Heterocycles
Heterocycles:
Cyclic compounds in which one or more of the ring atoms are not carbon are termed heterocycles; the noncarbon atoms are referred to as heteroatoms.
- We shall limit our discussions to compounds in which the heteroatoms are nitrogen, oxygen, or sulfur.
- To study and understand their properties, heterocycles are conveniently grouped into two classes, i.e. non-aromatic and aromatic.
Non-Aromatic Heterocycles:
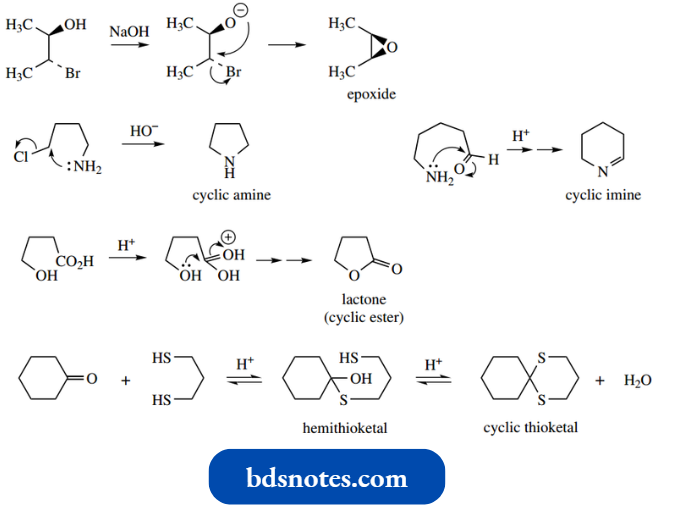
We have already met many examples of nonaromatic heterocycles in earlier chapters, for example., cyclic ethers, including epoxides, and cyclic amines, as well as lactones, lactams, and cyclic acetals and ketals.
- From the familiar examples shown below, it should be clear that the standard approach to generating heterocyclic systems requires a difunctional compound containing a leaving group or elec aerophilic center, together with a nucleophilic species that provides the heteroatom.
- The nomenclature of simple heterocyclic ring systems containing one heteroatom is indicated overleaf.
These form a useful reference, but there is little to be gained in committing them to memory.
Note, however, that the most important of these structures tend to have a trivial rather than systematic name, a consequence of long-standing common usage.
- Some of these, for example., tetrahydrofuran and tetrahydropyran, are derived from the name of the corresponding aromatic heterocycle by the concept of reduction.
A few examples of commonly encountered heterocycles with two heteroatoms are also shown.
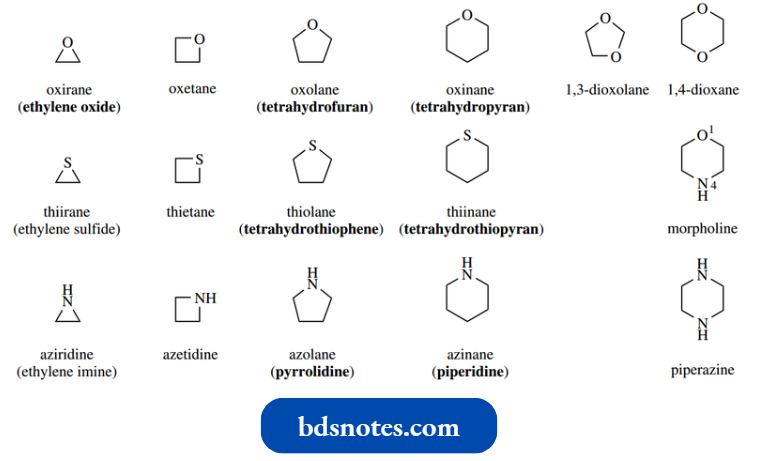
Numbering always begins at the heteroatom; in the case of morpholine, numbering starts at oxygen, the heteroatom of a higher atomic number.
- Remember that an accepted alternative in nomenclature is to indicate a heteroatom by the prefix aza-, oxo, or thia in the appropriate carbocycle.
- Thus, we could name piperidine as azacyclohexane, and ethylene oxide tetrahydrofuran as oxacyclopentane.
The chemistry of these non-aromatic heterocycles differs little from the chemistry of their acyclic counterparts and we emphasize only the relative reactivity of the three-membered ring systems towards ring opening, thus achieving relief of ring strain.
We have already noted the ring opening of epoxides, and similar reactivity is found with aziridines and iranes.
- Four-membered systems are also considerably strained and reactive towards nucleophiles, though not as readily as the three-membered compounds.
- Some of these heterocycles provide us with valuable laboratory solvents, for example., the ethers tetrahy drofuran and dioxane (1,4-dioxane). Others are useful as organic bases, for example., piperidine, pyrrolidine, and morpholine.
The basicities of the nitrogen derivatives are comparable to those of similar acyclic amines, but physical properties, for example., higher boiling point, make them more versatile than the simple amines.
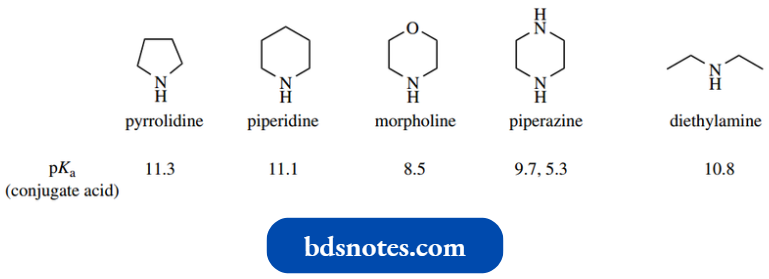
From the pKa values shown, there is relatively little difference in basicities for diethylamine, pyrrolidine, or piperidine. Note, however, that morpholine and piperazine are weaker bases than piperidine.
