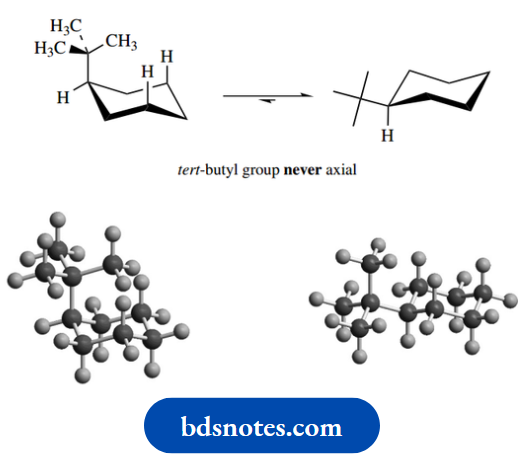
Now, as the substituent gets bigger, the proportion of axial conformer will diminish even further.
With a substituent as big as a tert-butyl group, the equilibrium is such that essentially all molecules are in the equatorial conformation; in general terms, we can consider that a tert-butyl group will never be axial.
Although analysis of the consequences of ring flip in monosubstituted cyclohexane is pretty straightforward, the presence of two or more substituents requires careful consideration to decide which conformer, if any, is the more favoured.
Let us illustrate the approach using 1,4- dimethylcyclohexane. Now, two configurational isomers of this structure can exist, namely trans and cis.
The terms trans and cis are used to describe the configuration, not conformation, of the isomers; in the trans isomer, the two methyl substituents are on opposite sides (faces) of the ring (Latin: trans = across).
Whereas in the cis isomer, they are on the same side of the ring (Latin: cis = on this side). These concepts will become clear when we reach them.
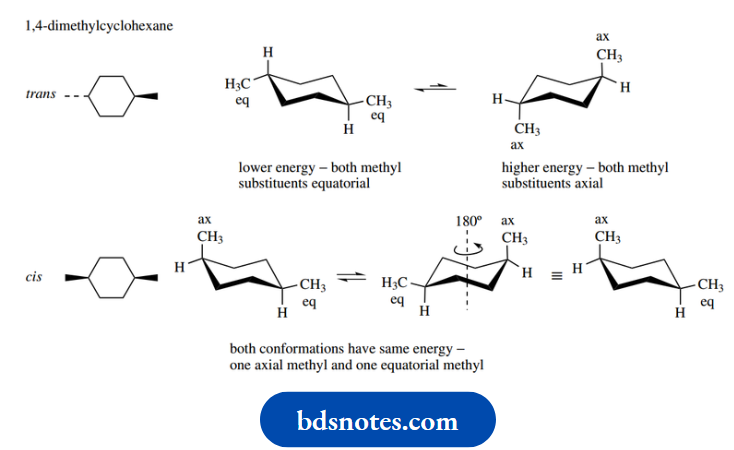
In the trans isomer, one methyl is written down (dotted bond) whilst the other is written up (wedged bond).
If we transform this to a chair conformation, as shown in the left-hand structure, the down methyl will be equatorial and the up methyl will also be equatorial.
With ring flip, both of these substituents then become axial as in the right-hand conformer. From what we have learned about monosubstituted cyclohexanes, it is now easily predicted that the equatorial conformer will be very much favoured over the diaxial conformed.
In the cis isomer, both methyls are written with wedges, i.e. up. In the left-hand chair conformation, one methyl is therefore axial and the other is equatorial.
With a ring flip, the axial methyl becomes equatorial and the equatorial methyl becomes axial. Both conformers have one equatorial methyl and one axial methyl; they must, therefore, be of the same energy, so form a 50:50 equilibrium mixture.
In fact, it is also easy to see that the rotation of either structure about its central axis produces the other structure, a clear illustration that they must be energetically equivalent.
Note that the cis isomer with both methyls down is actually the same compound viewed from the opposite side. This type of reasoning may be applied to other dimethylcyclohexanes, as indicated in the figure.
There is no easy way to predict the result; it must be deduced in each case. One conformer is of much lower energy in the cases of trans-1,2-, cis-1,3-, and trans-1,4-dimethylcyclohexane; both conformers have equal energy in the cases of cis-1,2-, trans-1,3-, and cis-1,4-dimethylcyclohexane.
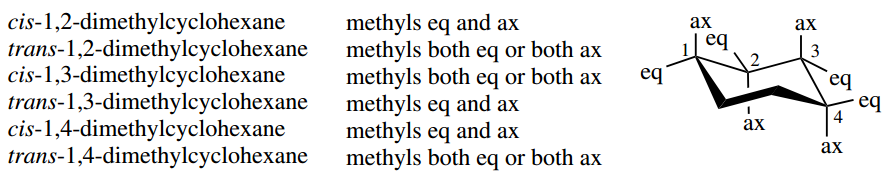
Should the two substituents be different, and especially of different sizes, then the simple reasoning used above with two methyl substituents will need adapting; the larger substituent will prefer to be equatorial.
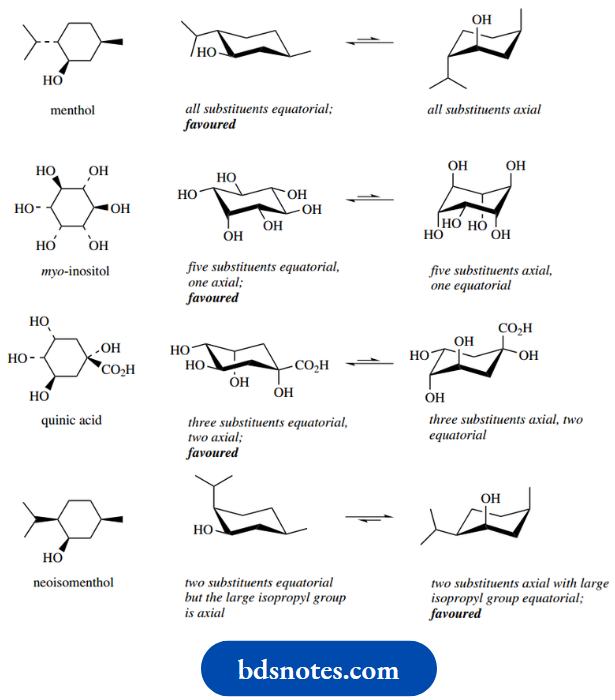
Where we have three or more substituents, the most favoured conformer is going to be the one with the maximum number of equatorial substituents, or perhaps where we have the largest substituents equatorial. This is seen in the following examples.
How To Draw Conformational Isomers And To Flip Cyclohexane Rings
Interpreting a two-dimensional stereochemical structure, converting it into a conformational drawing, and considering the consequences of ring flip can cause difficulties.
The process can be quite straightforward if you approach it systematically. We saw early that, if we draw cyclohexane in typical two-dimensional form.
The bonds to the ring could be described as ‘up’ or ‘down’, according to whether they are wedged or dotted. This is how we would see the molecule if we viewed it from the top.
When we look at the molecule from the side, we now see the chair conformation; the ring is not planar as the two-dimensional form suggests.
Bonds still maintain their ‘up’ and ‘down’ relationship, but this means bonds shown as ‘up’ alternate axial–equatorial around the ring; they are not all axial or all equatorial.
Whilst the ring flip process changes equatorial bonds to axial bonds, and vice versa, it does not change the ‘up’–‘down’ relationship.
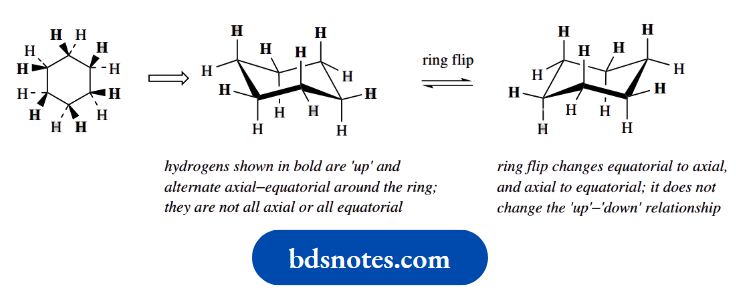
Let us consider the trimethylcyclohexane isomer shown below. All three substituents are ‘up’. We need to use one of the carbons as a reference marker; let us choose the top one.
I like to make this the left-hand carbon in the chair; to make the process more obvious, we could turn the structure so that our reference carbon is also on the left.
It is most important to have this reference carbon, so that as we put the various substituents in we put them on the correct carbons.
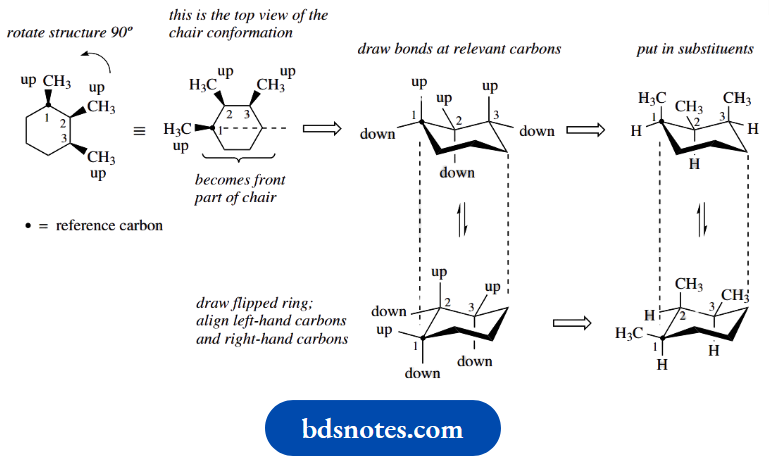
Now draw the two chair conformations of cyclohexane, both having the reference carbon on the left. The carbons opposite our reference point must be furthest right.
If we draw the structures one above the other, lefthand carbons and right-hand carbons should be aligned.
Draw axial and equatorial bonds at the relevant carbons where we have the substituents and identify them as ‘up’ or ‘down’.
Since we are interpreting the structure as though we are looking down on it from the top, the lower part of the ring represents the near-most part of the conformational drawing.
It can also help to number the carbons. Then fill in the substituents as necessary.
In this example, our three methyl groups are all ‘up’, which means that in one conformer the groups will be axial, equatorial, and axial, whereas in the other they will be equatorial, axial, and equatorial.
The latter conformer, with the most equatorial substituents, will be the favoured one. A word of warning is appropriate here.
As we shall see in due course (see Box 3.11), merely changing a substituent from, say, equatorial to axial without flipping the ring changes the configuration, and can produce a different molecule. It would also destroy the ‘up’ or ‘down’ identifier.
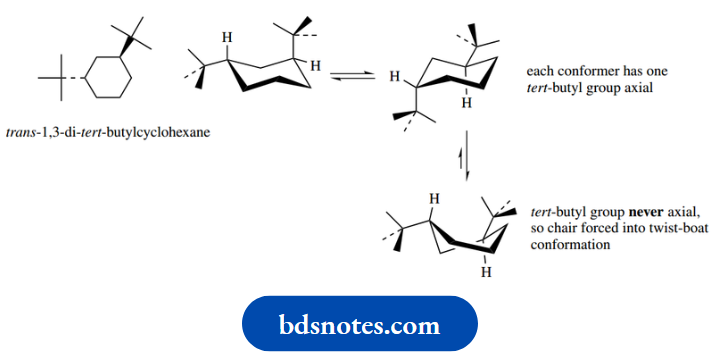
To take this general principle to its extreme, we noted above that tert-butyl groups are sufficiently large that they never occupy an axial position.
It is possible to make di-tert-butylcyclohexanes where conformational mobility would predict that one of these groups would have to be axial, namely cis- 1,2-, trans-1,3- or cis-1,4-derivatives.
As a result, in these cases, we do not see an axial tert-butyl, but instead, the ring system adopts the less favourable twist-boat conformation.
It follows, therefore, that there must be a greater energy difference between chair conformations carrying axial and equatorial tert-butyl substituents than there is between chair and twist-boat conformations.
These conformational changes are shown for trans-1,3-di-tert-butylcyclohexane.
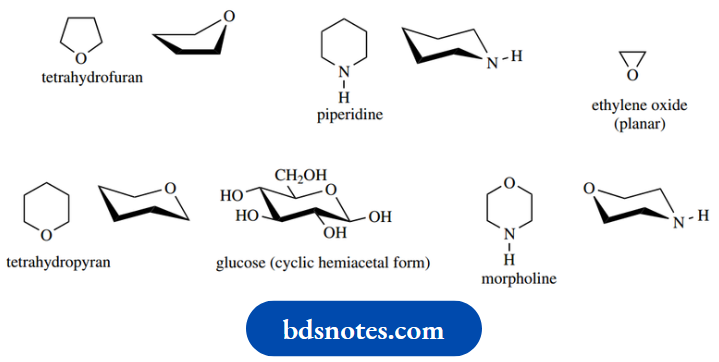
We noted earlier that bonds around nitrogen and oxygen atoms occupied some of the tetrahedral arrays, with lone pairs taking up other orbitals.
This means that we can use essentially the same basic principles for predicting the shape and conformation of heterocycles as we have used for carbocycles.
A substituent on the heteroatom is considered to be larger than the lone pair electrons. Some common examples are shown below.
As we shall see the heteroatom may have other influences, and there are sometimes unexpected effects involving a substituent adjacent to the heteroatom.
Conformationo Flindane
Chlorination of benzene gives an addition product that is a mixture of stereoisomers known collectively as hexachlorocyclohexane (HCH). At one time, this was incorrectly termed benzene hexachloride.
The mixture has insecticidal activity, though activity was found to reside in only one isomer, the so-called gamma isomer, γ-HCH.
γ-HCH, sometimes under its generic name lindane, has been a mainstay insecticide for many years and is about the only example of the chlorinated hydrocarbons that has not been banned and is still available for general use.
Although chlorinated hydrocarbons have proved very effective insecticides, they are not readily degraded in the environment, they accumulate and persist in animal tissues, and have proved toxic to many bird and animal species.
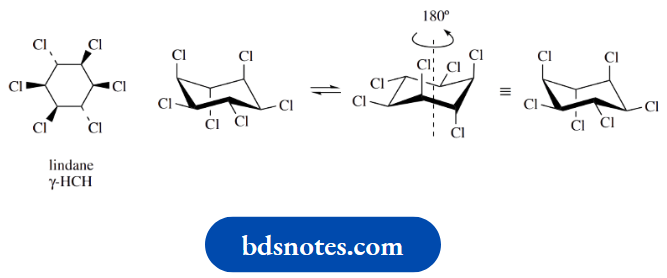
The stereochemistry of the γ-isomer is shown in the diagram, and when converted into a conformational stereo drawing it can be seen that there are three axial chlorines and three equatorial ones.
Ring flip produces an alternative conformation of equal energy, but it can be seen that this is identical to the first structure; rotation through 180◦ produces an identical and, therefore, superimposable structure.
It can be seen that conformational change will not stop the compound from interacting with the insect receptor site.
Configurational Isomers
As we have now seen, conformational isomers interconvert easily by rotation about single bonds. Configurational isomers, on the other hand, are isomers that interconvert only with difficulty, and it usually requires bond breaking if they do interconvert.
Optical Isomers: Chirality And Optical Activity
If tetrahedral carbon has four different groups attached, it is found that they can be arranged in two different ways. These molecules are not superimposable and they have a mirror-image relationship to each other. This is most easily seen with models.
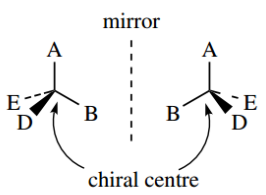
Such an arrangement is called chiral (Greek: cheir = hand), and the carbon atom is termed a chiral centre or stereogenic centre. Look at your two hands.
You will see that they appear identical (allowing for minor blemishes or broken fingernails). However, do what you will, it is not possible to superimpose them, and you should be able to appreciate the mirror image relationship.
The two different arrangements – non-superimposable mirror images – are called enantiomers (Greek: enantiosis = opposite), and we say that enantiomers have different configurations.
The configuration is thus the spatial sequence about a chiral centre. It is also apparent that enantiomers are not going to interconvert readily, and to achieve interconversion we would have to break one of the bonds and then remake it so as to get the other configuration.
Note that the enantiomer of a particular compound can be drawn by reversing two of the substituents; this is actually much easier than drawing the mirror image compound, especially in more complicated structures. As an alternative, the wedge–dot relationship could be reversed.
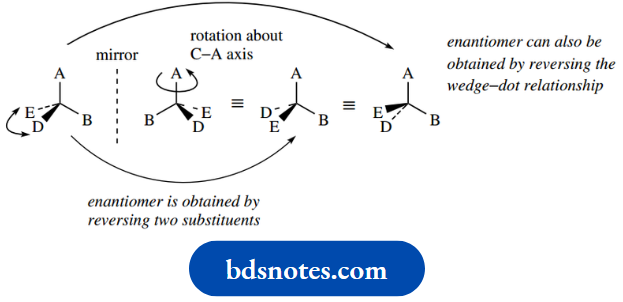
Molecules that are superimposable on their mirror images are said to be achiral. With tetrahedral carbon, this is typically the case when two or more of the attached groups are the same.
This introduces a plane of symmetry into the molecule; molecules with a plane of symmetry can be superimposed on their mirror images.

Note that chirality is not restricted to tetrahedral carbon; it can also be associated with other tetrahedral systems, such as quaternary nitrogen compounds.
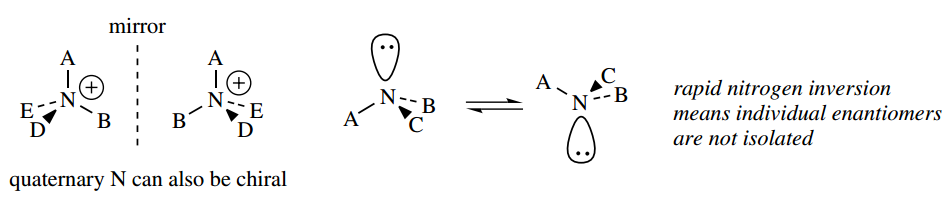
However, non-quaternary nitrogen, although tetrahedral, is not chiral. There is a rapid inversion that converts one enantiomer into the other; effectively, the lone pair does not maintain its position.
The energy barrier to interconversion is about 25 kJ mol-1, which is sufficiently low that inversion occurs readily at room temperature.
This usually makes it impossible to obtain neutral amines in optically active form; quaternization stops this inversion. We shall later need to introduce a related term, prochiral. The concept of prochirality is discussed.
Manipulating Stereostructures
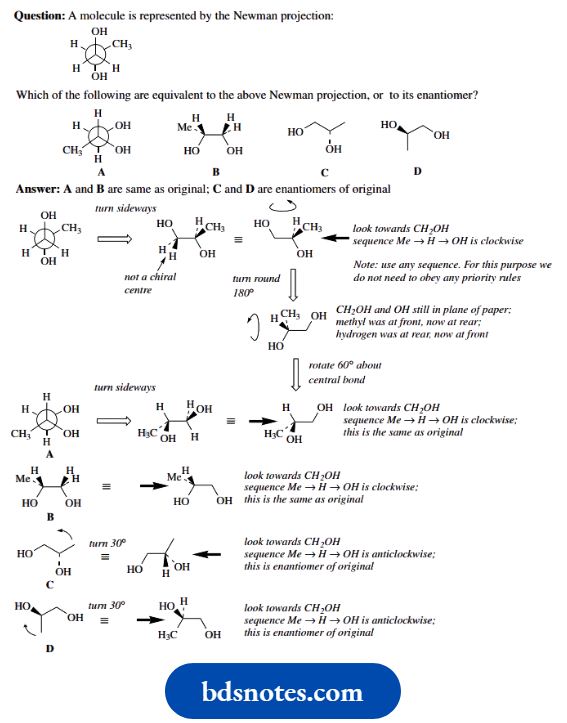
It is not always easy to look at stereo structures – two-dimensional representations of three-dimensional molecules – and decide whether two separate representations are the same or different.