- This is the result of an electron-withdrawing inductive effect from the second heteroatom, making the nitrogen atom both less basic and also less nucleophilic. This makes morpholine a useful base with basicity between that of piperidine and pyridine (pKa 5.2).
- The second pKa value for the diamine piperazine is substantially lower than the first since the inductive effect from the protonated amine will withdraw electrons away from the unprotonated amine.
Aromaticity And Heteroaromaticity:
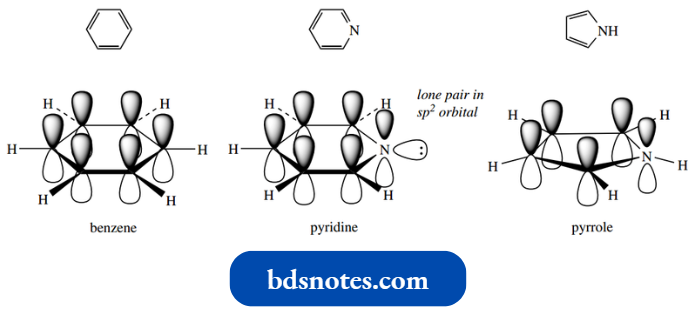
Pyridine is structurally related to benzene: one CH unit has been replaced by N. If we consider the constitutions of the two compounds in more detail, we shall see even closer similarity.
- Thus, we have seen that the ring atoms in benzene are sp2 hybridized. The remaining singly occupied p orbitals are oriented at right angles to the plane of the ring, and overlap to form a delocalized π system, extending to form a closed loop above and below the ring.
- Compared with what we might expect for the hypothetical cyclohexatriene, this results in a considerable stabilization, with significantly modified structure and reactivity in benzene. We termed this aromaticity.
- Benzene conforms to Huckel’s rule, which predicts that planar cyclic polyenes containing 4n + 2 π electrons show enhanced stability associated with aromaticity.
- Pyridine is also aromatic: nitrogen contributes one electron in a p orbital to the π electron system, and its lone pair is located in an sp2 orbital that is in the plane of the ring and perpendicular to the π electron system.
It also conforms to Huckel’s rule, in that we still have an aromatic ¨ sextet of π electrons.
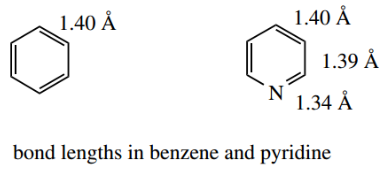
One of the structural features of benzene that derives from aromaticity is the equal length of the C–C bonds (1.40 A), which lies between the conformal single (1.54 A) and double (1.34˚A) bonds.
- Nevertheless, we continue to draw benzene with single and double bonds because this allows us to represent reaction mechanisms in terms of electron movements.
Pyridine does not have a perfect hexagon shape; the symmetry is distorted because the C–N bonds are slightly shorter (1.34 A) than the C–C bonds (1.39–1.40 A).
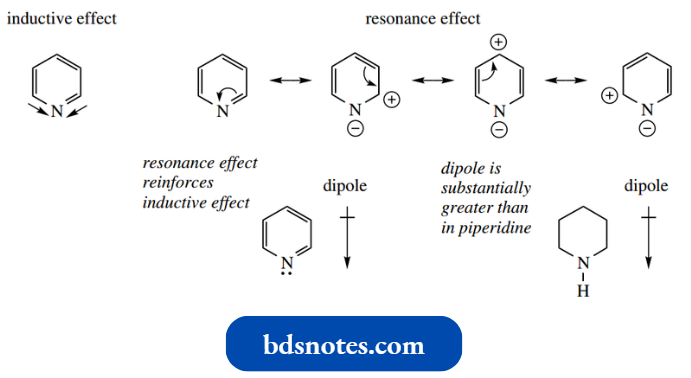
Nitrogen is more electronegative than carbon, and this influences the electron distribution in the π electron system in pyridine through inductive effects, such that nitrogen is electron-rich.
- In addition, the nitrogen will also become electron-rich through a resonance effect: several resonance forms may be drawn that have a negative charge on nitrogen. These effects thus reinforce each other.
- The heteroatom thus distorts the π electron cloud of the aromatic ring system, drawing electrons towards the nitrogen and away from the carbons.
The consequences of this are that we can predict that the pyridine nitrogen will react readily with electrophiles, whereas the remainder of the ring system will be resistant to electrophilic attack.
An experimental probe for aromaticity is the chemical shift of the hydrogen signals in NMR spectroscopy.
- The substantially greater δ values for benzene protons (δ 7.27 ppm) compared with those in alkenes (δ 5–6 ppm) have been ascribed to the presence of a ring current that creates its magnetic field opposing the applied magnetic field.
- This ring current is the result of circulating electrons in the π system of the aromatic ring.
- The hydrogen NMR signals for pyridine also appear at relatively large δ values, in the range of 7.1–8.5 ppm, typical of aromatic systems.
The signals do not all appear at the same chemical shift; the heteroatom distorts the π electron distribution and affects the 2/6, 3/4, and 5 positions to different extents.
- Now let us consider pyrrole, where we have a five-membered ring containing nitrogen. Pyrrole is also aromatic.
- This is somewhat unexpected: how can we get six π electrons from just five atoms? The answer is that each carbon contributes one electron as before, but nitrogen now contributes two electrons, its lone pair, to the π electron system.
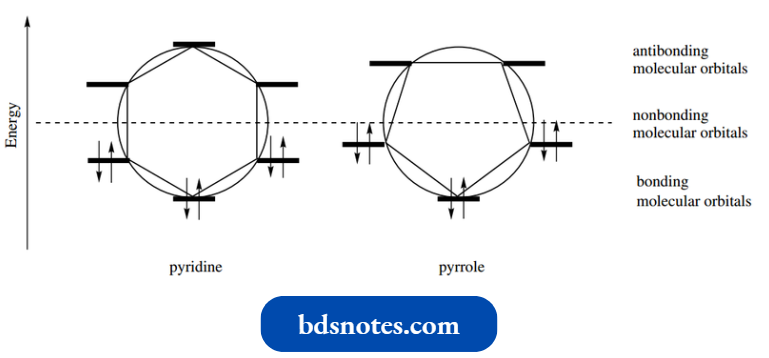
- We can draw Frost circles to show the relative energies of the molecular orbitals for pyridine and pyrrole.
The picture for pyridine is essentially the same as for benzene, with six π electrons forming an energetically favorable closed shell.
- For pyrrole, we also get a closed shell, and there is considerable aromatic stabilization over electrons in the six atomic orbitals.
- However, the contribution of the nitrogen lone pair to the aromatic sextet in pyrrole makes the nitrogen atom relatively electron deficient. The nitrogen atom should create an inductive effect, as in pyridine, drawing electrons towards the heteroatom.
- However, a consideration of the resonance structures leads to several resonance forms with a positive charge on nitrogen. The resonance effect is opposite to the inductive effect and of greater magnitude.
Overall, the heteroatom distorts the π electron cloud of the aromatic ring system by pushing electrons away from the nitrogen and towards the carbons.
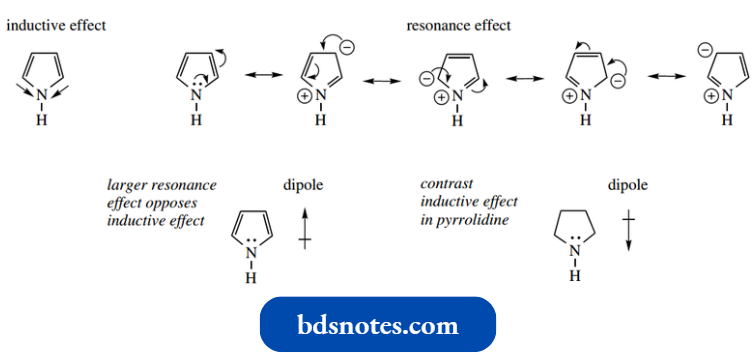
The difference in electron distribution in pyridine and pyrrole manifests itself via the measured dipole moments. More importantly, we shall see that this electron distribution influences the chemical reactivity of the two systems.
- In broad terms, ring systems where the carbons are electron deficient because of the electron-withdrawing effect of the heteroatom, for example., pyridine, are more reactive towards nucleophiles than benzene.
- On the other hand, ring systems where the carbons are electron-rich because of the electron-donating heteroatom, for example., pyrrole, are more reactive towards electrophiles than benzene.
- Note the deliberate choice of terminology here: ring systems where the carbons are electron deficient or electron rich.
- You may meet the older terminology of π-deficient heterocycles and π-excessive heterocycles, but these can give a false impression.
- Each heterocycle contains six π electrons, so it is not the heterocycle that is electron deficient or electron-rich, but the carbons that receive less or more than their equal share because of the effect of the heteroatom.
- Though we shall return to this again, one critical difference between pyridine and pyrrole to note here relates to basicity. Pyridine is a base because its nitrogen still carries a lone pair able to accept a proton.
Pyrrole is not basic: it has already used up its lone pair in contributing to the aromatic sextet.
Six-Membered Aromatic Heterocycles Pyridine:
From our discussions in the last section, we might expect pyridine to display properties associated with the nitrogen function and the aromatic ring.
Not surprisingly, it turns out that the aromatic ring affects the properties of the amine; but, more significantly, the aromatic properties are greatly influenced by the presence of the heteroatom.
Based on our earlier knowledge, and from a simple inspection of its structure, we might expect to observe three types of general reactivity in pyridine. We might expect to see:
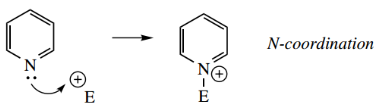
Reaction at the heteroatom – the non-bonding electrons on the nitrogen might coordinate to H+ or another suitable electrophile.
Reaction of the aromatic π system – typical elec trophilic substitution as seen for benzene might be expected.
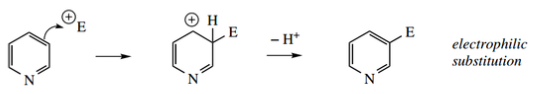
Reaction of the C=N ‘imine’ function – though this is not an isolated imine function but is part of the aromatic ring, its polarization might make it susceptible to nucleophilic attack.
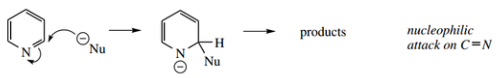
Reassuringly, our predictions turn out to be well-founded.
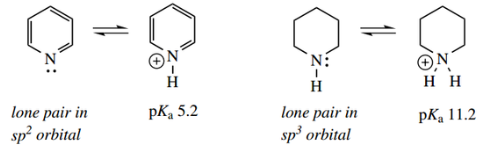
Pyridine is a base (pKa pyridinium cation 5.2), but it is a considerably weaker base than a typical non-aromatic heterocyclic amine such as piperidine (pKa piperidinium cation 11.2).