To compare structures, it is usually necessary to manipulate one or both so that they can be compared directly. Here are a few demonstrations of how to approach the problem on paper.
Of course, constructing models for comparison is the easiest method, but there will always be occasions when we have to figure it out on paper.
Optical activity is the ability of a compound to rotate the plane of polarized light.
This property arises from an interaction of the electromagnetic radiation of polarized light with the unsymmetric electric fields generated by the electrons in a chiral molecule.
The rotation observed will clearly depend on the number of molecules exerting their effect, i.e. it depends upon the concentration.
Observed rotations are thus converted into specific rotations that are characteristic of the compound according to the formula below.
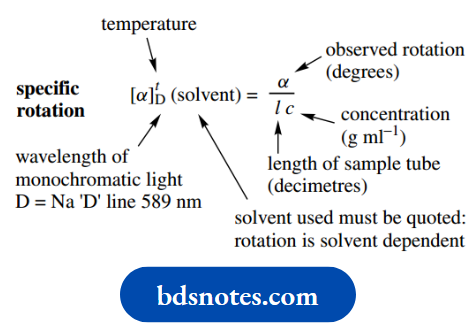
The observed rotation in degrees is divided by the sample concentration (g ml−1 ) and the sample tube length (decimetres). The unusual units used transform the measured small rotations into more manageable numbers.
The specific rotation is then usually in the range 0–1000◦; the degree units are strictly incorrect but are used for convenience.
The polarized light must be monochromatic, and for convenience and consistency, the D line (589 nm) in the sodium spectrum is routinely employed.
Both the temperature and solvent may influence the rotation somewhat, so must be stated.
Enantiomers have equal and opposite rotations. The (+)- or dextrorotatory enantiomer is the one that rotates the plane of polarization clockwise (as determined when facing the beam).
And the (−)- or laevorotatory enantiomer is the one that rotates the plane anticlockwise. In older publications, d and l were used as abbreviations for dextrorotatory and laevorotatory respectively.
But these are not now employed, thus avoiding any possible confusion with D and L.
An equimolar mixture of enantiomers is optically inactive since the individual effects from the two types of molecule are cancelled out.
This mixture is called a racemic mixture or racemate and can be referred to as the (±)-form.
A mixture of enantiomers in unequal proportions has a rotation numerically less than that of either enantiomer; this measurement could be used to determine the proportions of each.
Note that it is not possible to predict the sign or magnitude of the optical activity for a particular enantiomer; it must be measured experimentally.
The presence of more than one chiral centre in a molecule results in an optical rotation that reflects a contribution from each centre, though this is unlikely to be a simple summation.
It must also be appreciated that a positive contribution from one centre may be reduced, countered, or cancelled out by a negative contribution arising from another centre or centre.
Optical Purity And Enantiomeric Excess
A racemic mixture contains equal amounts of the two enantiomeric forms of the compound and has an optical rotation of zero: the optical rotations arising from each of the two types of molecule are cancelled out.
It follows that a mixture of enantiomers in unequal proportions will have a rotation that is numerically less than that of an enantiomer.
Here, we see how to use the measured optical activity to determine the proportions of each enantiomer in the mixture, and therefore its optical purity.
Optical purity is a measure of the excess of one enantiomer over the other in a sample of a compound.
There are a number of occasions when optical purity is of interest. We shall see later that many drugs are chiral compounds, and that biological activity often resides in just one enantiomer.
To minimize potential side effects, it is desirable to supply the drug in a single enantiomeric form.
This might be achieved by devising a synthetic procedure that produces a single enantiomer, an enantiospecific synthesis.
However, syntheses that are enantiospecific can be difficult to achieve, and it is more likely that the procedure is only enantioselective, i.e. it produces both enantiomers but with one predominating.
Alternatively, it is possible to separate the racemic mixture into the two enantiomers. This might not be achieved in a single step.
In both cases, it is usually necessary to monitor just how much of the desired enantiomer is present in the product mixture.
To illustrate the calculation of optical purity, we shall consider another type of reaction of interest, racemization.
This is the conversion of a single enantiomer into a racemic mixture of the two enantiomers. It depends upon the chemical nature of the compound and whether this is easily achievable.
One compound that racemizes readily is hyoscyamine, a natural alkaloid found in deadly nightshade, which is used as an anticholinergic drug.
The natural compound is laevorotatory,\([\alpha]_{\mathrm{D}}^{20}\) − 21° (EtOH), and the enantiomer is almost devoid of biological activity.
Upon heating with a dilute base such as 1% NaOH for about an hour, hyoscyamine racemizes, and the solution becomes optically inactive.
At shorter times, racemization is incomplete and the solution will still be optically active. Consider first a very simple situation in which exactly half of the material has racemized.
Half of the material is now optically inactive, consisting of equal amounts of each enantiomer, whilst the other half is still unchanged.
Since the concentration of the unchanged part is half of the original concentration, the optical rotation will also have dropped to half its original value.
The solution will contain 50% laevorotatory isomer and 50% racemate. However, the racemate is itself a 50 :
50 mixture of the two enantiomers, so the solution actually contains 25% dextrorotatory and 25 + 50% = 75% laevorotatory enantiomers.
Now let us consider when measurements indicate \([\alpha]_{\mathrm{D}}^{20}-9.2^{\circ}\). Calculations now tell us that the sample is 56.2% racemic, and contains 71.9% laevorotatory enantiomer and 28.1% dextrorotatory enantiomer. These figures are derived as follows:
The optical purity (%) = \(\frac{\text { specific rotation of sample }}{\text { specific rotation of pure enantiomer }} \times 100\)
= -9.2/ = 21 x 100 = 43.8%
The sample thus contains 43.8% of laevorotatory enantiomer and 100 − 43.8% = 56.2% of the racemate, the latter contributing no overall optical activity.
The racemate contains equal amounts of laevorotatory and dextrorotatory enantiomers, i.e. it contributes 28.1% of each isomer to the overall mixture.
Therefore, we have 43.8 + 28.1 = 71.9% of laevorotatory enantiomer, and 28.1% of dextrorotatory enantiomer in the partially racemized mixture.
Many workers use the equivalent term percentage enantiomeric excess rather than optical purity:
% Enantiomeric excess = \(\frac{\begin{array}{l}
\text { moles of one enantiomer}-\text { moles of other enantiomer }
\end{array}}{\text { total moles of both enantiomers }} \times 100\)
but this is exactly equivalent to optical purity. From the above calculations, one can see that the laevorotatory enantiomer (71.9%) is in excess of the dextrorotatory enantiomer (28.1%) by 43.8%.
The physical properties of enantiomers and racemates, except for optical rotation and melting points, are usually the same.
The melting points of (+)- and (−)- enantiomers are the same, though that of the racemate is usually different and can be greater or less than the melting point of the enantiomers.
Most spectral properties, For Example. NMR, mass spectrometry, etc., of (+)-, (−)-, and (±)-forms are indistinguishable.
However, pharmacological properties are frequently different, because they may depend upon the overall shape of the compound and its interaction with a receptor.
Pharmacological Properties Of Enantiomers
Although most physical properties of enantiomers are identical, pharmacological properties may be different.
There are examples of compounds where:
- Only one enantiomer is active;
- Both enantiomers show essentially identical activities;
- Both enantiomers have similar activity, but one enantiomer is more active;
- Enantiomers show different pharmacological activities.
These observations may reflect the proximity of the chiral centre to the part of the molecule that binds with the receptor site.
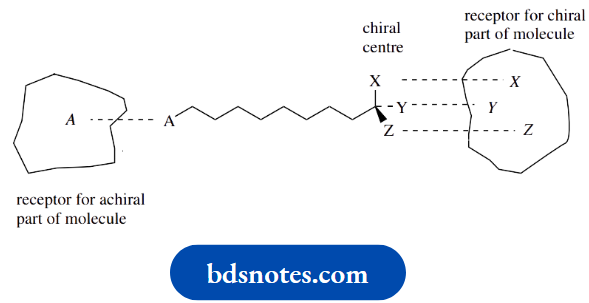
If binding to the receptor involves the chiral centre, then we may see activity in only one enantiomer.
But if binding does not involve the chiral centre, then there may be similar activities for each enantiomer.
Binding close to the chiral centre may cause the same type of activity but of a different magnitude. A different pharmacological activity for each enantiomer almost certainly reflects different receptors.
Further, drug absorption, distribution, and elimination from the body may vary due to differences in protein binding, enzymic modification, etc, since proteins are also chiral entities.
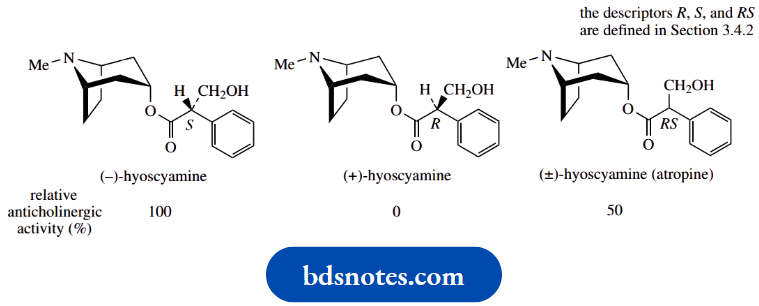
Thus, the anticholinergic activity of the alkaloid hyoscyamine is almost entirely confined to the (−)-isomer, and the (+)-isomer is almost devoid of activity.
The racemic (±)-form, atropine, has approximately half the activity of the laevorotatory enantiomer.
An anticholinergic drug blocks the action of the neurotransmitter acetylcholine and thus occupies the same binding site as acetylcholine.
The major interaction with the receptor involves that part of the molecule that mimics acetylcholine, namely the appropriately positioned ester and amine groups.
The chiral centre is adjacent to the ester and also influences binding to the receptor.
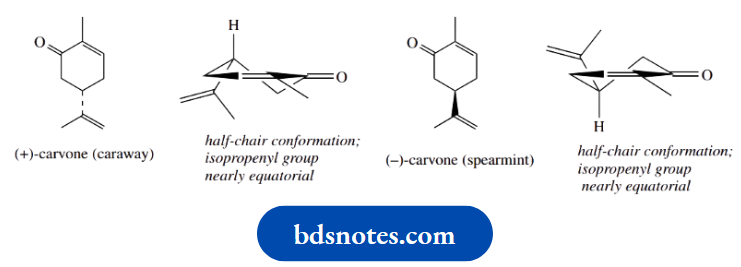
The major constituent of caraway oil is (+)-carvone and the typical caraway odour is mainly due to this component.
On the other hand, the typical minty smell of spearmint oil is due to its major component, (−)- carvone.
These enantiomers are unusual in having quite different smells, i.e. they interact with nasal receptors quite differently. The two enantiomeric forms are shown here in their half-chair conformations.
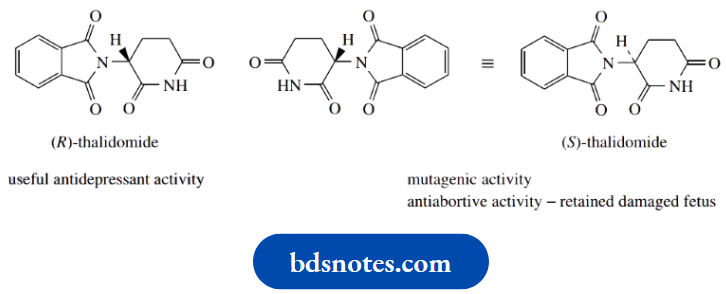
One of the most notorious and devastating examples of a drug’s side effects occurred in the early 1960s when thalidomide was responsible for many thousands of deformities in newborn children.
Thalidomide was marketed in racemic form as a sedative and antidepressant and was prescribed to pregnant women.
Although one enantiomer, the (R)-form, has useful antidepressant activity, it was not realized at that time that the (S)-form.
Thought to be inactive, actually has mutagenic activity and causes defects in the unborn fetus.
Furthermore, the (S)-isomer also has antiabortive activity, facilitating retention of the damaged fetus in the womb, so that any natural tendency to abort a damaged fetus is suppressed.
It is now general policy in the pharmaceutical industry to release new drugs as optically pure isomers, rather than as racemates.
It is desirable to minimize the amount of foreign chemicals a patient is subjected to since even the inactive portion of a drug has to be metabolized and removed from the body.
Such tragedies as occurred with thalidomide may also be avoided. Where a drug is supplied as a single enantiomer, the optical isomer is often incorporated into the drug name, For Example. dexamfetamine, dexamethasone, levodopa, levomenthol, levothyroxine.
Nevertheless, many racemic compounds are currently used as drugs, including atropine, mentioned above, and the analgesic ibuprofen.
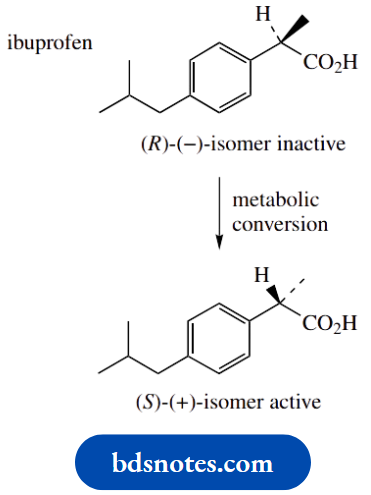
Ibuprofen is an interesting case, in that the (S)-(+)-form is an active analgesic, but the (R)-(−)-enantiomer is inactive.
However, in the body, there is some metabolic conversion of the inactive (R)-isomer into the active (S)-isomer, so that the potential activity from the racemate is considerably more than 50%.
It shows a mechanism to account for this isomerism. There are two approaches to producing drugs as a single enantiomer. If a synthetic route produces a racemic mixture, then it is possible to separate the two enantiomers by a process known as resolution.
This is often a tedious process and, of course, half of the product is then not required.
The alternative approach, and the one now favoured, is to design a synthesis that produces only the required enantiomer, i.e. a chiral synthesis.
Note, the descriptors R and S for enantiomers and RS for racemates are defined.
Cahn–Ingold–Prelog System To Describe Configuration At Chiral Centres
The arrangement of groups around a chiral atom is called its configuration, and enantiomers have different configurations.
Therefore, it is necessary for us to have a means of describing configuration so that we are in no doubt about which enantiomer we are talking about.
Although enantiomers have equal and opposite optical rotations, the sign of the optical rotation does not tell us anything about the configuration.
The system adopted by IUPAC for describing configuration was devised by Cahn, Ingold, and Prelog, and is often referred to as the R, S convention.
The approach used is as follows:
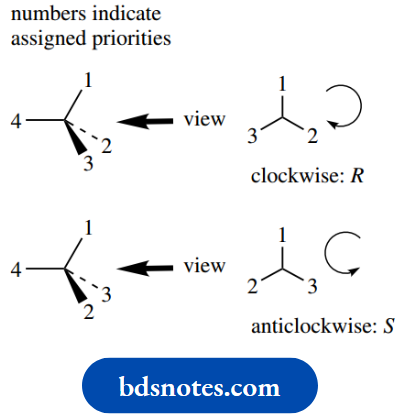
- Assign an order of priority, 1, 2, 3, and 4, to the substituents on the chiral centre.
- View the molecule through the chiral centre towards the group of lowest priority, i.e. priority 4.
- Now consider the remaining groups in order of decreasing priority. If the sense of decreasing priority 1 → 2 → 3 gives a clockwise sequence, then the configuration is described as R (Latin: rectus = right); if the sequence is anticlockwise, then the configuration is described as S (Latin: sinister = left).
The remaining part of the procedure is to assign the priorities. The IUPAC priority rules form a rather long document in order to encompass all possibilities.
Here is a very short version suitable for our requirements. Note that it applies to both acyclic and cyclic compounds.
- A higher atomic number precedes a lower one, for example. Br > Cl > S > O > N > C > H.
- For isotopes, higher atomic mass precedes lower, for example. T > D > H.
- If atoms have the same priority, then secondary groups attached are considered. If necessary, the process is continued to the next atom in the chain.
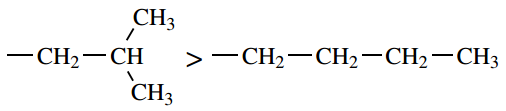
the first atom is carbon in both cases;
consider the second atom:
the second atom is carbon in both cases;
consider the next atom(s):
carbon directly bonded to two further
carbons have higher priority than carbon
directly bonded to just one further carbon
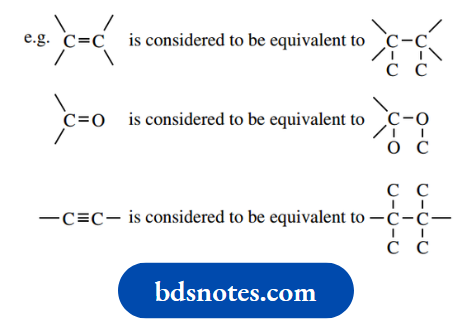
- Double and triple bonds are treated by assuming each atom is duplicated or triplicated.
As simple examples of the approach, let us consider the amino acid (−)-serine and the Krebs cycle intermediate (+)-malic acid.
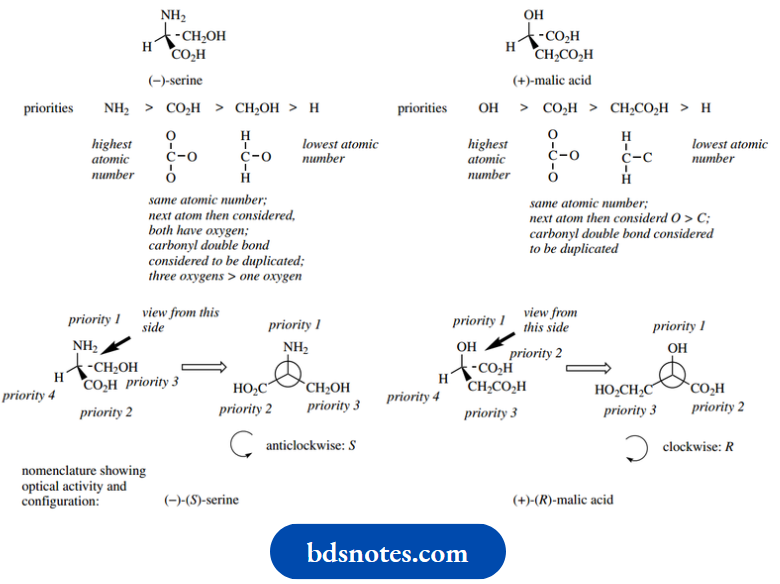
It is now possible to incorporate the configuration of the compound into its nomenclature to give more detail. (−)-Serine becomes (−)-(S)-serine, whilst (+)- malic acid becomes (+)-(R)-malic acid.
Because there is no relationship between (+)/(−) and configuration (R)/(S), it is necessary to quote both optical activity and configuration to convey maximum information.
The descriptor (RS ) is used to indicate a (±) racemic mixture.
Note also that the configuration (R) or (S) is defined by the priority rules, and configuration (R) could easily become (S) merely by altering one substituent.
For instance, all the amino acids found in proteins can be represented by the formula
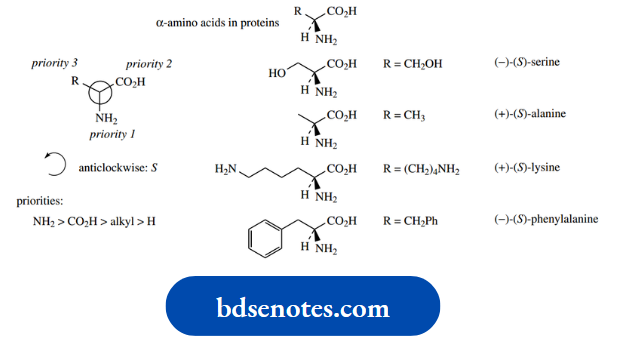
Now all these amino acids that are chiral (glycine, R = H is achiral) have the (S) configuration except for cysteine, which is (R).
Just looking at the structures, one might imagine that they would all have the same configuration.
And indeed one can consider that they have; they differ only in the nature of the R group, but are all arranged around the chiral centre in the same manner.
However since (R) and (S) are only descriptors of configuration, the designation depends upon the nature of the R group.
In most cases, R is an alkyl or substituted alkyl, so it has a lower priority than carboxyl.
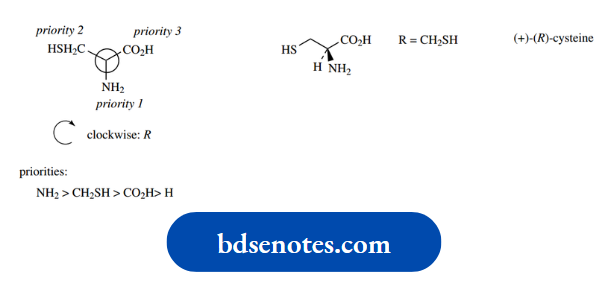
In the case of cysteine, R = CH2SH, and since S has a higher atomic number than any of the other atoms under consideration, this group will have a higher priority than the carboxyl. The net result is that cysteine is (R)- cysteine.
Configurations in cyclic compounds are considered in the same way as those in acyclic compounds.
If you cannot get an answer with the first atom, move on to the next, even though this may mean working around the ring system. Consider, for example, the stereoisomer of 3-methylcyclohexanol.
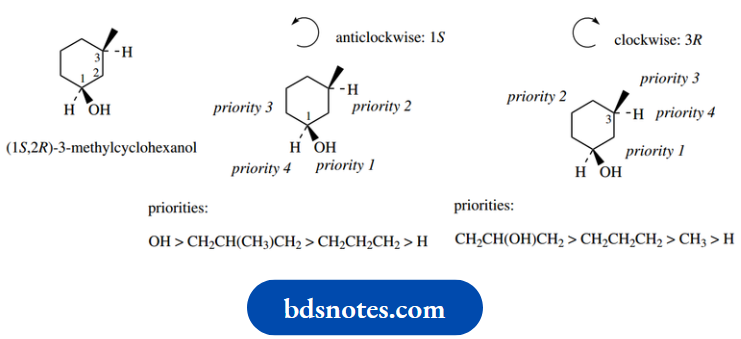
This has two chiral centres, C-1 and C-3. It can readily be deduced that this isomer is actually (1S,2R)- 3-methylcyclohexanol.
At both centres, two of the groups under consideration for priority assignment are part of the ring system.
These are only differentiable when one comes to the ring substituent, the methyl group when one considers C-1 and the hydroxyl when one considers C-3.
In each case, the substituted arm is going to take precedence over the unsubstituted arm. A more interesting example (6-aminopenicillanic acid) containing heterocyclic rings is discussed.
Configurations in 6-aminopenicillanic acid
Let us look at the common substructure of penicillin antibiotics, namely 6-aminopenicillanic acid.
To illustrate some aspects of working out whether a chiral centre is allocated the R or S configuration.
First of all, there are three chiral centres in this molecule, carbons 3, 5 and 6; note that carbon 2 is not chiral, since two of the groups attached are methyls. Only the three carbons indicated have four different groups attached.
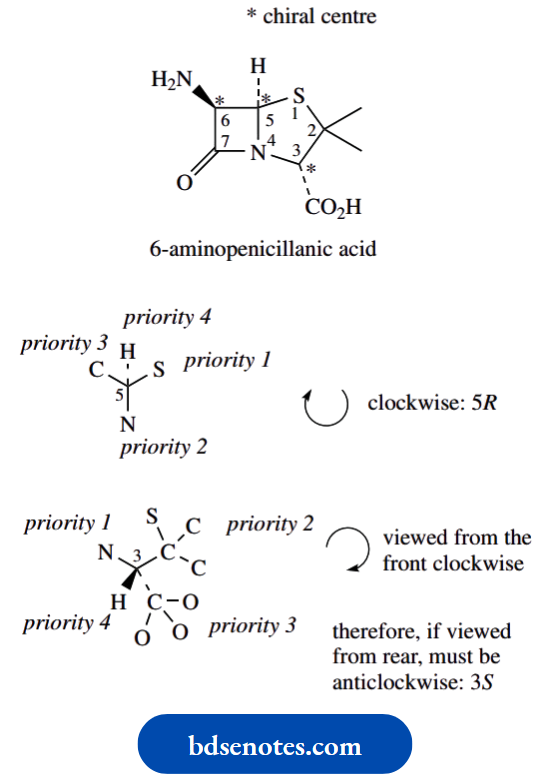
The chirality at C-5 is assigned in the usual way. The groups attached have easily assigned priorities, with S > N > C > H. The configuration is thus 5R.
For the chirality at position 3, the priorities are assigned N > C–S > C–O > H.
Now a very useful hint. Since the group of lowest priority is wedged/up, it is rather difficult to imagine the sequence when viewed from the rear.
Accordingly, view the sequence from the front, which is easy, and reverse it.
From the front, the sequence for C-3 looks clockwise, so if viewed from the rear, it must be anticlockwise, and the descriptor is 3S.
Note how we consider substituents in the standard way even if they are part of a ring system. If you cannot get an answer with the first atom, move on to the next around the ring system.
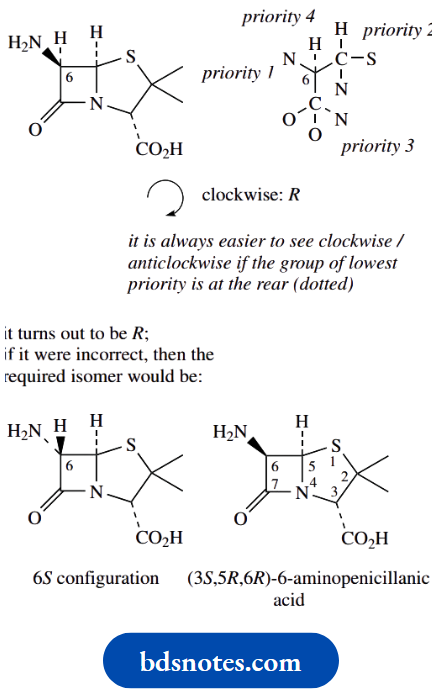
Lastly, suppose one is asked to draw a particular configuration at C-6, namely 6R.
There is no way one can visualize a particular configuration, so the approach is to draw one and see if it is correct; if it is not correct, then change it by reversing wedged/dotted bonds.
And which to try first? Well, always put the group of lowest priority, usually H, away from you, i.e. dotted or down. Then you can see the clockwise/anticlockwise relationship easily from the front.
In this case, the version with H down gave the 6R configuration; but, if it were to be wrong, then the alternative configuration at this centre would be the required one, i.e. a wedged bond to the hydrogen.
Geometric Isomers
Restricted rotation about double bonds or due to the presence of ring systems leads to configurational isomers termed geometric isomers.
Thus, we recognize two isomers of but-2-ene, as shown below, and we term these cis and trans isomers. We have met these terms earlier.
With a double bond, rotation would destroy the π bond that arises from the overlap of p orbitals; consequently, there is a very large barrier to rotation.
It is of the order of 263 kJ mol-1, which is very much higher than any of the barriers to rotation about single bonds that we have seen for conformational isomerism.
Accordingly, cis and trans isomers do not interconvert under normal conditions. Ring systems can also lead to geometric isomerism.
And cis and trans isomers of cyclopropane-1,2-dicarboxylic acid similarly do not interconvert; interconversion would require the breaking of bonds.
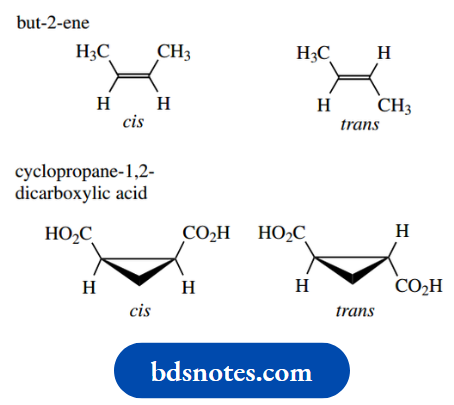
The terms cis and trans are used to describe the configuration, which is considered to be the spatial sequence about the double bond or the spatial sequence relative to a ring system.
The cis isomer has substituents on the same side of the double bond or ring system (Latin: cis = on this side), whereas the trans isomer has substituents on opposite sides (Latin: trans = across).
With simple compounds, like the isomers of but-2- ene, the descriptors cis and trans are quite satisfactory, but a compound such as 3-methylpent-2-ene causes problems.
Do we call the isomer below cis because the methyls are on the same side, or trans because the main chain goes across the bond?
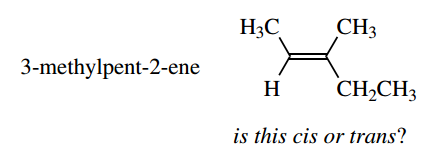
For double bonds, the configuration is now usually described via the non-ambiguous E,Z nomenclature, assigned using the Cahn–Ingold–Prelog priority rules for substituents on each carbon.
First, consider each carbon of the double bond separately, and assign priorities to its two substituents. Then consider the double bond with its four substituents.
If the two substituents of higher priority are on the same side of the double bond, the configuration is Z (German: zusammen = together), whereas if they are on opposite sides, the configuration is E (German: entgegen = across).
Thus, for the 3-methylpent-2-ene isomer we can see that, for C-2, the substituents are methyl and hydrogen with priorities methyl > hydrogen.
For C-3, we have substituents methyl and ethyl, with ethyl having the higher priority.
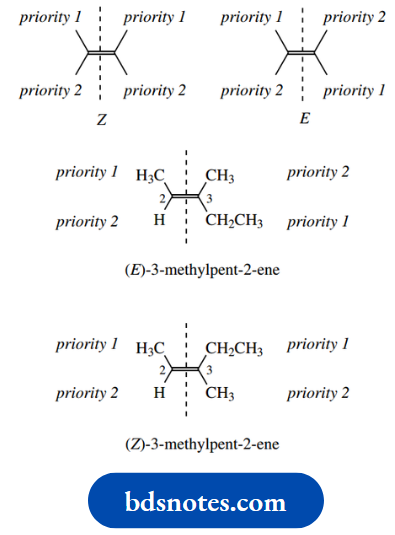
Higher priority. Thus, the high-priority groups are on opposite sides of the double bond, and this isomer has the E configuration.
The alternative arrangement with high-priority substituents on the same side of the double bond has the Z configuration.
Configurations Of Tamoxifen, Clomifene And Triprolidine
The oestrogen-receptor antagonist tamoxifen is used in the treatment of breast cancer and is highly successful.
Clomifene is also an oestrogen-receptor antagonist but is principally used as a fertility drug, interfering with feedback mechanisms and leading to ova release, though this often leads to multiple pregnancies.
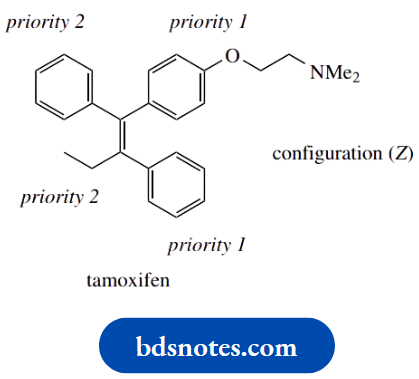
As can be deduced from the application of the Cahn–Ingold–Prelog priority rules, high-priority groups are positioned on the same side of the double bond in each case.
Note that the substituted aromatic ring has higher priority than the unsubstituted ring. Both tamoxifen and clomifene thus have the Z configuration.
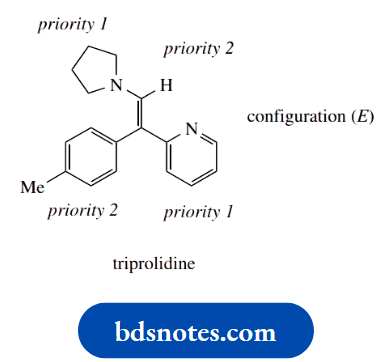
The antihistamine drug triprolidine has the E configuration; note that the heterocyclic pyridine ring takes priority over the benzene ring, even though the latter has a substituent.
Priority is deduced by working along the carbon chain towards the first atom that provides a decision, in this case, the nitrogen atom in the pyridine.
Configurational Isomers With Several Chiral Centres
Configurational isomerism involving one chiral centre provides two different structures, the two enantiomers.
If a structure has more than one chiral centre, then there exist two ways of arranging the groups around each chiral centre.
Thus, with n chiral centres in a molecule, there will be a maximum number of 2n configurational isomers. Sometimes, as we shall there are less.
Starting with two chiral centres, there should, therefore, be four stereoisomers.
And this is nicely exemplified by the natural alkaloid (−)-ephedrine, which is employed as a bronchodilator drug and decongestant.
Ephedrine is (1R,2S)-2-methylamino-1-phenylpropan- 1-ol, so has the structure and stereochemistry shown.
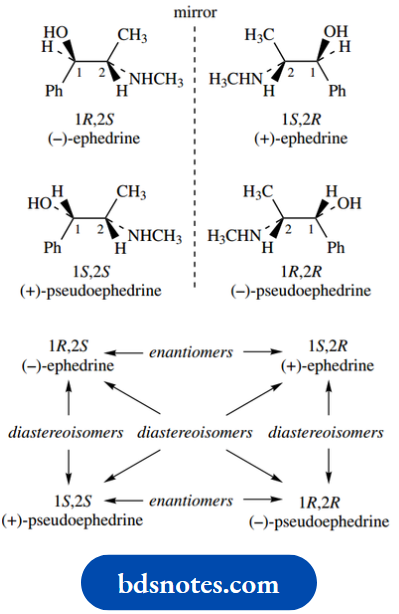
Now the other three of the possible four stereoisomers are the (1S,2S), (1R,2R), and (1S,2R) versions. These are also shown, and mirror-image relationships are emphasized.
The (1S,2R) isomer is the mirror image of (−)-ephedrine, which has the (1R,2S) configuration. Therefore, it is the enantiomer of (−)-ephedrine, and can be designated (+)-ephedrine.
Note that the enantiomeric form has the opposite configuration at both chiral centres.
The other two isomers are the (1 S,2S) and (1R,2R) isomers, and these two also share a mirror image relationship, have the opposite configuration at both chiral centres, and are, therefore, a pair of enantiomers.
From a structure with two chiral centres, we thus have four stereoisomers that consist of two pairs of enantiomers.
Stereoisomers that are not enantiomers we term diastereoisomers, or sometimes diastereomers. Thus, the (1S,2S) and (1R,2R) isomers are diastereoisomers of the (1R,2S) isomer.
Other enantiomeric or diastereomeric relationships between the various isomers are indicated in the figure.
We have seen earlier that enantiomers are chemically identical except in optical properties, although biological properties may be different.
On the other hand, diastereoisomers have different physical and chemical properties, and probably different biological properties as well.
As a result, they are considered a completely different chemical entity and are often given a different chemical name.
The (1S,2S) and (1R,2R) isomers are thus known as (+)- pseudoephedrine and (−)-pseudoephedrine respectively.
Interestingly, (+)-pseudoephedrine has similar biological properties to (−)-ephedrine and it is used as a bronchodilator and decongestant drug in the same way as ephedrine.
One more useful piece of terminology can be introduced here. This is the term epimer. An epimer is a diastereoisomer that differs in chirality at only one centre.
Thus, (−)-pseudoephedrine is the 2-epimer of (−)-ephedrine, and (+)-pseudoephedrine is the 1-epimer of (−)-ephedrine.
The epimer terminology is of greater value when there are more than two chiral centres in the molecule.
Suppose we have a compound with three chiral centres, at positions 2, 3, and 4 in some unspecified carbon chain, with configurations 2R,3R,4S. There would thus exist a total of 23 = 8 configurational isomers.
The enantiomer would have the configuration 2S,3S,4R, i.e. changing the configuration at all centres.
The 2S,3R,4S diastereoisomer could then refer to as ‘the 2-epimer’, and the 2R,3S,4S diastereoisomer as ‘the 3-epimer’, since we have changed the stereochemistry at just one centre, keeping other configurations the same.
Drawing Enantiomers And Epimers: 6-aminopenicillanic Acid
The structure of the natural isomer of 6-aminopenicillanic acid is shown. You are asked to draw the structure of its enantiomer and its 6-epimer.
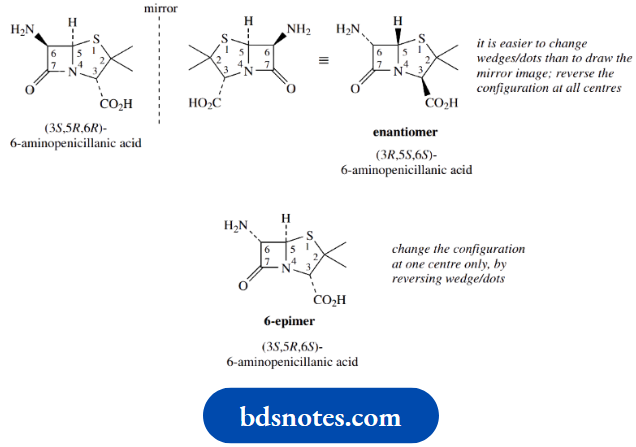
The enantiomer will have the configuration changed at all chiral centres, whereas the 6-epimer retains all configurations except for that at position 6.
Note that it is not necessary to draw the mirror image compound for the enantiomer, just reverse the wedge–dot relationship for the bonds at each chiral centre.
This is much easier and less prone to errors whilst transcribing the structure. Now for a rather important point.
In a compound such as (−)-ephedrine there are going to be many different conformations as a result of rotation about the central C–C bond.
Three of them are shown here, the energetically most favourable staggered conformer with all large groups anti, a less favourable staggered conformer, and a high-energy eclipsed version.
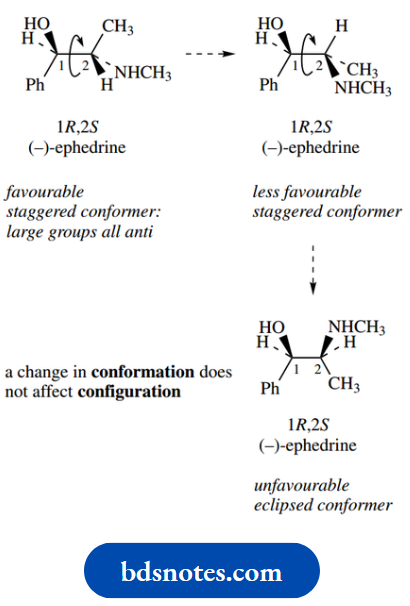
However, note carefully that changing the conformation does not affect the spatial sequence of the chiral centres, i.e. it does not change the configuration at either chiral centre.
This seems a trivial and rather obvious statement, and indeed it probably is in the case of acyclic compounds.
It is when we move on to cyclic compounds that we need to remember this fundamental concept because a common mistake is to confuse conformation and configuration.
The same stereochemical principles are going to apply to both acyclic and cyclic compounds. With simple cyclic compounds that have little or no conformational mobility, it is easier to follow what is going on.