- This is because the lone pair electrons in pyridine are held in an sp2 orbital.
- The increased s character of this orbital, compared with the sp3 orbital in piperidine, means that the lone pair electrons are held closer to the nitrogen, and are consequently less available for protonation.
- The lower basicity of pyridine compared with piperidine is thus a hybridization effect.
Although pyridine is a weak base, it can form salts with acids and is widely used in chemical reactions as an acid scavenger and as a very good polar solvent.
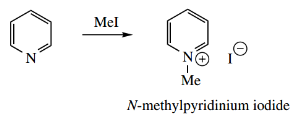
Just as pyridine is a weaker base than piperidine, it is also a poorer nucleophile. Nevertheless, it reacts with electrophiles to form stable pyridinium salts.
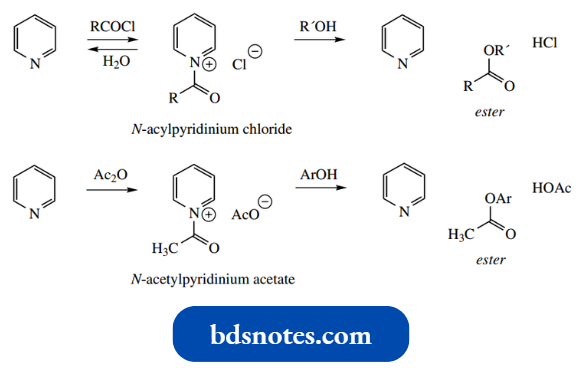
- In the examples shown, primary alkyl halides form N-alkylpyridinium salts, whereas acyl halides and anhydrides react to give N-acylpyridinium salts.
- We have already seen the latter compounds involved in esterification reactions, and see the value of pyridine in removing acidic by-products, for example., HCl.
Of course, N-acyl pyridinium salts will easily be hydrolyzed under aqueous conditions.
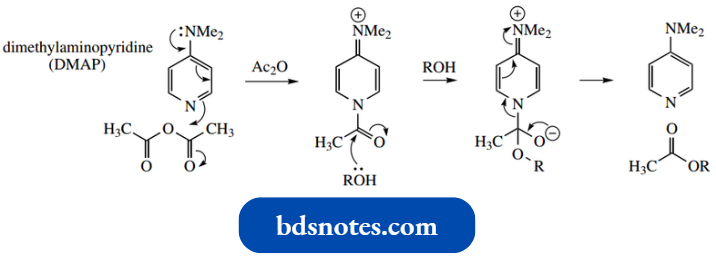
Even better than pyridine in such reactions is the derivative 4-N, N-dimethylamino pyridine (DMAP), where a resonance effect from the dimethylamino substituent reinforces the nucleophilicity of the pyridine nitrogen.
It then also promotes the acylation step by improving the nature of the leaving group. The example shows its function in a typical esterification process, i.e. acylation of an alcohol.
There are other gains, as well. Pyridine as a solvent is difficult to remove from the products, and it smells quite awful.
- In this reaction, a catalytic amount of DMAP is all that is necessary, and a more acceptable solvent can be employed.
- The pre-eminent reactivity associated with aromatic compounds is the ease of electrophilic substitution.
- As we have already predicted, the pyridine ring is rather unreactive towards electrophilic reagents, and these tend to be attacked by nitrogen instead, making the ring even less reactive.
It is readily seen from the intermediate addition cations and their resonance structures that attack at C-2 or C-4 will be unfavorable, in that one of the resonance forms features an unstable electron-deficient nitrogen cation.
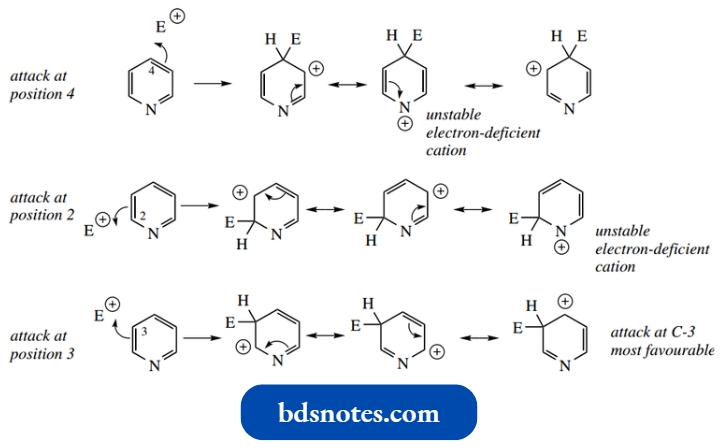
Attack at C-3 is the more likely, simply based on an inspection of resonance structures for the addition cation.
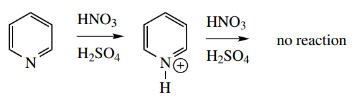
- However, elec trophilic attack still tends to be unfavourable, because many electrophilic reagents, for example., HNO3 –H2SO4, are strongly acidic, and the first effect is protonation on nitrogen.
- The attack of E+ on a positively charged pyridinium cation is even less favorable. Under acidic conditions, we require an attack on free pyridine, the concentration of which will be very small.
- Thus, under equivalent conditions, pyridine undergoes electrophilic substitution very much more slowly than benzene, by a factor of about 106.
Even Friedel–Crafts acylations are inhibited, because the nitrogen complexes with the Lewis acid, again leading to a cationic nitrogen.
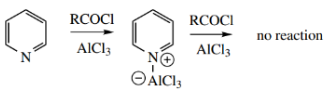
A striking demonstration of the reduced activity towards electrophiles for the pyridine ring compared with the benzene ring will be seen later when we consider the fused heterocycles quinoline and isoquinoline.
- These contain a benzene ring fused to a pyridine ring; electrophilic substitution occurs exclusively in the benzene ring.
- To facilitate electrophilic substitution, it is possible to first convert pyridine into pyridine N-oxide by the action of a peracid such as peracetic acid or m-chloroperbenzoic acid (MCPBA;).
- N-oxide formation is not peculiar to pyridine, but it is a general property of tertiary amines. There is no overall charge in the molecule, but it is not possible to draw the structure without charge separation.
Although the introduced oxygen atom causes electron withdrawal through an inductive effect, there is a greater and opposing resonance effect that donates electrons into the ring system.
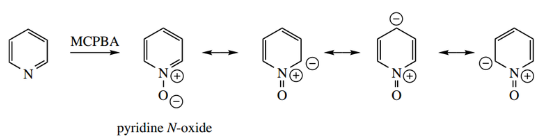
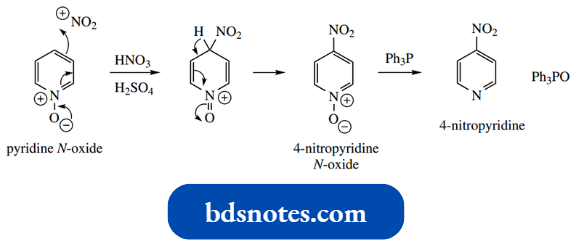
This improves reactivity towards electrophiles. Consideration of resonance structures shows positions 2, 4, and 6 are now electron-rich.
Nitration of pyridine N-oxide occurs at C-4; very little 2-nitration is observed. The pyridine compound can then be regenerated by deoxygenation with triphenylphosphine.
Pyridine, on the other hand, is more reactive than benzene towards nucleophilic aromatic substitution
- This is effectively a reaction towards the C=N ‘imine’ function, as described above.
- The attack is attacked at principally at positions 2 and 4, as predictable from resonance structures of reaction intermediates.
Attack at the 3 position does not allow the nitrogen to help stabilize the negative charge.
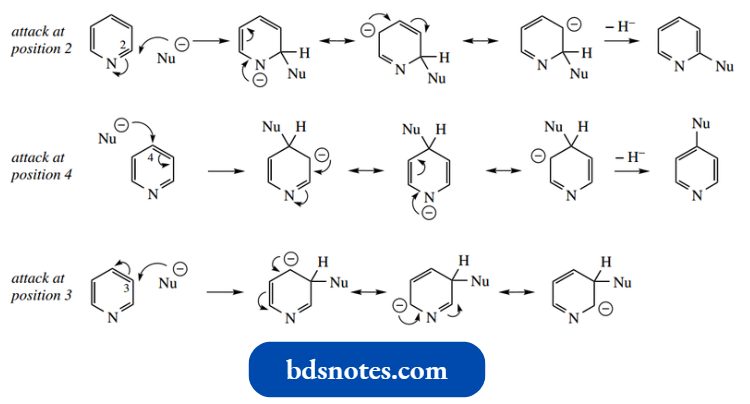
However, for an unsubstituted pyridine, the leaving group to finish off this reaction is hydride, which is a strong base and thus a poor leaving group.
- It may be necessary to use an oxidizing agent to function as a hydride acceptor to the Chichibabin reaction to facilitate this type of hydride transfer.
Nevertheless, there is a classic example of this process, known as the Chichibabin reaction, in which pyridine is converted into 2-aminopyridine through heating with sodium amide.
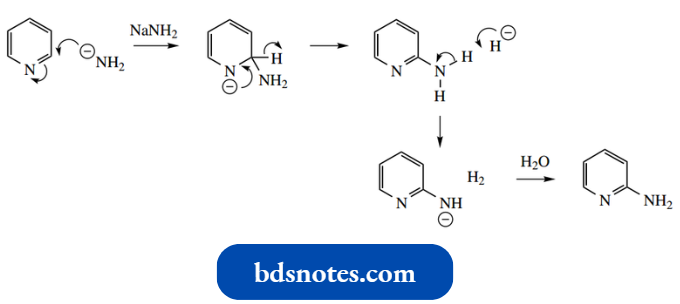
The hydride released appears to abstract a proton from the product since the other product of the reaction is gaseous hydrogen. The aminopyridine anion is finally quenched with water.
- The product is mainly 2-aminopyridine, probably the result of the enhanced inductive effect on carbons immediately adjacent to the electronegative nitrogen.
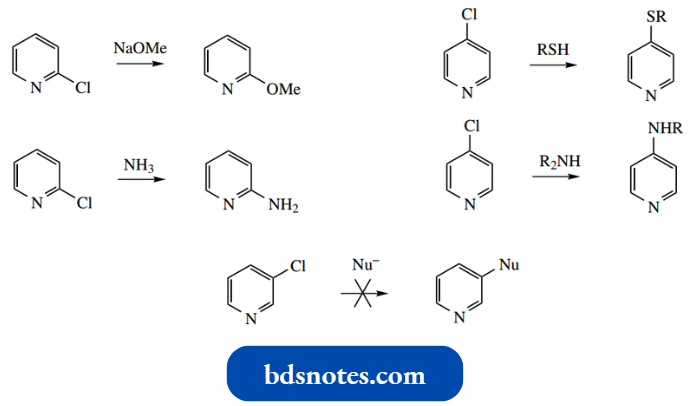
- It is much more effective to have a better-leaving group in the pyridine system. Thus 2- or 4-chloropyridines react with several nucleophiles to generate substituted products.