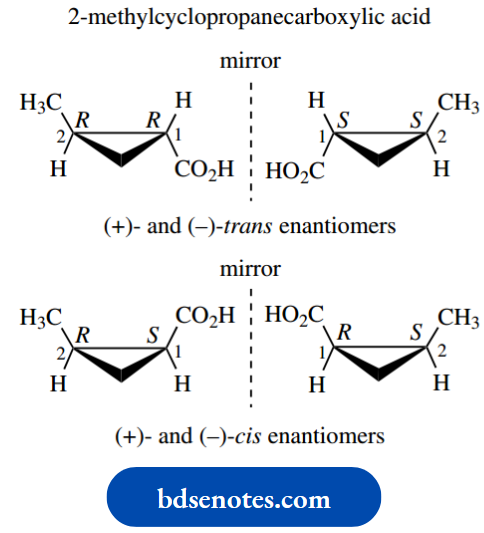
Consider a disubstituted cyclopropane system. As in the acyclic examples, there are four different configurational stereoisomers possible, comprising two pairs of enantiomers. No conformational mobility is possible here.
However, in a cyclohexane system, we also need to consider the conformational mobility that generates two different chair forms of the ring.
Let us consider 3-methylcyclohexanecarboxylic acid. This has two chiral centres, and thus there are four configurational stereoisomers. These are the enantiomeric forms of the trans and cis isomers.
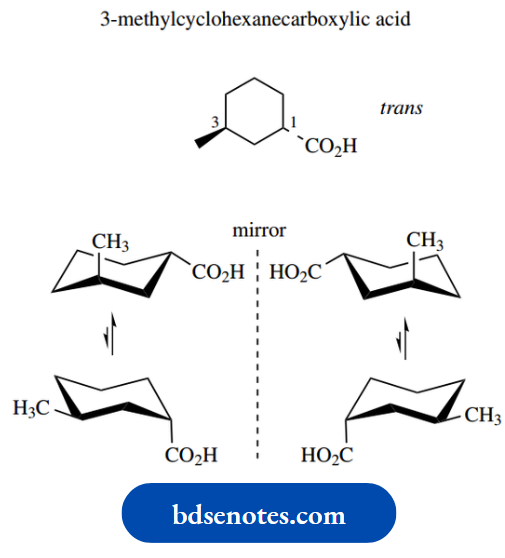
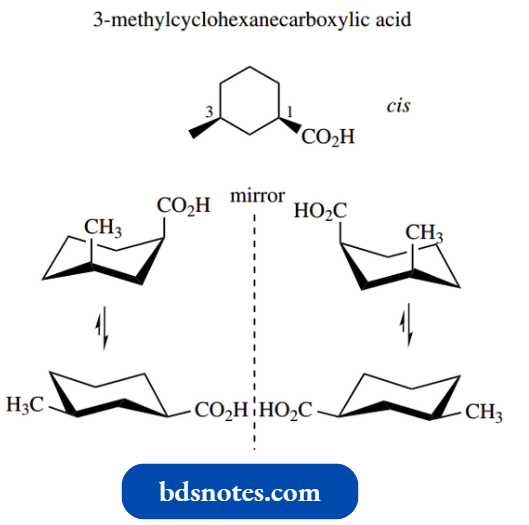
Each isomer can also adopt a different chair conformation as a consequence of ring flip.
We thus can write down eight possible stereoisomers, comprised of two interconvertible conformers for each of the four non-interconvertible configurational isomers.
Put another way, there are four configurational isomers (22 = 4), but each can exist as two possible conformational isomers.
Note that you can also see the mirror image relationship in the conformational isomers.
Of course, in practice, some conformers are not going to be energetically favourable. The cis compound has favoured equatorial and unfavoured diaxial conformers.
The trans compound has one equatorial and one axial substituent; we can assume that the larger carboxylic acid group will prefer to be equatorial.
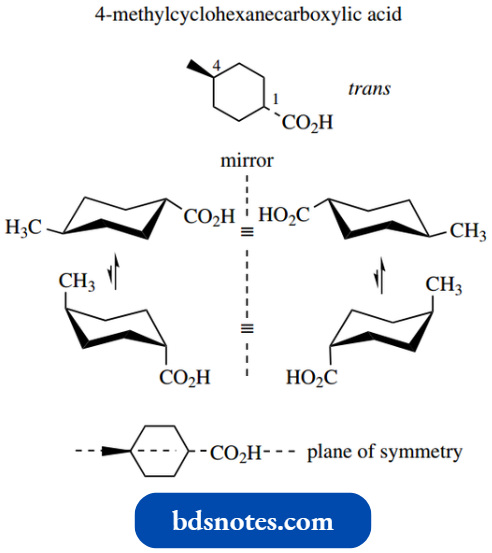
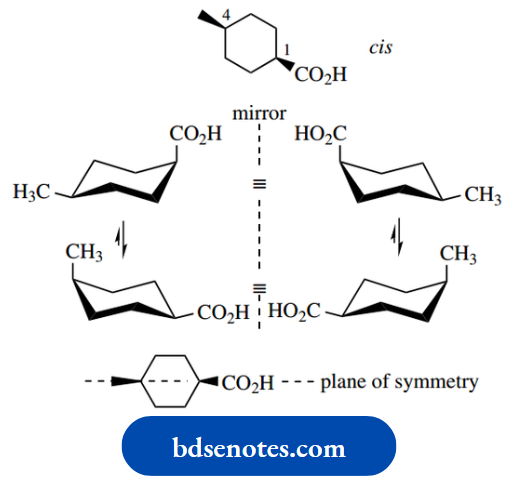
Do appreciate that cyclohexane rings with 1,2- or 1,3-substitution fit into the above discussions; however, if we have 1,4-substitution there are no chiral centres in the molecule.
Since two of the groups are the same at each possible site! However, cis and trans forms still exist; these are geometric isomers and can still be regarded as diastereoisomers.
We can spot this type of situation by looking for symmetry in the molecule.
Both cis- and trans-4- methylcyclohexanecarboxylic acid isomers have a plane of symmetry, and, as we saw for simple tetrahedral carbons, this symmetry means the molecule is achiral.
Configurations A Nd Conformations: Avoiding Confusion
At this stage, a word of caution: do not confuse conformation with configuration. Different conformations interconvert easily; different configurations do not interconvert without some bond-breaking process.
We commented above that changing the conformation did not affect the spatial sequence of chiral centres and used ephedrine as a rather trivial and obvious example.
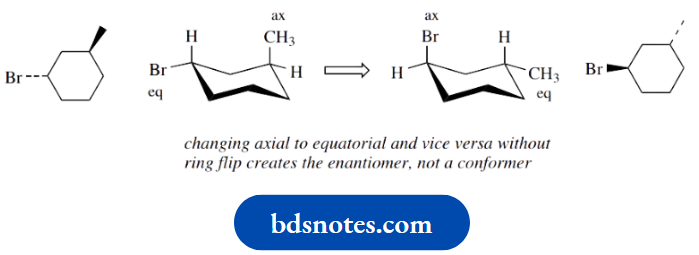
Rotation about single bonds did not change the configuration at either chiral centre. To emphasize this point, look at the following relationships for trans-3-methylcyclohexyl bromide.
Don’t confuse conformation with configuration
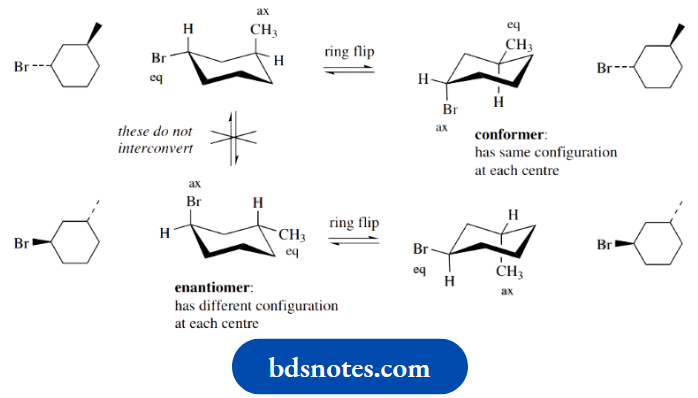
Ring flip of the upper left structure produces an alternative conformer. Ring flip does not change the configuration.
The axial–equatorial relationship (conformation) is modified, but the up–down relationship (configuration) is still there.
The enantiomer of this structure has the alternative configuration at both chiral centres, but it cannot be produced from the first structure by any simple isomerization process.
However, it is still conformationally mobile. The figure thus shows the conformational isomerism for two different configurational isomers, the enantiomeric pair.
A common mistake that can be made when one is trying to draw the different conformers that arise from ring flip in a cyclohexane compound is to remember vaguely that axial groups become equatorial, and vice versa, and to apply this change without flipping the ring.
Of course, as can be seen from looking at the compounds below, transposing the equatorial bromine to axial and the axial methyl to equatorial changes the configuration at both centres.
So we have produced the enantiomer. This is a configurational isomer and not a conformer.
Meso Compounds
Now for a rather unexpected twist. We have seen that if there are n chiral centres there should be 2n configurational isomers, and we have considered each of these for n = 2 (For example. ephedrine, pseudoephedrine).
It transpires that if the groups around chiral centres are the same, then the number of stereoisomers is less than 2n. Thus, when n = 2, there are only three stereoisomers, not four.
As one of the simplest examples, let us consider in detail tartaric acid, a component of grape juice and many other fruits. This fits the requirement, since each of the two chiral centres has the same substituents.
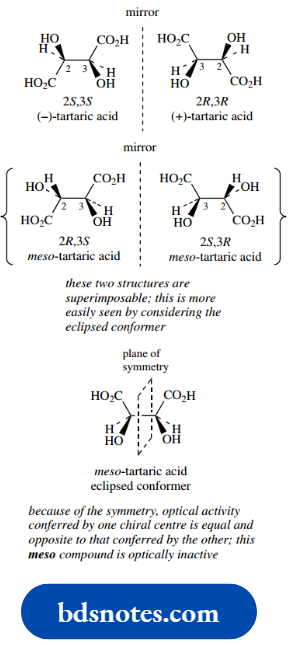
We can easily draw the four predicted isomers, as we did for the ephedrine–pseudoephedrine group and two of these represent the enantiomeric pair of (−)-tartaric acid and (+)-tartaric acid.
Now let us consider the other pair of isomers, and we shall see the consequences of the substituent groups being the same.
Because these two structures are actually superimposable and, therefore, only represent a single compound.
This is not so easily seen with the staggered conformers drawn, so it is best to rotate these about the 2,3-bond to give an eclipsed conformer.
They can both be rotated to give the same structure, so they represent only a single compound. This is called meso-tartaric acid (Greek: mesos = middle).
Furthermore, since we have superimposable mirror images, there can be no optical activity. We can see why a compound with chiral centres should end up optically inactive by looking again at the eclipsed conformer.
The molecule itself has a plane of symmetry, and because of this symmetry, the optical activity conferred by one chiral centre is equal and opposite to that conferred by the other and, therefore, is cancelled out.
It has the characteristics of a racemic mixture but as an intramolecular phenomenon. A meso compound is defined as one that has chiral centres but is itself achiral.
Note that numbering is a problem in tartaric acid because of the symmetry and that positions 2 and 3 depend on which carboxyl is numbered as C-1.
It can be seen that (2R,3S) could easily have been (3R,2S) if we had numbered from the other end, a warning sign that there is something unusual about this isomer.
The same stereochemical principles apply to both acyclic and cyclic compounds.
With simple cyclic compounds that have little or no conformational mobility, it can even be easier to follow what is going on. Let us first look at cyclopropane-1,2-dicarboxylic acid.
These compounds were considered as examples of geometric isomers, and cis and trans isomers were recognized.
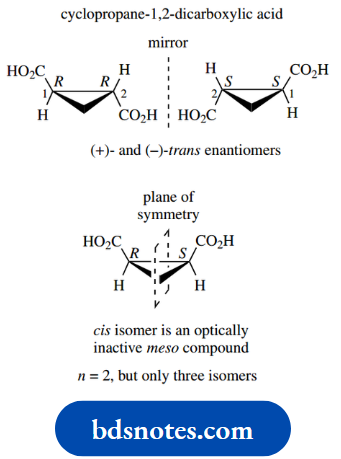
This is essentially the same as the tartaric acid example, without the conformational complication. Thus, there are two chiral centres, and the groups around each centre are the same.
Again, we get only three stereoisomers rather than four, since the cis compound is an optically inactive meso compound.
There is a plane of symmetry in this molecule, and it is easy to see that one chiral centre is mirrored by the other so we lose optical activity.
Conformational mobility, such as we get in cyclohexane rings, makes the analysis more difficult, and manipulating molecular models provides the clearest vision of the relationships.
Let us look at 1,2- dimethylcyclohexane as an example. Again, we have met the cis and trans isomers when we looked at conformational aspects. Here, we need to consider both configuration and confirmation.
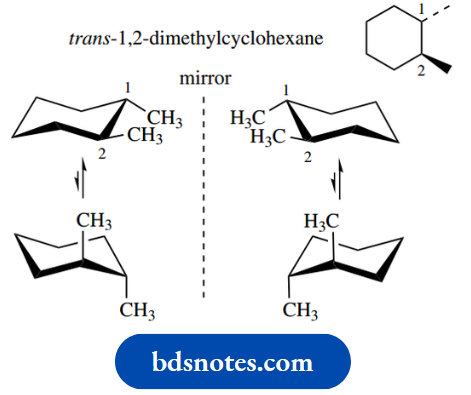
In the trans compound, two mirror-image enantiomeric forms can be visualized. These will be the (+)- and (−)-trans isomers.
Note particularly that conformational changes may also be considered, but these do not change configuration, so we are only seeing different conformers of the same compound.
The above scheme thus shows two interconvertible conformers (upper and lower structures) for each of the two non-interconvertible enantiomers (left and right structures).
The cis compound provides the real challenge, however. If we draw version A, together with its mirror image C, they do not look capable of being superimposed.
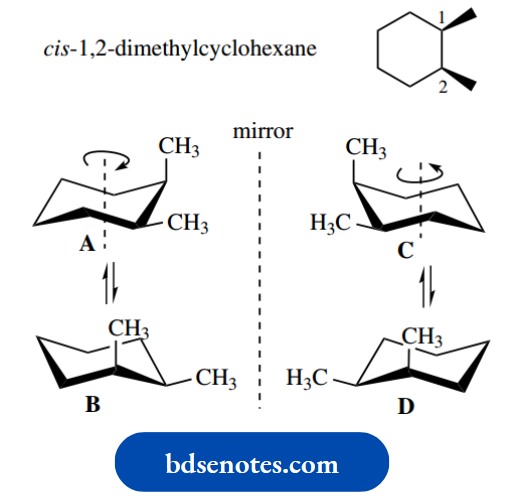
However, conformer A may be ring flipped to an equal-energy conformer B, and this will have a corresponding mirror image version D.
Now consider a 120° rotation of version A about the central axis; this will give D.A. A similar 120° rotation of version C about the central axis will give B.
It follows, therefore, that if a simple rotation of one structure about its axis gives the mirror image of a conformational isomer, then we cannot have enantiomeric forms but must have the same compound.
These are thus two different conformers of an optically inactive meso compound. It may require manipulation of models to really convince you about this!
Now, although the cyclohexane ring is not planar, the overall consequences for trans- and cisdimethylcyclohexane can be predicted by looking at the two-dimensional representations.
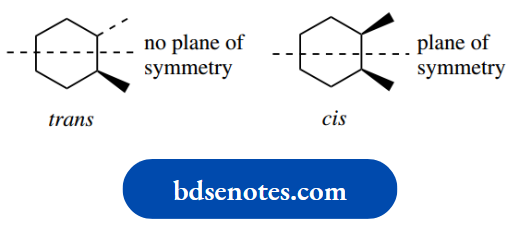
It is clear that this representation of cis-dimethylcyclohexane shows a plane of symmetry, and we can deduce it to be a meso compound.
No such plane of symmetry is present in the representation of trans-methylcyclohexane.
Why does this approach work? Simply because the transformation of planar cyclohexane (with eclipsed bonds) into a non-planar form (with staggered bonds) is a conformational change achieved by rotation about single bonds.
The fact that cyclohexane is non-planar means we may have to invoke the conformational mobility to get the three-dimensional picture.
Our consideration of meso compounds leads us to generalize:
- A molecule with one chiral centre is chiral;
- A molecule with more than one chiral centre may be chiral or achiral.
Now let us extend this generalization with a further statement:
- A molecule may be chiral without having a chiral centre.
This is the subject of the next section.
Chirality Without Chiral Centres
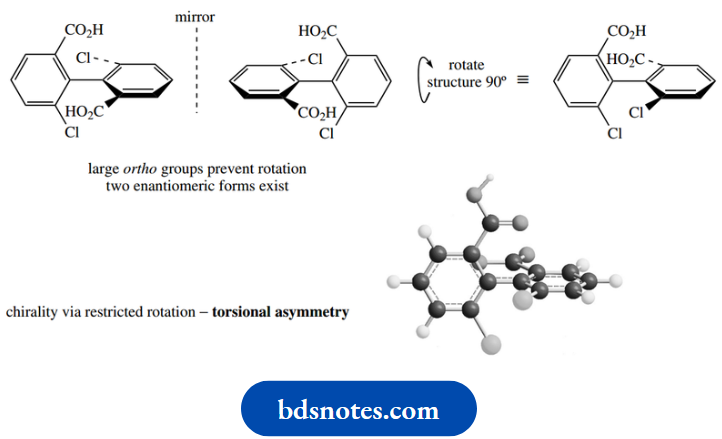
We shall restrict discussions here to three types of compounds. In the first, we get what is termed torsional asymmetry, where chirality arises because of restricted rotation about single bonds.
The commonest examples involve two aromatic rings bonded through a single bond (biphenyls).
If large groups are present in the ortho positions, these prevent rotation about the interring single bond.
The most favourable arrangement to minimize interactions is when the aromatic rings are held at right angles to each other. As a result, two enantiomeric forms of the molecule can exist.
Because of the size of the ortho groups, it is not possible to interconvert these stereoisomers merely by rotation. Even when we only have two different types of substituent, as shown, we get two enantiomeric forms.
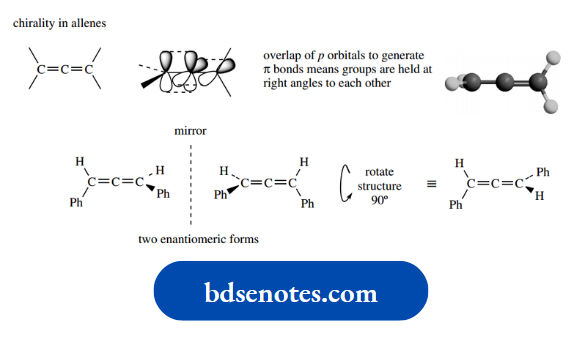
The second type of compound is called an allene; these compounds contain two double bonds involving the same carbon. These compounds exist but are often difficult to prepare and are very reactive.
It is the concept of chirality which is more important here than the chemistry of the compounds. If a carbon atom is involved in two double bonds, it follows that the π bonds created must be at right angles to each other.
The consequence of this is that the substituents on the other carbons of the allene are also held at right angles to each other. Again, two enantiomeric forms of the molecule can exist.
The third example of chirality without a chiral centre is provided by s p i r o compounds, which we shall meet later when we consider the stereochemistry of polycyclic systems.
But at this stage, it is worth noting that they provide a third example of chirality without a chiral centre.
Spiro compounds contain two ring systems that have one carbon in common, and it is easy to see this carbon could be chiral if four different groupings are present.