- Note that one can predict from the resonance structures that 3-chloropyridine, despite having a satisfactory leaving group, would not be susceptible to nucleophilic substitution at position 3.
It is not possible in the addition of anion to share the charge with nitrogen.
Methylpyridines are called picolines. 2-picoline and 4-picoline may be deprotonated by treatment with a strong base, giving useful anions.
- The methyl acidity results because of resonance stabilization in the conjugate base, providing an enolate anion analog.
However, pKa values for 2-picoline (32) and 4-picoline (34) show that they are somewhat less acidic than ketones.
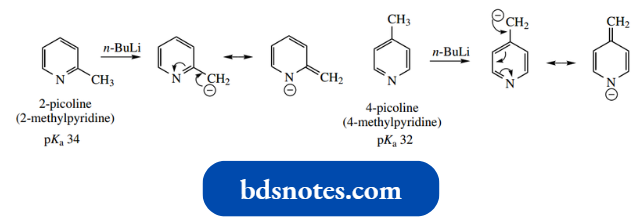
These anions can now be used as nucleophiles in several familiar reactions, for example., SN2 reactions with alkyl halides, or aldol reactions with carbonyl compounds.
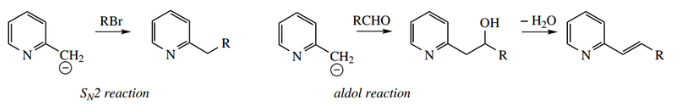
It is worthwhile here to relate the behavior of 2-chloropyridine and 2-methylpyridine to carbonyl chemistry.
- If we consider the pyridine ring as an imine, and therefore a carbonyl analog, then with 2-chloropyridine we are seeing reactions that parallel nucleophilic substitution of an acyl halide through an addition–elimination mechanism.
- With 2-methylpyridine we are seeing typical aldol reactions with activated methyl derivatives.
Nicotine, Nicotinic Acid, And Nicotinamide:
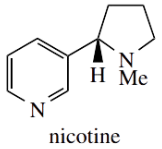
Nicotine is an oily, volatile liquid and is the principal alkaloid found in tobacco (Nicotiana tabacum). It can be seen to be a combination of two types of heterocycle, i.e. the aromatic pyridine and the non-aromatic N methylpyrrolidine.
In small doses, nicotine can act as a respiratory stimulant, though in larger doses it causes respiratory depression. Nicotine is the only pharmacologically active component in tobacco, and it is highly addictive.
- On the other hand, tobacco smoke contains several highly carcinogenic chemicals formed by incomplete combustion.
- Tobacco smoking also contributes to atherosclerosis, chronic bronchitis, and emphysema, and is regarded as the single most preventable cause of death in modern society.
- Nicotine, in the form of chewing gum, nasal sprays, or trans-dermal patches, is available for use by smokers who wish to stop the habit.
- Nicotine affects the nervous system, interacting with the nicotinic acetylcholine receptors, and the tight binding is partially accounted for by the structural similarity between acetylcholine and nicotine.
- Curare-like antagonists also block nicotinic acetylcholine receptors. There are other acetylcholine receptors, termed muscarinic, that are triggered by the alkaloid muscarine.
The tropane alkaloid hyoscyamine (see Box 10.9) binds to muscarinic acetylcholine receptors.
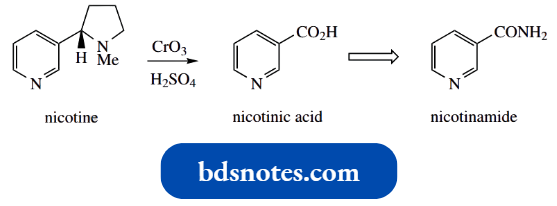
Oxidation of nicotine with chromic acid led to the isolation of pyridine-3-carboxylic acid, which was given the trivial name nicotinic acid.
- We now find that nicotinic acid derivatives, especially nicotinamide, are biochemically important.
- Nicotinic acid (niacin) is termed vitamin B3, though nicotinamide is also included under the umbrella term vitamin B3 and is the preferred material for dietary supplements.
- It is common practice to enrich many foodstuffs, including bread, flour, corn, and rice products.
Deficiency in nicotinamide leads to pellagra, which manifests itself in diarrhea, dermatitis, and dementia.
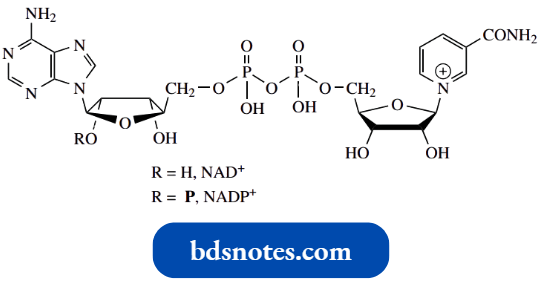
Nicotinic acid and nicotinamide are precursors of the coenzymes NAD+ and NADP+, which play a vital role in oxidation-reduction reactions and are the most important electron carriers in intermediary metabolism.
We shall look further at the chemistry of NAD+ and NADP+ shortly, but note that, in these compounds, nicotinamide is bound to the rest of the molecule as an N-pyridinium salt.
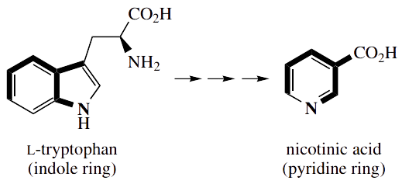
An intriguing feature of nicotinic acid formation in animals is that it is a metabolite produced from the amino acid tryptophan.