A nice natural example, the antibiotic griseofulvin, is shown here.
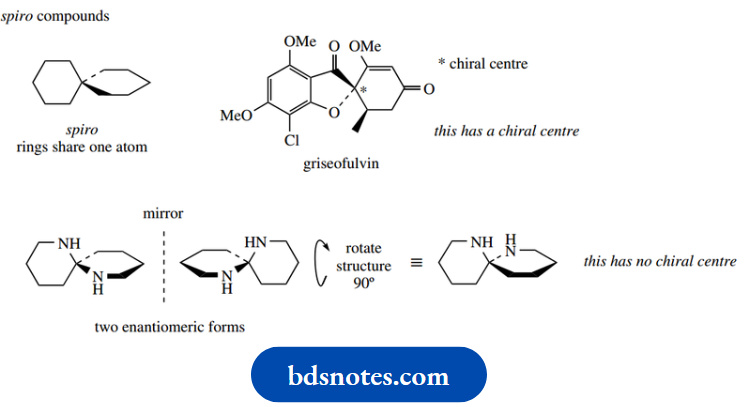
However, it is also possible to visualize spiro compounds with groupings that are not all different, where enantiomeric forms exist because mirror image compounds are not superimposable.
The diamine shown is chiral, in that the mirror image forms are not superimposable, even though only two types of substituent are attached to the spiro centre.
Both rings in this compound will have the chair conformation, but it is not easy to draw these because one ring will always be viewed face-on.
The solution is to ensure the spiro centre is not on the left or right tip of either ring.
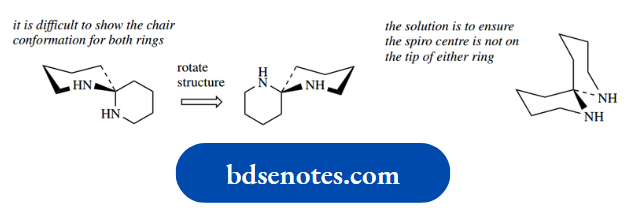
With biphenyls, allenes, and spiro compounds, groups are held at right angles by a rigid system, and this feature allows the existence of non-superimposable mirror image stereoisomers, i.e. enantiomers.
It is useful to think of this arrangement as analogous to a simple chiral centre, where the tetrahedral array also holds pairs of groups at right angles.
In contrast to tetrahedral carbon, it is not even necessary for all the groups to be different to achieve chirality, as can be seen in the examples above.
Torsionalasymmetry: Gossypol
The concept of torsional asymmetry is not just an interesting abstract idea. Some years ago, fertility in some Chinese rural communities was found to be below normal levels, and this was traced back to the presence of gossypol in dietary cottonseed oil.
Gossypol acts as a male contraceptive, altering sperm maturation, spermatozoid motility, and inactivation of sperm enzymes necessary for fertilization.
Extensive trials in China have shown the antifertility effect is reversible after stopping the treatment, and it has potential, therefore, as a contraceptive for men.
Gossypol is chiral due to restricted rotation, and only the (−)-isomer is pharmacologically active as an infertility agent. The (+)-isomer has been found to be responsible for some toxic symptoms.
Most species of cotton (Gossypium) produce both enantiomers of gossypol in unequal amounts, with the (+)-enantiomer normally predominating over the (−)-isomer.
It has proved possible to separate racemic (±)-gossypol from this type of mixture – the racemate complexes with acetic acid.
Whereas the separate enantiomers do not. The racemic form can then be resolved to give the useful biologically active (−)-isomer.
Prochirality
Enantiotopic Groups
We have defined chirality in terms of ‘handedness’, such that mirror image stereoisomers are not superimposable.
In the case of tetrahedral carbon, chirality is a consequence of having four different groups attached to it.
If two or more groups were the same, then the compound would be termed achiral. Now we introduce another term, prochiral. Achiral molecules that can become chiral by one simple change are called prochiral.
The simplest example we could include under this definition would be an achiral molecule in which two groups are the same.
The two like groups are termed enantiotopic, in that separate replacement of each would generate enantiomers.
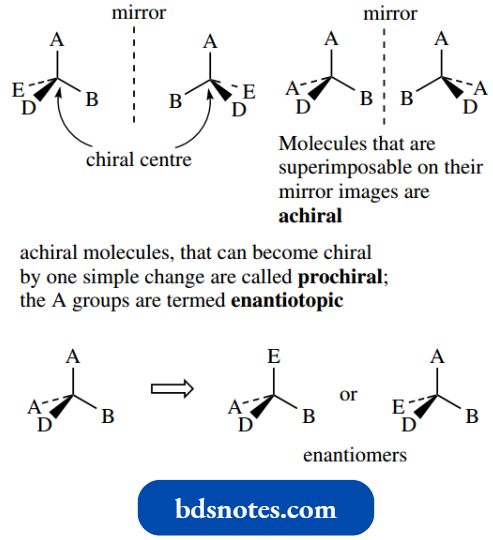
This seems an unnecessary complication. Why do we want to call an achiral centre prochiral? What benefits are there?
Well, remember that the Cahn–Ingold–Prelog system allowed us to describe a particular chiral arrangement of groups at a chiral centre; prochirality now allows us to distinguish between the two like groups at an achiral centre.
When might we want to do that? The following example from biochemistry shows the type of occasion when we might need to identify one or other of the like groups.
The enzyme alcohol dehydrogenase oxidizes ethanol to acetaldehyde, passing the hydrogen to the coenzyme nicotinamide adenine dinucleotide NAD+.
This is the enzyme that restores normal service after excessive consumption of alcoholic drinks.
By specifically labelling each hydrogen in turn, and then observing whether the substrate loses or retains the label in the enzymic reaction, it has been determined which hydrogen is lost from the methylene group of ethanol.
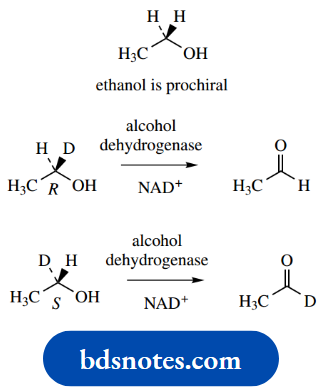
How then, in an unambiguous fashion, can we describe which hydrogen is lost? We define the two hydrogens as pro-R and pro-S.
By considering the effect of increasing their effective priorities according to the Cahn–Ingold–Prelog system; this is simply achieved if we consider having deuterium instead of protium (normal hydrogen).
Then, if replacing a particular hydrogen with deuterium produces a chiral centre with the R configuration, that hydrogen is termed the pro-R hydrogen.
Similarly, increasing the priority of the other hydrogen should generate the S configuration, so that that hydrogen is termed the pro-S hydrogen.
We can also label hydrogens in a structure as HR and HS according to this procedure.
We can thus deduce that alcohol dehydrogenase stereospecifically removes the pro-R hydrogen from the prochiral methylene.
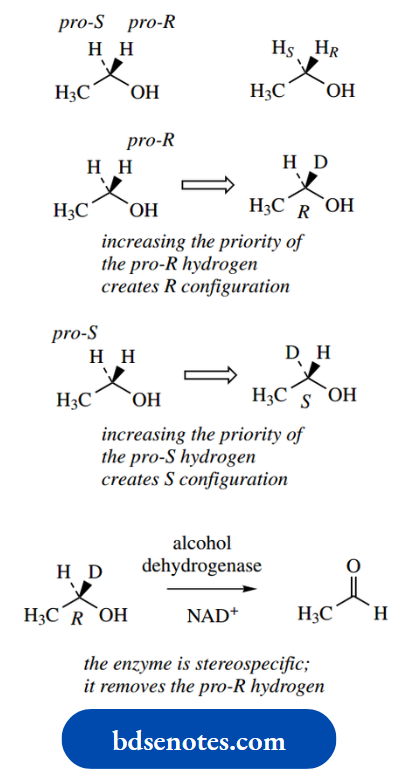
This example is from biochemistry. It is a feature of biochemical reactions that enzymes almost always catalyse reactions in a completely stereospecific manner.
They are able to distinguish between enantiotopic hydrogens because of the three-dimensional nature of the binding site.
There are also occasions where chemical reactions are stereospecific; refer to the stereochemistry of E2 eliminations for typical examples.
Citric Acid Has Three Prochiral Centres
The Krebs cycle is a process involved in the metabolic degradation of carbohydrates.
It is also called the citric acid cycle because citric acid was one of the first intermediates identified.
Once formed, citric acid is modified by the enzyme aconitase through the intermediate cis-aconitic acid to give the isomeric isocitric acid.
This is not really an isomerization, but the result of dehydration followed by rehydration. Both steps feature stereospecific anti-processes, i.e. groups are removed or added from opposite sides of the molecule.
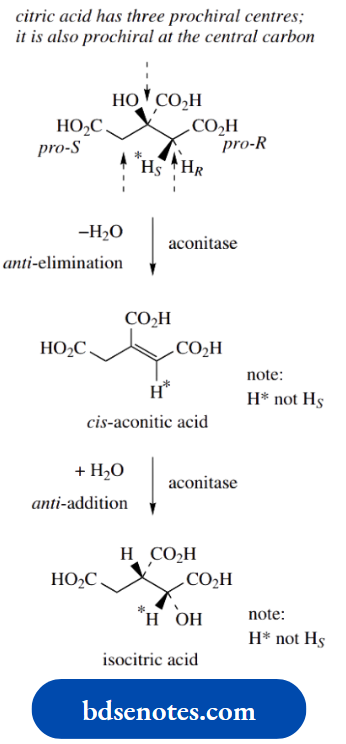
First, let us look closely at the structure of citric acid. It has three prochiral centres. Two of these are the methylenes, but note that the central carbon is also prochiral.
It has two groups the same, namely the –CH2CO2H groups. The loss of water from citric acid is an anti-elimination, so the hydroxyl is lost together with one of the methylene hydrogens.
The hydrogen lost has been found to be the pro-R hydrogen from the pro-R–CH2CO2H group.
This is followed by an anti-addition reaction in which water is added to the new double bond but in the reverse sense. The hydrogen retained throughout the process is shown with an asterisk.
Note that we can only label this hydrogen as pro-S in citric acid; in cis-aconitic acid and isocitric acid, it is no longer attached to a prochiral centre, and we must resort to some other labelling system, namely the asterisk.
This is a nice example of enzymic stereospecificity. It involves the specific removal of one hydrogen atom from a substrate that appears to have four equivalent hydrogens.
Because of the three-dimensional characteristics of both the enzyme and the substrate, the apparently equivalent side chains on the central carbon are going to be positioned quite differently and the enzyme is able to distinguish between them.
Further, it also distinguishes between the two hydrogens of a methylene group.
An interesting consequence of this stereospecificity is that because only one of the citric acid side chains is modified in the aconitase reaction, it takes further turns of the cycle before the material entering the cycle (acetyl-CoA) is actually degraded.
A reaction that gives a mixture of isomeric products with one isomer predominating would be termed stereoselective.
Enantiotopic Faces
We have thus seen that there could be a need to distinguish between two similar groups attached to tetrahedral carbon, and have exploited the Cahn–Ingold–Prelog priorities to label the separate groups.
We also need to consider another way in which a chiral centre might be generated, and that is by the addition of a group to a planar system.
For example, if we reduce a simple ketone that has two different R groups with lithium aluminium hydride we shall produce a racemic alcohol product.
This is because hydride can be delivered to either face of the planar carbonyl group with equal probability.
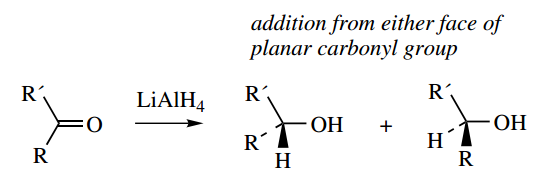
In marked contrast, nature’s reducing agent, reduced nicotinamide adenine dinucleotide (NADH), delivers hydride in a stereospecific manner because it is a cofactor in an enzyme-catalysed reaction.
For example, reduction of pyruvic acid to lactic acid in vertebrate muscle occurs via attack of hydride to produce just one enantiomer, namely (S)-lactic acid.
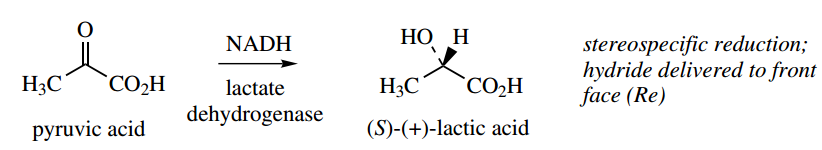
We can see from the diagram that hydride must be delivered from the front face as shown, but it makes sense to have a more precise descriptor for faces than front or back.
Once again, the Cahn–Ingold–Prelog system can help us out. We assign priorities to the three groups attached to the planar carbon.

We then consider the descending sequence and decide whether this is clockwise or anticlockwise; the face that provides a clockwise sequence is then labelled Re and the face that provides an anticlockwise sequence is labelled Si.
These are simply variants on R and S, in fact, the first two letters of rectus and sinister. Note that there is no correlation between Re or Si and the chirality R or S of the tetrahedral product formed.
It can now be seen that, in the enzymic reduction of pyruvic acid to lactic acid, hydride is delivered to the Re face of the pyruvic acid.
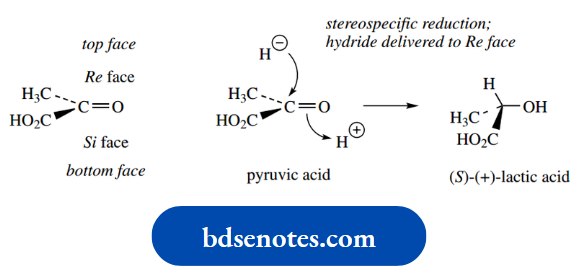
A molecule such as pyruvic acid is said to have two enantiotopic faces. An attack of a reagent onto the Reface yields one enantiomer, whereas an attack onto the Si face will produce the other enantiomer.
The Re and Si descriptors are similarly applied to the carbon atoms making up C=C bonds.
This gets a little more complex, in that a C=C bond generates four faces to be considered, two at each carbon. It is necessary to systematically deduce the descriptor for each, as shown below.
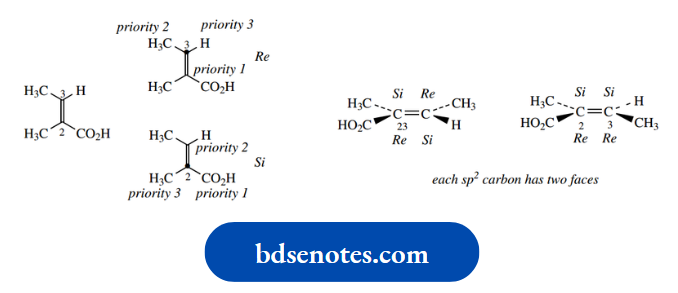
NADH Delivers Hydride From A Prochiral Centre; NAD+ Has Enantiotopic Faces
NADH (reduced nicotinamide adenine dinucleotide) is utilized in biological reductions to deliver hydride to an aldehyde or ketone carbonyl group.
A proton from water is used to complete the process, and the product is thus an alcohol. The reaction is catalysed by an enzyme called dehydrogenase.
The reverse reaction may also be catalysed by the enzyme, namely the oxidation of an alcohol to an aldehyde or ketone. It is this reverse reaction that provides the dehydrogenase nomenclature.
During the reduction sequence, NADH transfers a hydride from a prochiral centre on the dihydropyridine ring.
And is itself oxidized to NAD + (nicotinamide adenine dinucleotide) that contains a planar pyridinium ring.
In the oxidation sequence, NAD+ is reduced to NADH by acquiring hydride to an enantiotopic face of the planar ring. The reactions are completely stereospecific.
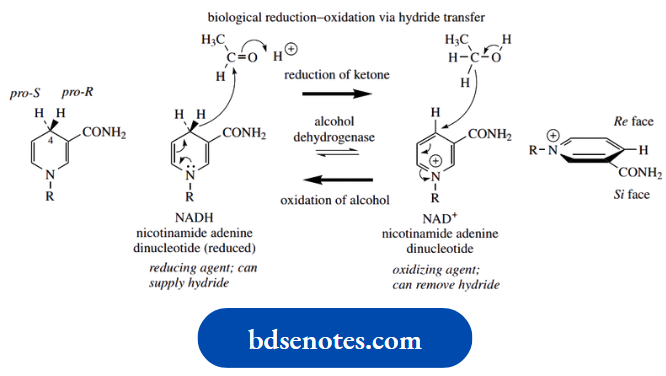
The stereospecificity depends upon the enzyme in question. Let us consider the enzyme alcohol dehydrogenase, which is involved in the ethanol-to-acetaldehyde interconversion.
It has been deduced that the hydrogen transferred from ethanol is directed to the Re face of NAD+, giving NADH with the 4R configuration.
In the reverse reaction, it is the 4-pro-R hydrogen of NADH that is transferred to acetaldehyde.
Note also that the transfer of hydride to the carbonyl compound is also stereospecific, as is the removal of hydrogen from the prochiral centre of ethanol in the reverse reaction.
We should note that prochiral molecules have the potential to become chiral if we make certain changes.
We have used the term enantiotopic to identify the groups at sp3-hybridized carbon or the faces of sp2– 2-hybridised carbon where alternative changes lead to the production of enantiomers.
However, if there is also a chiral centre in the molecule, then the same changes would lead to the formation of diastereoisomers, not enantiomers. Such groups or faces are now correctly termed diastereotopic.
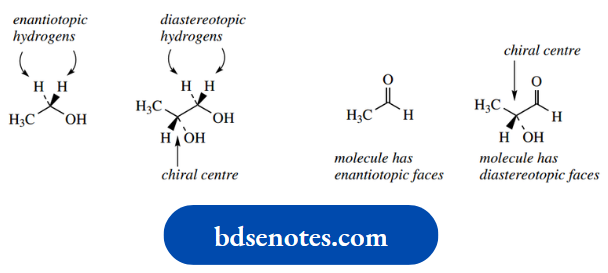
Separation Of Enantiomers: Resolution
We saw that enantiomers have the same physical and chemical properties, except for optical activity, and thus they behave in exactly the same manner.
We also saw, however, that this generalization did not extend to biological properties, and that there were compelling reasons for administering drugs as a single enantiomer rather than a racemate.
At some stage, therefore, it might be necessary to have the means of separating individual enantiomers from a racemic mixture. This is termed resolution.
The traditional method has been to convert enantiomers into diastereoisomers.
Because diastereoisomers have different physical and chemical properties and can, therefore, be separated by various methods.
Provided one can convert the separated diastereoisomers back to the original compound, this offers a means of separating or resolving enantiomers.
The simplest method has been to exploit salt formation by reaction of a racemic acid (or base) with a chiral base (or acid).
For example, treating a racemic acid with a chiral base will give a mixture of two salts that are diastereoisomeric.
Although there is no covalent bonding between the acid and base, the ionic bonding is sufficient that the diastereoisomeric salts can be separated by some means, typically fractional crystallization.
Although fractional crystallization may have to be repeated several times, and, therefore, is tedious, it has generally been an effective means of separating the diastereoisomeric salts.
Finally, the salts can separately be converted back to the acid, completing the resolution
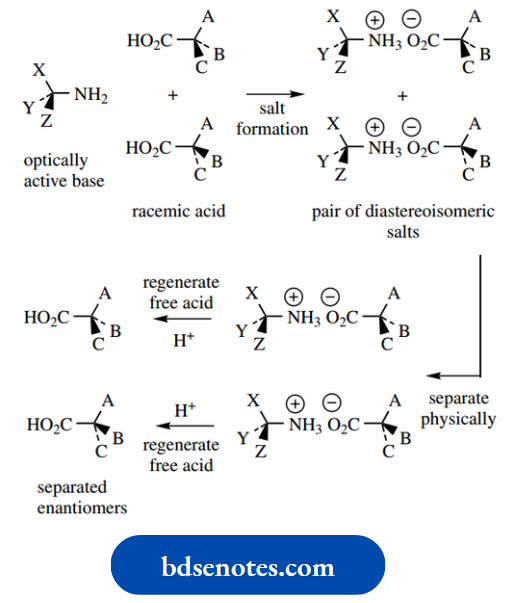
The bases generally employed in such resolutions have been natural alkaloids, such as strychnine, brucine, and ephedrine.
These alkaloids are more complex than the general case shown in the figure, in that they contain several chiral centres.
Tartaric acid has been used as an optically active acid to separate racemic bases.
Of course, not all materials contain acidic or basic groups that would lend themselves to this type of resolution. There are ways of introducing such groups, however, and a rather neat one is shown here.
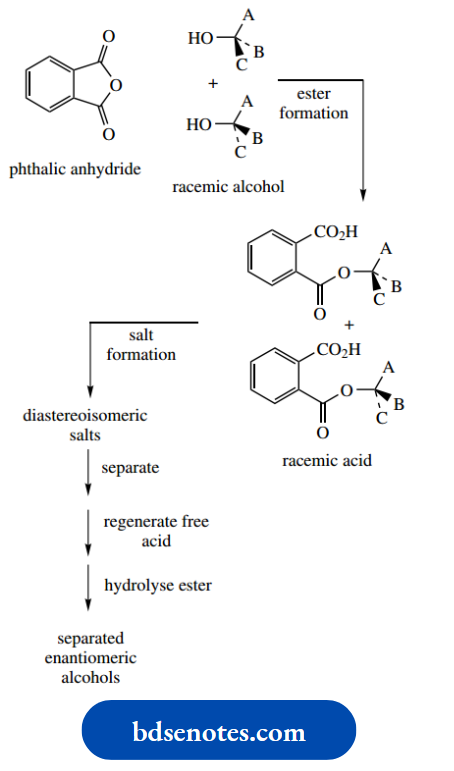
Racemic alcohol may be converted into a racemic acid by reaction with one molar equivalent of phthalic anhydride; the product is a half ester of a dicarboxylic acid.
This can now be subjected to the resolution process for acids and, in due course, the alcohols can be regenerated by hydrolysis of the ester.
A significant improvement in the fractional crystallization process came with the introduction of chiral phases for column chromatography. This allows simple chromatographic separation of enantiomers.
In practice, it is effectively the same principle, that of forming diastereoisomeric complexes with the chiral material comprising the column.
One enantiomer binds more tightly than the other and, therefore, passes through the column at a different rate. The two enantiomers thus emerge from the column as separate fractions.
It has also proved possible to exploit the enantiospecific properties of enzymes to achieve the resolution of a racemic mixture during chemical synthesis.
Enzymes are proteins that catalyse biochemical reactions with outstanding efficiency and selectivity.
This is a consequence of the size and shape of the enzyme’s binding site, a feature that is determined by the sequence of amino acid residues in the protein.
The selectivity of enzymes means that they carry out reactions on one functional group in the presence of others that might be affected by a chemical reagent.
It also means that they can be stereoselective, either performing reactions in a stereospecific manner or only reacting with substrates with a particular chirality.
As a simple example, racemic ester structures may be resolved by the use of ester hydrolysing enzymes called lipases.
With the appropriate choice of enzyme, it has been found that only one enantiomer of the racemic mixture is hydrolysed, whilst the other remains unreacted.
It is then a simple matter to separate the unreacted ester from the alcohol. The unreacted ester may then be hydrolysed chemically, thus achieving resolution of the enantiomeric alcohols.
Fischer Projections
Fischer projections provide a further approach to the two-dimensional representations of three-dimensional formulae.
They become particularly useful for molecules that contain several chiral centres and are most frequently encountered in discussions of sugars.
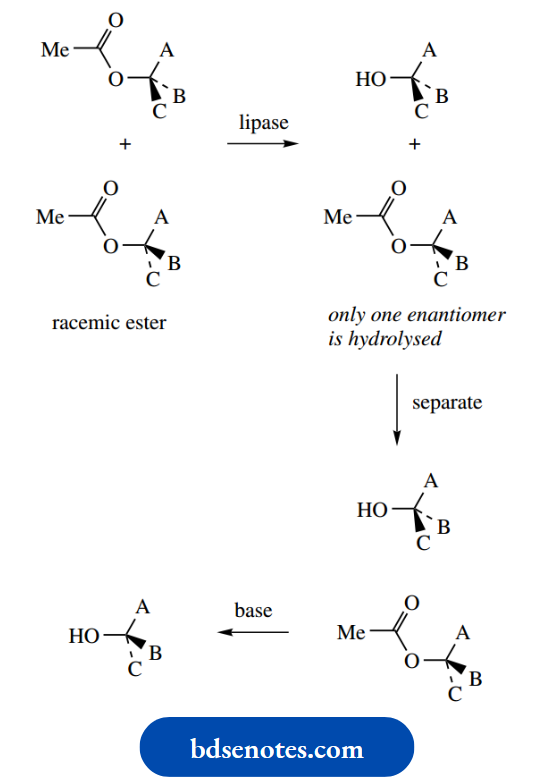
To start, though, let us consider just one chiral centre, and choose the amino acid we met earlier, (−)-(S)-serine.
The Fischer projection is drawn with groups on horizontal and vertical lines, but without showing the chiral carbon atom.
Should you put in this carbon atom, it can no longer be considered that you are representing stereochemistry.
The Fischer projection then implies that horizontal bonds are wedged, whilst vertical bonds are dotted, and it thus speeds up the drawing of stereochemical features.
For (−)-(S)-serine, the wedge–dot version is what one would see if one looked down on the right-hand stereostructure as indicated.
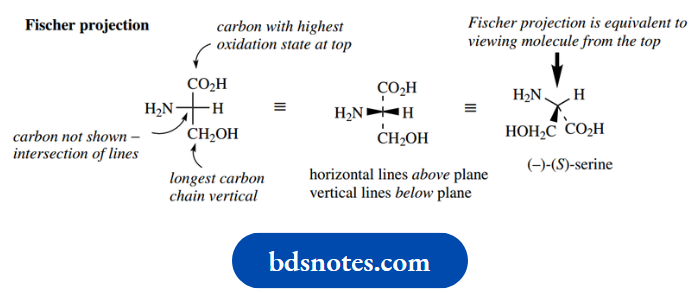
Accordingly, we can now transform stereostructures into Fischer projections, and vice versa. The only significant restrictions are
- We should draw the longest carbon chain vertical;
- We should place the carbon of the highest oxidation state at the top.
However, when we come to manipulate Fischer’s projections, we may need to disregard these restrictions in the interests of following the changes.
Manipulations we can do to a Fischer projection may at first glance appear confusing, but by reference to a model of a tetrahedral array, or even a sketch of the representation, they should soon become quite understandable, perhaps even obvious.
The molecular manipulations shown are given to convince you of the reality of the following statements.
- Rotation of the formula by 180° gives the same molecule.
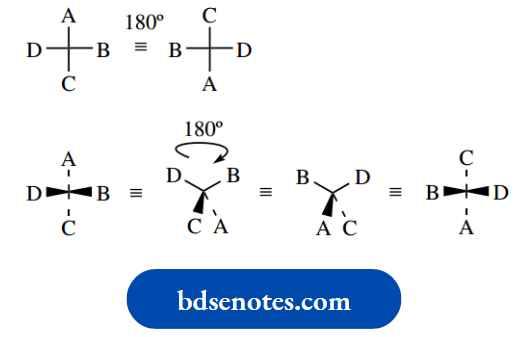
- Rotation of any three groups clockwise or anticlockwise gives the same molecule.
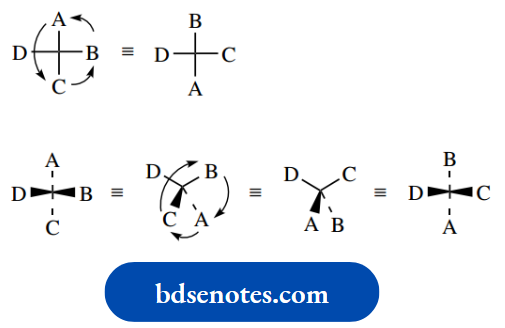
- The exchange of any two groups gives the enantiomer.
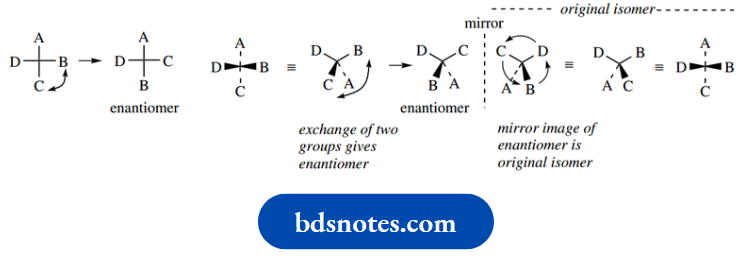
- Rotation of the formula by 90◦ gives the enantiomer.
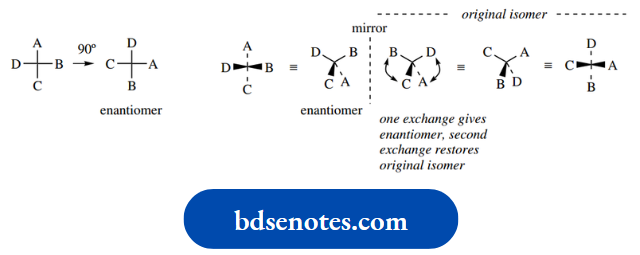
It is also surprisingly easy to assign R or S configurations to chiral carbons in the Fischer projections.
But, because horizontal lines imply wedged bonds (towards you) and vertical lines imply dotted bonds (away from you), there are important guidelines to remember:
- If the group of lowest priority is on the vertical line, a clockwise sequence gives the R configuration;
- if the group of lowest priority is on the horizontal line, a clockwise sequence gives the S configuration.
These do not represent a different set of rules from the clockwise = R, anticlockwise = S conventions we already use It is merely a consequence of the lowest priority group being down (dotted bond) on the vertical line, but up (wedged) on the horizontal line.
We have noted that, if the lowest priority group is wedged, it is easier to look at the sequence from the front, and then reverse it to give us the sequence as viewed from the rear, i.e. towards the group of lowest priority.
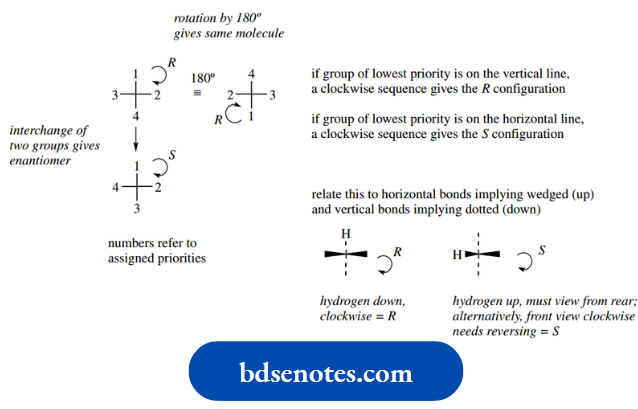
Let us apply these principles to tartaric acid. This compound has two chiral centres; but, as we saw previously, only three stereoisomers exist, since there is an optically inactive meso compound involved.
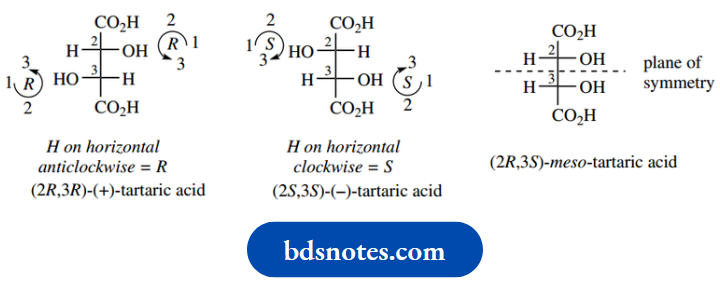
We can draw these three stereoisomers as Fischer projections, reversing the configurations at both centres to get the enantiomeric stereoisomers, whilst the Fischer projection for the third isomer, the meso compound, is characterized immediately by a plane of symmetry.
For (+)-tartaric acid, the configuration is (2R,3R), and for (−)-tartaric acid it is (2S,3S).
For both chiral centres, the group of lowest priority is hydrogen, which is on a horizontal line. In fact, this is the case in almost all Fischer projections, since, by convention, the vertical line is the longest carbon chain.
Thus, we have to reverse our normal configurational thinking: a clockwise sequence of priorities gives S and an anticlockwise sequence gives R.
The configuration of the meso isomer can be deduced by abstracting the appropriate portions from the other two structures and assigning equivalent configurations.
It should be appreciated that a Fischer projection involving more than one chiral centre actually depicts an eclipsed conformer, which is naturally a high-energy state and is normally an unlikely arrangement of atoms.
We need to bear this in mind when we transpose Fischer projections into wedge–dot stereochemical drawings.
Further manipulations are necessary to give lower energy staggered conformers. This is illustrated here with the five-carbon sugar (−)-ribose.
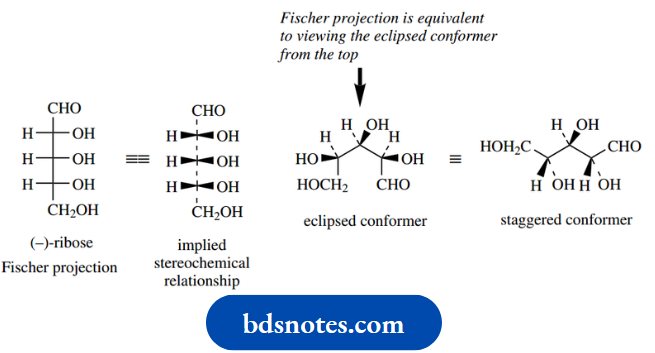
However, as we shall see shortly, Fischer-projection-derived eclipsed conformers are particularly useful in deducing the stereochemistry in cyclic forms of sugars.
D And L configurations
The concept of D and L as configurational descriptors is well established, particularly in amino acids and sugars; frankly, however, we could live without them and save ourselves a lot of confusion.
Since they are so widely used, we need to find out what they mean, but in most cases, the information conveyed is less valuable than sticking with R and S.
D and L sugars
The simplest of the sugars is glyceraldehyde, which has one chiral centre. Long before R and S were adopted as descriptors, the two enantiomers of glyceraldehyde were designated as D and L.
D-(+)-Glyceraldehyde is equivalent to (R)-(+)-glyceraldehyde, the latter configuration being fully systematic.
Configurations in other compounds were then related to the configurations of Dand L-glyceraldehyde by direct comparison of Fischer projections.
For example, (+)-glucose (= dextrose) is represented by a Fischer projection that defines the configuration at all four chiral centres.
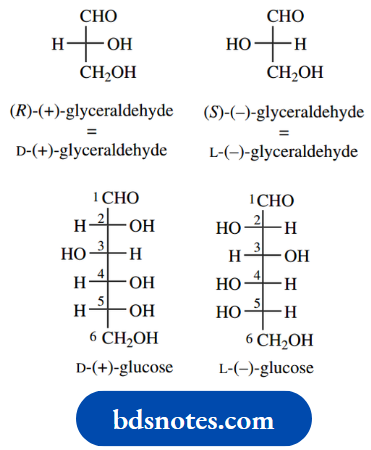
Since the configuration at position 5 in (+)-glucose can be directly related to that in D-(+)-glyceraldehyde, (+)-glucose is said to have the D configuration and is thus termed D-(+)-glucose.
By similar reasoning, the enantiomer of glucose has the L configuration and is termed L-(−)-glucose. Now the limitations of this system become obvious when one realizes that D and L refer to the configuration at just one centre.
By convention, the highest numbered chiral centre, and the remaining configurations are not specified, except by the name of the sugar.
Fischer Projections Of Glucose And Stereoisomers
The sugar glucose has four chiral centres; therefore, 24 = 16 different stereoisomers of this structure may be considered. These are shown below as Fischer projections

The 16 stereoisomers are divided into D and L groups, which reflect only the configuration at the highest numbered chiral centre, namely C-5.
The chirality at other centres is defined solely by the name given to the sugar, so we have eight different names for particular configurational combinations.
Note that although D and L strictly refer to the configuration at only one centre, L-glucose is the enantiomer of D-glucose and, therefore, must have the opposite configuration at all chiral centres.
A change in configuration at only one centre produces a diastereoisomer that has different chemical properties and is accordingly given a different name.
Whilst this system of nomenclature has some obvious shortcomings, it is analogous to the ephedrine and pseudoephedrine example where we were considering just two chiral centres.
A more systematic approach (though not one that is used) might give all the above sugars the same name, for example. hexose.
But specify the chirality at each centre, for example. D-(+)-glucose would be (+)-(2R,3S,4R,5R)-hexose and L-(−)-galactose would become (−)-(2S,3R,4R,5S)-hexose.
Instead, we have the eight different names in two configurational classes, D and L.
We can also use the term epimer to describe the relationship between isomers, where the difference is in the configuration at just one centre. This is shown for the four epimers of D-(+)-glucose.
An interesting observation with the 16 stereoisomers is that the optical activity of a particular isomer does not appear to relate to the configuration at any particular chiral centre.
Stereochemistry In Hemiacetal Forms Of Sugars From Fischer Projections
In solution, aldehyde sugars normally exist as cyclic hemiacetals through the reaction of one of the hydroxyls with the aldehyde group, giving a strain-free six- or five-membered ring.
The Fischer projection for the sugar is surprisingly useful in predicting the configuration and conformation of the cyclic form.
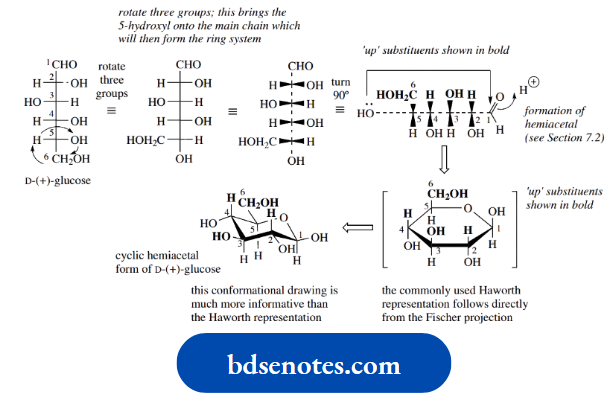
The approach is straightforward. Since cyclic hemiacetal formation requires a hydroxyl group as the nucleophile to attack the protonated carbonyl.
We put this hydroxyl group on the vertical, thus getting all the ring atoms onto the vertical. This requires the rotation of three groups attached to the appropriate atom, C-5 in the case of D-(+)-glucose.
Such rotation does not affect the configuration at C-5. Then put in the stereochemistry implied by the Fischer projection, using wedges and dots. This structure should then be turned on its side.
The ring formation is considered by joining up the C-5 hydroxyl and the carbonyl at the rear of the structure.
Note that, as drawn, this eclipsed conformer from the Fischer projection actually has these atoms quite close together, so that ring formation is easily achieved and, most importantly, easily visualized.
The net result is a cyclic system looking like the Haworth representation that is commonly used, especially in biochemistry books.
The Haworth representation nicely reflects the up–down relationships of the various substituent groups, but is uninformative about whether these are equatorial or axial.