This means the pyridine ring is formed by biochemical modification of the indole fused-ring system, and, as you might imagine, it involves a substantial sequence of transformations.
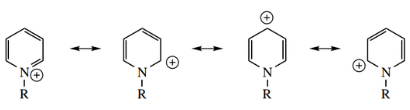
Nucleophilic Addition To Pyridinium Salts:
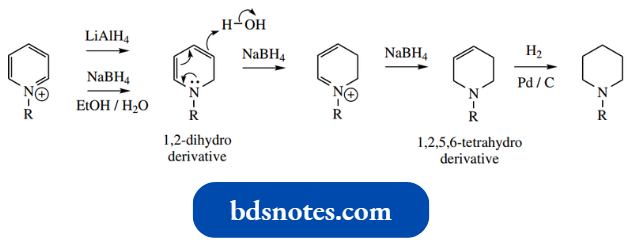
The reaction of nucleophiles with pyridinium salts leads to addition, giving dihydropyridines.
Attack is normally easier at positions 2 or 6, where the inductive effect from the positively charged nitrogen is greatest; however, if these sites are blocked, attack occurs at position 4. This is easily predicted from a consideration of resonance structures.
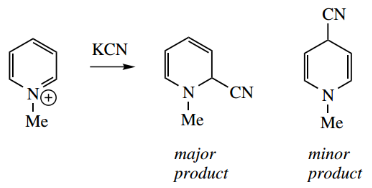
Thus, treatment of N-methyl pyridinium salts with cyanide produces a mixture of 2- and 4-cyanide hydropyridines, with the 2-isomer predominating.
It is quite difficult to reduce benzene or pyridine because these are aromatic structures. However, partial reduction of the pyridine ring is possible by using complex metal hydrides on pyridinium salts.
- Hydride transfer from lithium aluminum hydride gives the 1,2-dihydro derivative, as predictable from the above comments.
- Sodium borohydride under aqueous conditions achieves a double reduction, giving the 1,2,5,6-tetrahydro derivative, because protonation through the unsaturated system is possible.
- The final reduction step requires catalytic hydrogenation. The reduction of pyridinium salts is of considerable biological importance.
Note the way we can refer to the unsaturated heterocycle by considering it as a reduced pyridine, for example., a dihydropyridine (two double bonds) or tetrahydropyridine (one double bond).
Nicotinamide Adenine Dinucleotide: Reduction Of A Pyridinium Salt:
Nicotinamide adenine dinucleotide (NAD+) is a complex molecule in which a pyridinium salt provides the reactive functional group, hence the superscript + in its abbreviation.
- NAD+ acts as a biological oxidizing agent, and in so doing is reduced to NADH (reduced nicotinamide adenine dinucleotide).
- An enzyme, a dehydrogenase, catalyzes the process and NAD+ is the cofactor for the enzyme. The reaction can be regarded as directly analogous to the hydride reduction of a pyridinium system to a dihydropyridine, as described above.
- We have already seen that NADH can act as a reducing agent, delivering the equivalent of hydride to a carbonyl compound.
In the oxidizing mode, the enzyme can extract hydride from the substrate and use it to reduce the pyridinium salt NAD+, producing the dihydropyridine NADH.
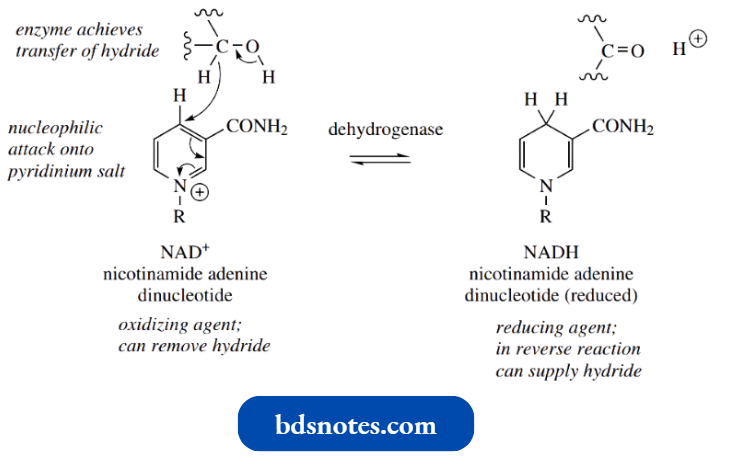
The substrate in most reactions of this type is alcohol, which becomes oxidized to an aldehyde or ketone, for example., ethanol is oxidized to acetaldehyde.
- Some reactions employ the alternative phosphorylated cofactor NADP+; the phosphate does not function in the oxidation step but is merely a recognition feature helping to bind the compound to the enzyme. The full structures of NAD+ and NADP+.
- Note that the attack of hydride is at position 4 of the dihydropyridine ring. This is controlled by the enzyme, but it is also probably the only site accessible since the rest of the complex molecule hinders the approach to positions 2 and 6.
Tautomerism: Pyridones
The pyridine ring system may carry substituents, just as we have seen with benzene rings.
- We have encountered several such derivatives in the previous section. Hydroxy or amino heterocycles, however, may sometimes exist in tautomeric forms.
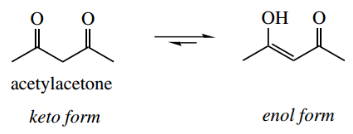
- We have met the concept of tautomerism primarily with carbonyl compounds, and have seen the isomerization of keto and enol tautomers.