The last step, therefore, is to transcribe this representation into a chair conformation, as shown, so that we see the conformational consequences.
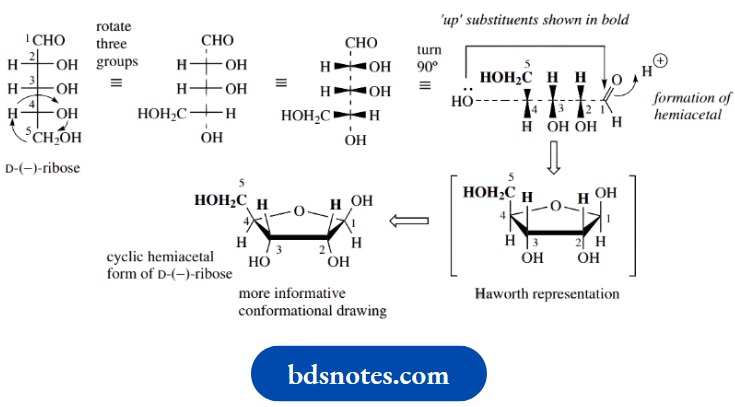
The alternative chair conformation, should we draw it instead, would be less favoured than that shown because of the increased number of axial substituents.
The conformation of D-glucose is the easily remembered one, in that all the substituents are equatorial.
A similar procedure is shown for D-(−)-ribose, which, although it is capable of forming a six-membered cyclic form, is found to exist predominantly as a five-membered ring.
D and L amino acids
There is a correlation between D- and L-glyceraldehyde and D- and L-amino acids, in that it is possible to convert one system chemically into another without affecting the integrity of the chiral centre.
The fine detail of the transformations need not concern us here. The net result is that D- and L-amino acids have the general configurations shown.
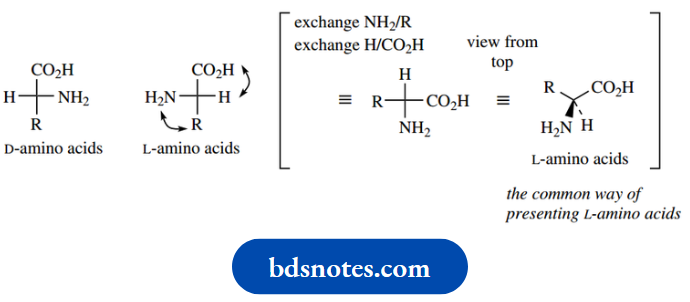
Note that all the amino acids found in proteins are of the L configuration (except the achiral glycine); D-amino acids are found in some polypeptide antibiotics.
As we pointed out this brings up an apparent anomaly in nomenclature. In all protein L-amino acids, except for cysteine, this represents an S configuration; cysteine, because of its high-priority sulfur atom has the R configuration.
One further point; as mentioned, the now obsolete descriptors d and l are abbreviations for dextrorotatory (+) and laevorotatory (−) respectively. They do not in any way relate to D and L.
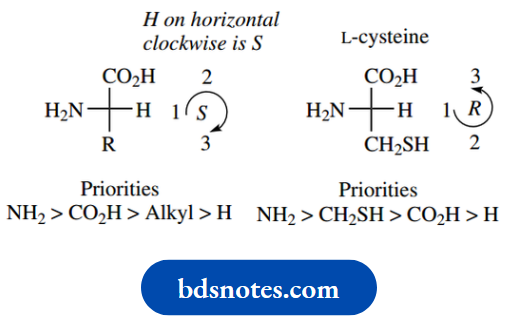
Polycyclicsy Stems
Many molecules of biological or pharmaceutical importance contain polycyclic ring systems, and we have already met some examples in other contexts, For Example. penicillins.
There are three main ways in which rings can be joined together, according to whether they share one atom, two atoms, or more than two atoms. These are termed spiro, fused, or bridged systems respectively.
Examples are shown where six-membered rings are joined in various ways, but the concepts apply equally to rings of other sizes.

Spiro Systems
Spiro systems have two rings sharing a single carbon atom, and since this has essentially a tetrahedral array of bonds, the bonds starting the two rings must be arranged perpendicular to each other.
If there is an appropriate substitution on the rings, then this can lead to the spiro centre becoming chiral.
Natural Spirocom Pounds
Spiro compounds are exemplified by several natural product structures. One of these is the antifungal agent griseofulvin produced by cultures of the mould Penicillium griseofulvin.
Griseofulvin is the drug of choice for many fungal infections, but it is ineffective when applied topically, so is administered orally.
Griseofulvin has two chiral centres, one of which is the spiro centre, so there are potentially four configurational isomers for the structure. Natural griseofulvin has the configurations shown.
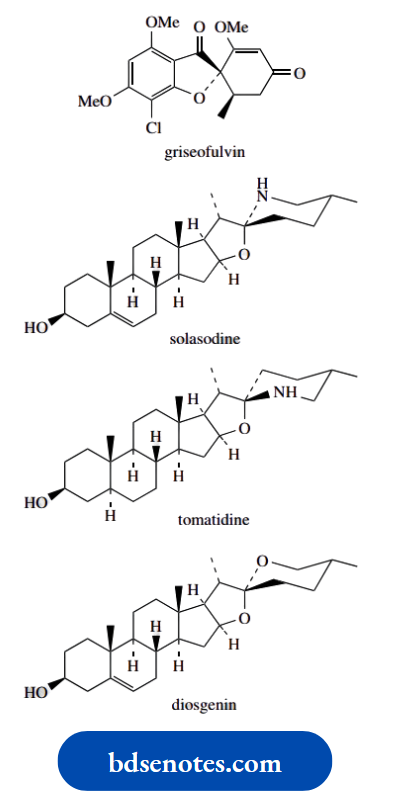
Solasodine and tomatidine are steroidal alkaloids produced by potatoes (Solanum tuberosum) and tomatoes (Lycopersicon esculente) respectively.
These compounds, as glycosides, are responsible for the toxic properties of the foliage and green fruits of these plants.
They are not present in potato tubers, unless green, or in ripe tomato fruits. Both compounds contain a spiro system, a nitrogen analogue of a ketal.
A spiroketal is present in diosgenin from Dioscorea species, a raw material used for the semi-synthesis of steroidal drugs.
Note that solasodine and tomatidine demonstrate the different configurations at the spiro centre; all natural spiroketals have the same stereochemistry at the spiro centre as in diosgenin.
Fused Ring Systems
Fused ring systems are particularly common. It is logical to suppose that fusing on one or more additional ring systems is going to have stereochemical consequences.
In particular, the conformational changes seen with single-ring systems are likely to be significantly modified. Initially, let us consider two cyclohexane rings fused together, giving a bicyclic system called decalin.
Two configurational isomers exist, trans- and cisdecalin, according to the stereochemistry of ring fusion.
The trans or cis relationship is most easily seen with the hydrogens at the ring fusion carbons, but it also follows that the bonds forming part of the second ring can be considered to share a trans or cis relationship to each other.
It is usual practice to show the stereochemistry in the former way, via the ring fusion substituents.
The situation is in many ways analogous to transand cis-1,2-methylcyclohexane and these afford useful comparisons as we consider conformational changes.
Now, trans-decalin forms a rather rigid system, and it transpires that the only conformational mobility possible is ring flip of chairs to very much less favourable boats.
Since both bonds of the second ring are equatorial with respect to the first ring, any other type of conformational change would require these to become axial.
It is impossible to join the two axial bonds into a ring system as small as six carbons; hence, there is no conformational mobility.
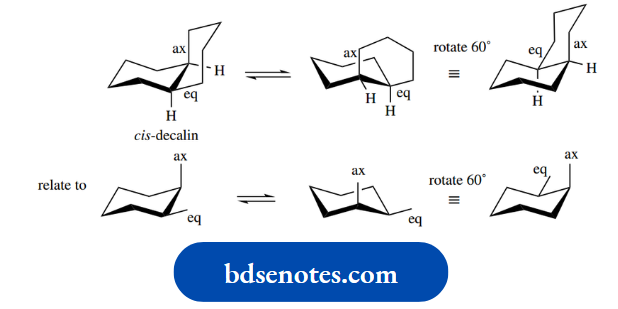
On the other hand, cis-decalin is conformationally mobile, and a simultaneous flipping in both rings produces a new conformer of equal energy.
This is not easy to visualize. In the scheme, the middle conformer has one ring viewed face-on, so we have resorted to the rotation of the structure to get an appreciation of the new conformer with its rings in chair form.
It is best to have models to appreciate this conformational flexibility.
It is quite clear, though, that an axial bond becomes equatorial and an equatorial one becomes axial, just as with substituents in the cis- 1,2-dimethylcyclohexane analogue.
However, it is probably reassuring to appreciate that this conformational flexibility in two cis-fused cyclohexane rings is lost when a third ring is fused, and in many of the fused ring systems of interest to us, it becomes of no further consequence.
Since the second ring in trans-decalin effectively introduces two equatorial substituents to the first ring, whilst cis-decalin.
It provides one equatorial and one axial substituent, it is logical to predict that transdecalin should have a lower energy than cis-decalin.
This is indeed the case, the energy difference being about 12 kJ mol-1. When we considered trans- and cis-1,2-dimethylcyclohexane.
We found that only three configurational isomers exist, enantiomeric forms of the trans isomer, together with the cis isomer, which is an optically inactive meso compound.
The meso relationship could be deduced from the plane of symmetry in the hexagon representation.
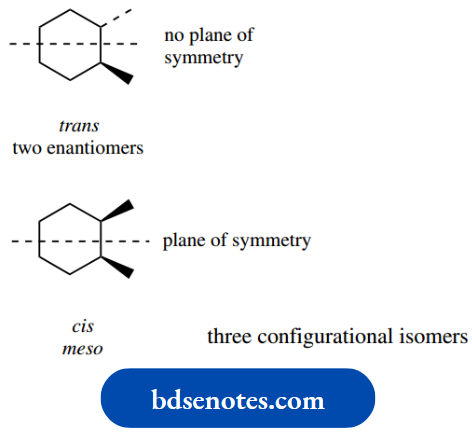
When we look at the structures of trans- and cisdecalin, it is apparent that a further plane of symmetry, through the ring fusion, is present in both structures.
This means that each isomer is superimposable on its mirror image; consequently, there are only two configurational isomers of decalin, one trans and one cis.
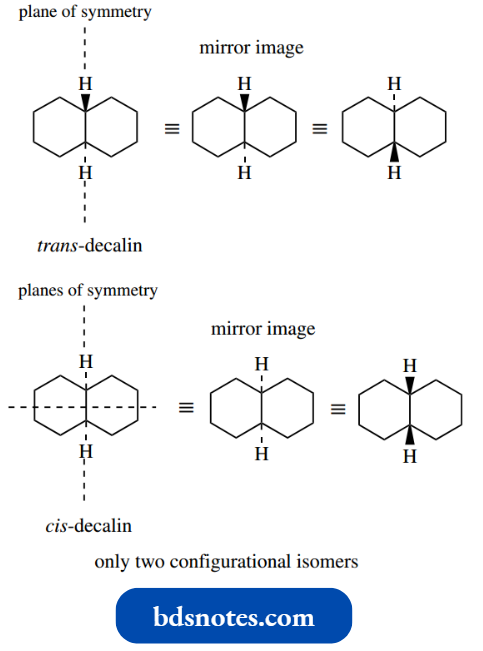
The situation in trans- and cis-decalin is complicated by the symmetry elements.
If this symmetry is destroyed, For Example. by introducing dimethyl substituents, we get back to reassuringly familiar territory in which two chiral centres lead to four configurational isomers.
The same is true in the trans- and cis-1,2- dimethylcyclohexane series. Fusing rings of different sizes can produce significant restraints, especially when rings of less than six carbons are involved.
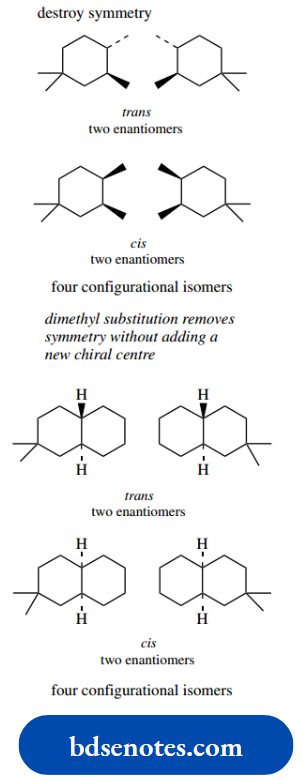
However, the characteristics of these fused systems can be deduced logically by applying our knowledge of single-ring systems.
Fusion of a five-membered ring to a six-membered ring gives a hydride system, and, as with decalins, cis and trans forms are possible.
Because the cyclopentane ring is more planar than a cyclohexane ring this causes deformation and increases strain at the ring fusion.
This deformation is more easily accommodated with the cis-fusion than the transfusion, and, in contrast to the decalins, the cis isomer has a lower energy than the trans isomer (by about 1 kJ mol-1).
As in the decalins though, the cis form is conformationally mobile, whereas the trans form is fixed.
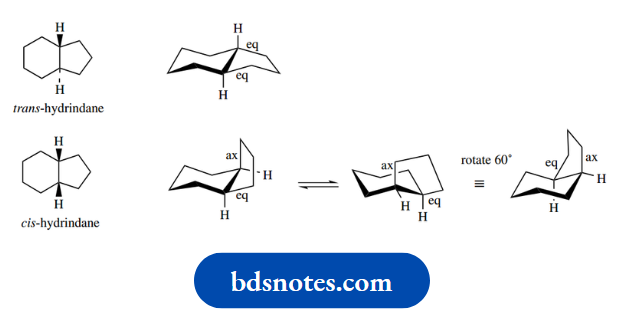
The fusion of rings of different sizes reduces symmetry in the structures; instead of the rather unusual situation with the decalins, where there are only two configurational isomers.
The hydrindanes exist in the anticipated three isomeric forms, two enantiomeric trans isomers and a meso cis isomer (compare 1,2- dimethylcyclohexane).
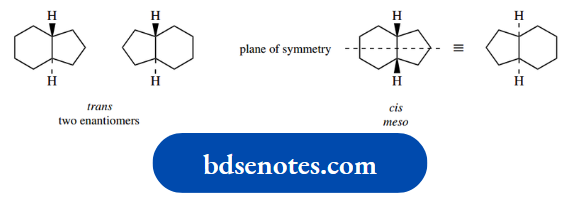
Isomerizations Influenced By Ring Fusions
Epimerization Of Cis-Decalone If cis-decalone is treated with a mild base, it is predominantly isomerized to trans-decalone. This can be rationalized by considering stereo drawings of the two isomers.
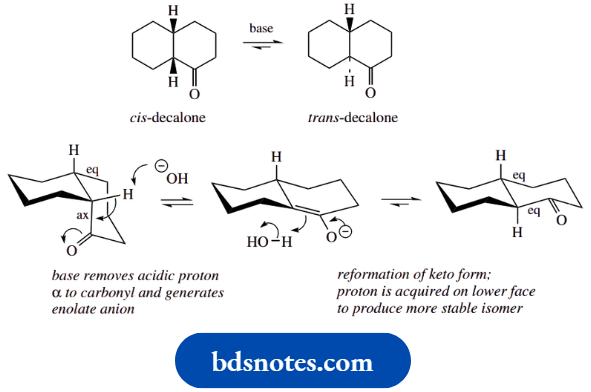
The ring fusion in cis-decline means that bonds forming the second ring have a relationship to the first ring in which one bond is equatorial and one axial.
In contrast, both such bonds in trans-decline are equatorial to the first ring. We can predict, therefore, that trans-decline has a lower energy than cis-decline.
The isomerization is brought about because the carbonyl group is adjacent (α) to the hydrogen at the ring fusion.
This hydrogen is relatively acidic and may be removed by the base, generating the enolate anion.
The enolate anion must now be planar around the site of ring fusion and, by a reversal of the process, may pick up a proton from either side of the double bond.
However, instead of getting a 1:1 mixture of the two possible isomers, this reaction very much favours the trans isomer because of its lower thermodynamic energy. The equilibrium mixture contains principally trans-decline.
Epimerization Of Etoposide The anticancer agent etoposide contains a five-membered lactone function that is significantly strained because it is trans-fused.
This material is readily converted into a relatively strain-free cis-fused system by treating with very mild alkali, For Example. traces of detergent, and produces an epimer called picroetoposide. This isomer has no significant biological activity.
The epimerization can be formulated as involving an enolate anion, as above. However, in contrast to the decalin example above, cis-hydride is of lower energy than trans-hydride.
In this particular case, on reverting back to a carbonyl compound, the planar enolate anion is presented with the alternatives of receiving.
A proton from one face to form a strained trans-fused system, or from the other face to form a strain-free cis-fused system.
The latter is very much preferred, so much so that the conversion of etoposide into its epimer is almost quantitative.
Although we can rationalize this behaviour simply by considering the hydride-type rings.
The fusion of this system to an aromatic ring causes additional distortion, and the effect becomes even more pronounced in favour of the cis-fused system.
This behaviour contrasts with the racemization of hyoscyamine to atropine, which also involves an enolate anion derived from an ester system.
As the term racemization implies, atropine is a 50:50 mixture of the two enantiomers. It shows how the proportion of each epimer formed can be influenced by other stereochemical factors.
The fusion of a three-membered ring into a six-membered ring has much more serious limitations.
A three-membered ring must be planar, so it will distort the ring it is being fused to, and this restricts stereochemical possibilities.
For example, epoxycyclohexane can, therefore, only be cis-fused, and the six-membered ring is forced to adopt the half-chair conformation we saw with cyclohexene.
There will be conformational mobility in this ring provided that there are no other ring fusions to prevent this.
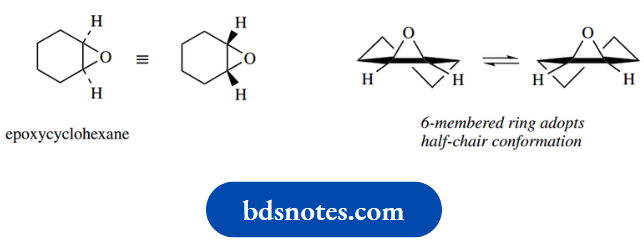
Note that, in situations where a ring fusion produces chiral centres, we can find the number of configurational isomers possible is less than that predicted from the 2n guidelines.
This may be the consequence of symmetry, in that an isomer is the same as its mirror image, as we have seen above.
However, it can also be the result of restrictions caused by the ring fusion, so that one centre effectively defines the chirality of another, thus reducing the number of combinations.
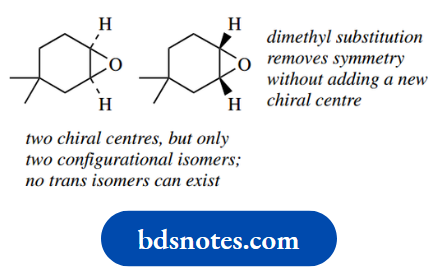
In epoxycyclohexanes, no trans-fused variants can exist. Note that a cyclohexane system will be forced into a similar half-chair conformation by fusing a planar aromatic ring onto a cyclohexane ring (a tetrahydronaphthalene system).

Shapes Of Steroids
Steroids all contain a tetracyclic ring system comprised of three six-membered rings and one five-membered ring fused together. Cholesterol is the best known of the steroids.
It is an essential structural component of animal cells, though the presence of excess cholesterol in the blood is definitely associated with the incidence of heart disease and heart attacks.
Whilst cholesterol typifies the fundamental structure, further modifications to the side chain and the ring system help to create a wide range of biologically important natural products.
For Example. sterols, steroidal saponins, cardioactive glycosides, bile acids, corticosteroids, and mammalian sex hormones.
Because of the profound biological activities encountered, many natural steroids, together with a considerable number of synthetic and semi-synthetic steroidal compounds, are routinely employed in medicine.
The markedly different biological activities observed emanating from compounds containing a common structural skeleton are, in part.
Ascribed to the functional groups attached to the steroid nucleus and, in part, to the overall shape conferred on this nucleus by the stereochemistry of ring fusions.
Let us start with cholestane, which is the basic hydrocarbon skeleton of cholesterol. This structure has all ring fusions trans.
By logical extension trans-decalin and trans-hydride can be deduced to have approximately the shape illustrated.
Because of the transfusions, there is no conformational mobility except for the unlikely flipping of ring A into a boat form, which we can ignore.
The overall shape of cholestane is a rather rigid and flattish structure. The rings are designated A–D as indicated.
Cholesterol has a double bond in ring B at the A–B ring fusion, so this distorts the rings by demanding that the arrangement around the double bond is planar.
It is not possible to depict this perfectly in a typical two-dimensional representation.
The natural progestogen hormone progesterone also has a double bond at the A–B ring fusion, but this time in ring A, so a similar distortion in ring A is required.
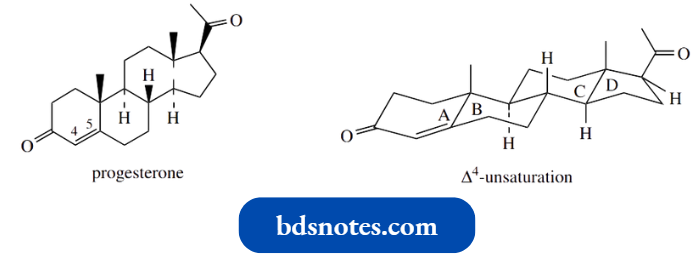
The fungal sterol ergosterol has double bonds at positions 5 and 7, both in the B ring, which consequently should become essentially planar.
The picture shown is a rough approximation. The antifungal effect of polyene antibiotics, such as amphotericin and nystatin.
depends upon their ability to bind strongly to ergosterol in fungal membranes. They do not bind significantly to cholesterol in mammalian cells, so this provides selective toxicity.
The binding to ergosterol is very much influenced by the changes in shape conferred by the extra double bond in ring B.
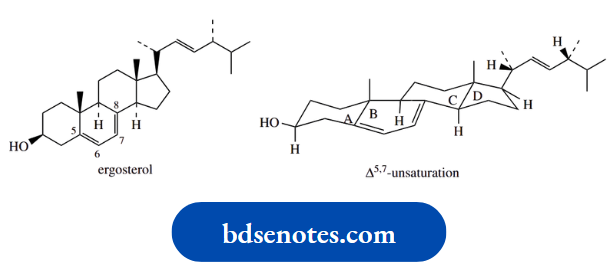
In oestrogens, such as estradiol, the A ring is aromatic. Consequently, this ring is planar and distorts ring B accordingly; again, it is difficult to draw this perfectly.
The stereochemical outcome makes oestrogens seem rather more flattened than the original all-trans arrangement in cholestane.
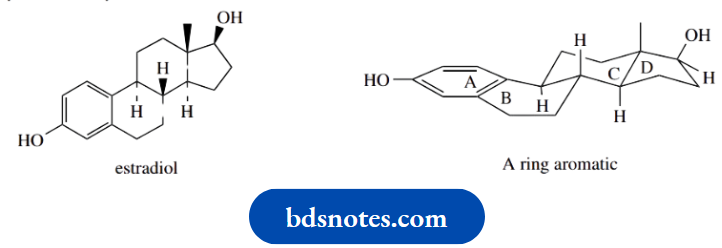
More dramatic changes are made to the shape of the steroid skeleton if ring fusions become cis rather than trans.
The most important examples involve the A–B and C–D ring fusions. It is not difficult to work out how the modified skeleton looks after these changes.
The approach is to start from the all-trans system and to delete the appropriate ring, though retaining the bonds to the unchanged part as a guide to putting in the new ring.
This provides us with three of the bonds in the new ring, and it is just necessary to fill in the rest, using earlier decalin or hydrindane templates.
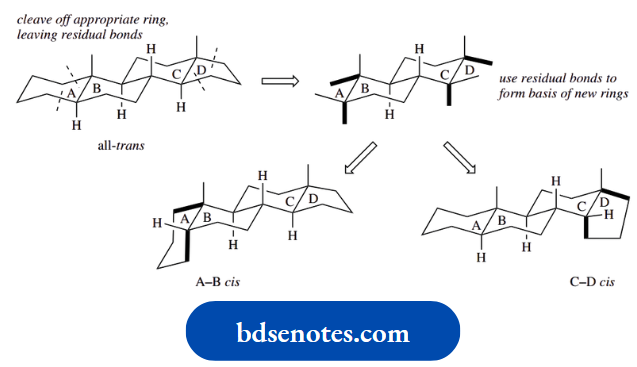
The approach is used to show the shape of cholic acid, one of the bile acids secreted into the gut to emulsify fats and encourage digestion. Cholic acid is characterized by a cis fusion of rings A and B.
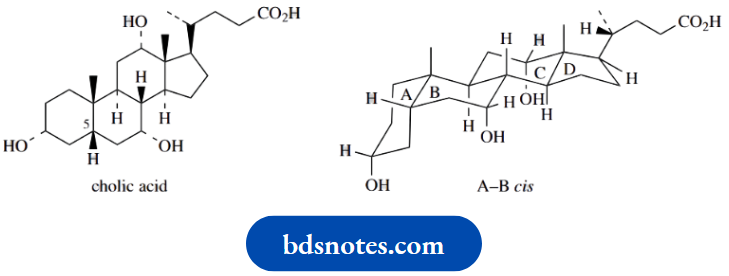
Digitoxigenin has cis fusions for both A–B and C–D rings. Glycosides of digitoxigenin are the powerful heart drugs found in the foxglove, Digitalis purpurea.
Note how a cis ring fusion changes the more-or-less flat molecule of cholestane into a molecule with a significant ‘bend’ in its shape; digitoxigenin has two such ‘bends’.
These features are important in the binding of steroids to their receptors and partially explain why we observe quite different biological activities from compounds containing a common structural skeleton.
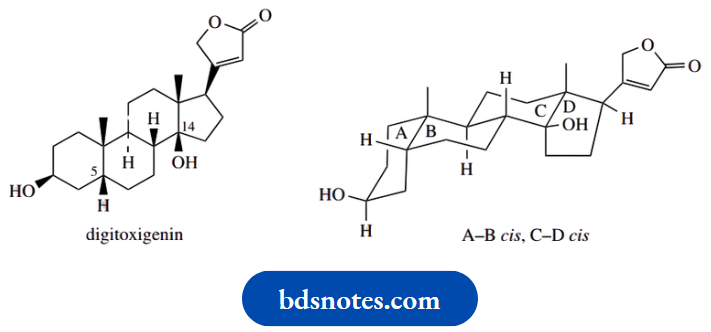
Most natural steroids have the stereochemical features seen in cholesterol, though, as we have seen, there may be some variations, particularly with respect to ring fusions affecting the A and D rings.
Note that transfusion at the hydride C–D ring junction is energetically less favourable than a cis fusion. but most natural steroid systems actually have this transfusion.
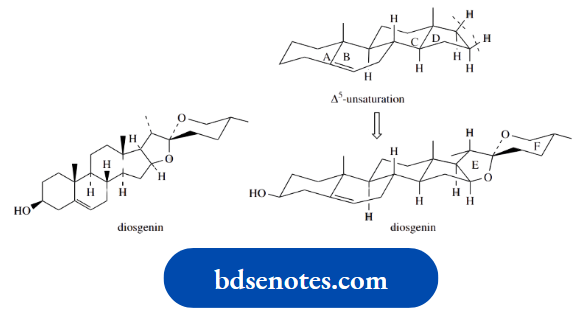
We have met diosgenin as an example of a natural spiro compound. and further examination of the structure shows the Δ5 double bond as in cholesterol, a second five-membered ring cis-fused onto the five-membered ring D, as well as the spiro fusion of a six-membered ring.
Before this structure dismays you, take it slowly and logically. It should not be too difficult to end up with the stereo drawing shown here.
The Shape Of Penicillins
Penicillins are the most widely used of the clinical antibiotics. They contain in their structures an unusual fused ring system in which a four-membered β-lactam ring is fused onto a five-membered thiazolidine.
Both rings are heterocyclic, and one of the ring fusion atoms is nitrogen. These heteroatoms do not alter our understanding of molecular shape, since we can consider that they also have an essentially tetrahedral array of bonds or lone pair electrons.
We have seen that, in cyclobutane and cyclopentane, a lower energy conformation is attained if the rings are not planar.
If one fuses a five-membered ring onto a four-membered ring, models demonstrate that it is only possible to have a cis fusion in such a structure.
And that conformational freedom in the four-membered ring disappears if we are to achieve this bonding; the four-membered ring reverts to a more planar shape.
It is still possible to have the five-membered ring non-planar, thereby reducing eclipsed interactions.
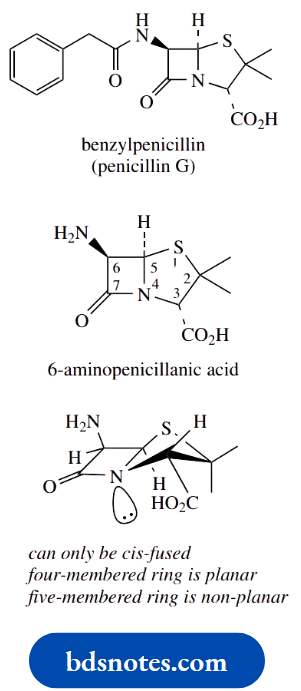
The cis fusion in which one of the fusion atoms is nitrogen merely indicates that the nitrogen lone pair electrons occupy the remaining part of the tetrahedral array.
It does, however, mean that inversion at the nitrogen atom is not possible, since that would hypothetically result in the formation of the impossible trans-fused system.
The ring fusion has thus frozen the nitrogen atom into one configuration. Fusion of a four-membered ring onto a six-membered ring is also only possible with a cis fusion.
Cephalosporins provide excellent examples of such compounds, and the comments made above for penicillins are equally valid for these compounds.
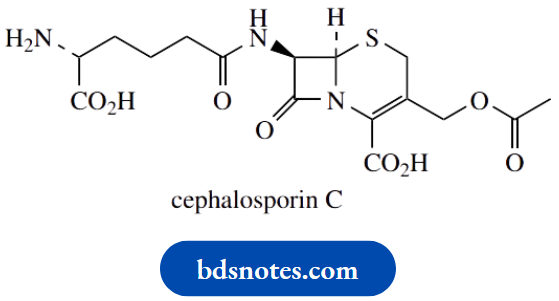
Bridged Ring Systems
In bridged ring compounds, rings share more than two atoms, and the bridge can consist of one or more atoms. We have already met an example in bornane.
which we used as an illustration of how a cyclohexane ring can be forced into a boat conformation to achieve the necessary bonding.
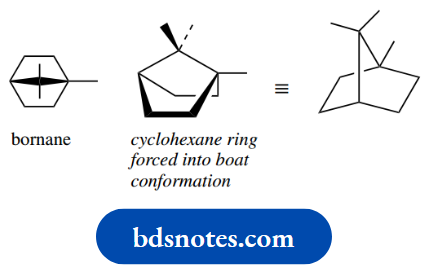
If we inspect the ring system of bornane, omitting the methyl groups, we can see that there are actually several bridges of different lengths spanning the bridgehead atoms, depending upon which atoms are considered.
This is used in nomenclature, as illustrated below, including in square brackets all the bridges, listed in decreasing lengths.
Numbering, when necessary, always starts from a bridgehead atom. A closer inspection of the shape of bicyclo[2,2,2]octane (best with a model), which has two-carbon bridges, shows that each ring system has the boat conformation.
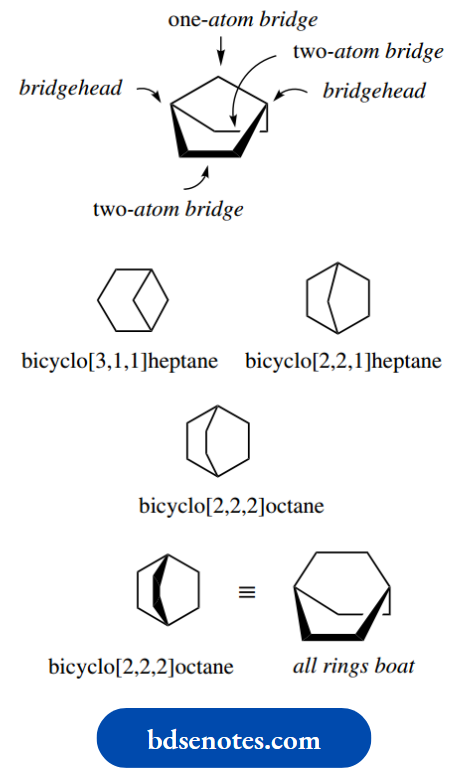
Note that the ring systems with small bridges illustrated here can have no conformational mobility, and are quite fixed. Bornane also has no configurational isomers.
If we are going to bridge a cyclohexane ring with a one-carbon bridge, there is only one way to achieve this; in other words, the configuration at the second bridgehead is fixed by that chosen at the first.
A similar situation confronted us with fused rings, in that, in order to achieve the fusion of a small ring, only a cis fusion was feasible.
Furthermore, bornane has a plane of symmetry and can be superimposed on its mirror image, so only one configurational isomer can exist.
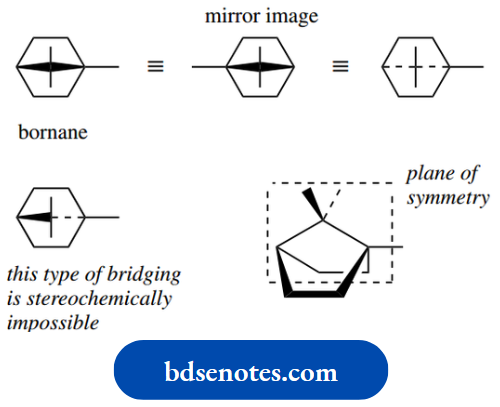
We should compare this system with a 1,4- disubstituted cyclohexane such as 4-methylcyclohexanecarboxylic acid.
There is a plane of symmetry in this molecule, so there are no chiral centres; but geometric isomers exist, allowing cis and trans stereoisomers.
The restrictions imposed by bridging have now destroyed any possibility of geometric isomerism.
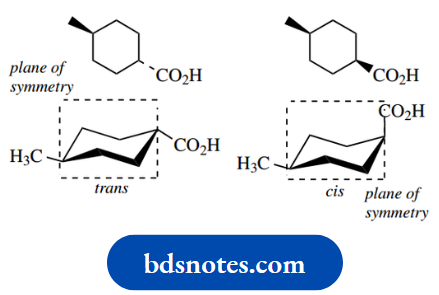
When we move on to camphor, a ketone derivative of bornane, we find this can exist in two enantiomeric forms because the plane of symmetry has been destroyed.
Nevertheless, there are only two configurational isomers despite the presence of two chiral centres; bridging does not allow the other two variants to exist.
β-Pinene is representative of a bicyclo[3,1,1]heptane system. This natural product has two chiral centres, but can exist only in the (+)- and (−)-enantiomeric forms shown.

Stereochemistry Of Tropane Alkaloids
The tropane alkaloids (−)-hyoscyamine and (−)-hyoscine are found in the toxic plants deadly nightshade (Atropa belladonna) and thornapple (Datura stramonium) and are widely used in medicine.
Hyoscyamine, usually in the form of its racemate atropine, is used to dilate the pupil of the eye, and hyoscine is employed to control motion sickness. Both alkaloids are esters of (−)-tropic acid.
The alcohol portion in hyoscyamine is tropine; in hyoscine, it is the epoxide scopine.
Tropine is an example of an azabicyclo[3,2,1]octane system with a nitrogen bridge, whereas scopine is a tricyclic system with a three-membered epoxide ring fused onto tropine.
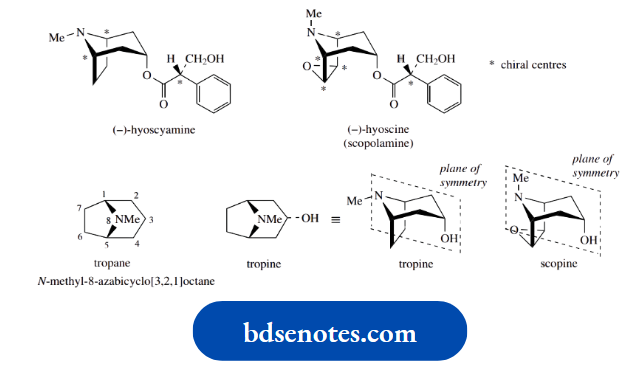
Note that systematic nomenclature considers an all-carbon ring system with one carbon replaced by nitrogen; hence, tropane is an azabicyclooctane.
There are several interesting stereochemical features accommodated within these structures.
First, both tropine and scopine are optically inactive meso compounds; despite the chiral centres, two for tropine and four for scopine,
Both compounds have a plane of symmetry so that optical activity conferred by one centre is cancelled out by its mirror image centre.
The optical activities of hyoscyamine and hyoscine are derived entirely from the chiral centre in the tropic acid portion. Atropine, the racemic form of hyoscyamine, is the ester of tropine with (±)-tropic acid.
Note also that, although we normally see rapid inversion at a nitrogen atom, the N-methyl group in hyoscyamine is preferentially in the lower energy equatorial position of the chair-like piperidine ring, as would be predicted.
However, in hyoscine, the N-methyl group has been found to be axial, not the expected equatorial. This seems to arise to minimize interaction with the extra epoxide ring in scopine.
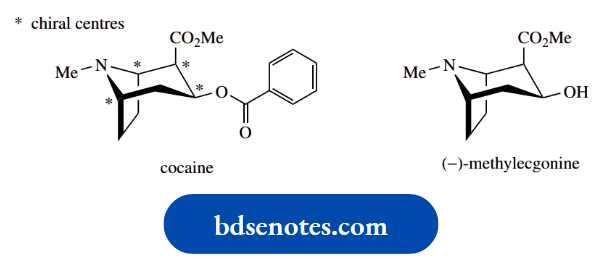
When we look at another tropane alkaloid, cocaine, we get a different scenario. Cocaine is obtained from the coca plant Erythroxylum coca and is a powerful local anaesthetic, but now known primarily as a drug of abuse.
There is no chiral centre in the acid portion, which is benzoic acid, but the optical activity of cocaine comes from the alcohol methylecgonine.
Because of the ester function in methylecgonine, the tropane system is no longer symmetrical, and the four chiral centres all contribute towards optical activity.
Now, you may have noticed that the hydroxyl group in methylecgonine is oriented differently from that in tropine.
In methylecgonine, it is easy to define the position of the hydroxyl, since this is a chiral centre and we can use the R/S nomenclature.
An alternative stereoisomer of tropine exists, and this is called pseudotropine.
How can we define the configuration for the hydroxyl when the plane of symmetry of the molecule goes through this centre and means this centre is not chiral but can exist in two different arrangements?
This is a situation allowed for in the IUPAC nomenclature rules, because if we are faced with two groups which are the same but have opposite chiralities, then the group with R chirality has a higher priority than the group with S chirality.
Applying this rule, tropine would have the S configuration and pseudotropine the R configuration at this centre. Because of the plane of symmetry.
These atoms are not strictly chiral, and this is taken into account by using lower-case letters; tropine is s and pseudotropine is r.
Leave a Reply