- In certain cases, for example., 1,3-dicarbonyl compounds, the enol form is a major component of the equilibrium mixture. amides.
- This was used to explain why amides are fragile bases. Note that such resonance forms of pyridones are favorable, having a positive charge on the nitrogen and a negative charge on the more electronegative oxygen.
- In addition, the structure gains further stabilization from the carbonyl group. The pyridone forms are very much favored over the phenol forms, and typical C=O peaks are seen in the infrared (IR) spectra.
In the example shown, liquid acetylacetone contains about 76% of the enol tautomer.
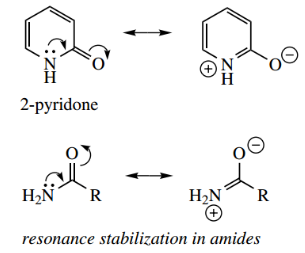
2-Hydroxy- and 4-hydroxy-pyridines are in equilibrium with their tautomeric ‘amide’ structures containing a carbonyl. These tautomers are called 2- pyridone and 4-pyridone respectively.
This type of tautomerism does not occur with the corresponding benzene derivative phenol, since it would destroy the stabilization conferred by aromaticity.
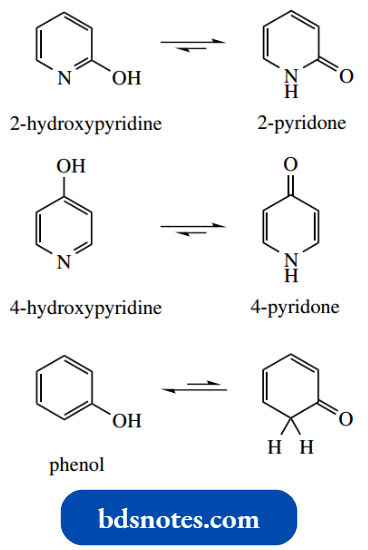
So why can tautomerism occur with a hydroxypyridiet? It is because 2-pyridone and 4-pyridone still retain aromaticity, with the nitrogen atom donating its lone pair electrons to the aromatic sextet.
- This is more easily seen in the resonance structures and should remind us of the resonance stabilization in amides. This was used to explain why amides are fragile bases.
- Note that such resonance forms of pyridones are favorable, having a positive charge on the nitrogen and a negative charge on the more electronegative oxygen. In addition, the structure gains further stabilization from the carbonyl group.
The pyridone forms are very much favored over the phenol forms, and typical C=O peaks are seen in the infrared (IR) spectra.
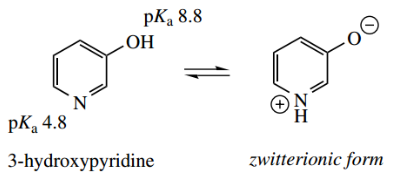
Note, however, that we cannot get the same type of tautomerism with 3-hydroxypyridine. In polar solvents, 3-hydroxypyridine may adopt a dipolar zwitterionic form.
- This may look analogous to the previous structure, but appreciate that there is a difference. With 3-hydroxyproline, the zwitterion is a major contributor and arises simply from acid-base properties.
- The hydroxyl group acts as an acid, losing a proton, and the nitrogen acts as a base, gaining a proton.
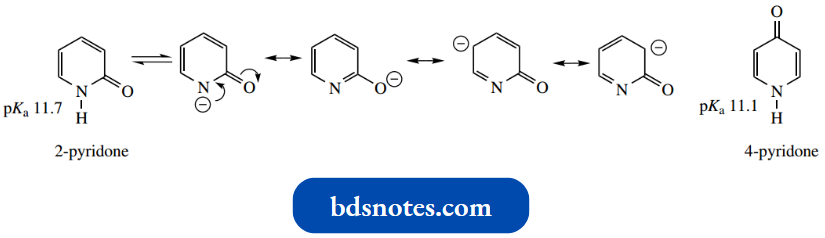
The structure from 2-pyridone is a minor resonance form that helps to explain charge distribution; the compound is almost entirely 2-pyridone.
Like amides, 2- and 4-pyridones are also very weak bases, much weaker than amines.
- Like amides, they protonate on oxygen rather than nitrogen. This further emphasizes that the nitrogen lone pair is already in use and not available for protonation.
- On the other hand, the N–H can readily be deprotonated; pyridones are appreciably acidic (pKaabout 11).
- The conjugate base benefits from considerable resonance stabilization, both via the carbonyl group and also via the ring.
The main structures in which charge is associated with the electronegative N or O atoms.
It is thus possible to N-alkylate a pyridone by exploiting its acidity. As with enolate anions, there is the possibility for O-alkylation and N-alkylation.
Although it depends upon the conditions and the nature of the electrophile, carbon electrophiles tend to react with nitrogen rather than oxygen.
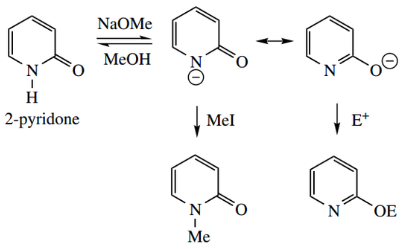
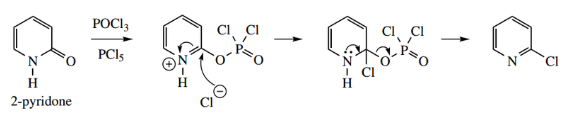
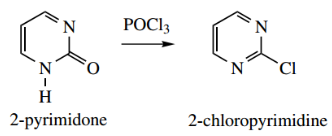
A useful reaction of pyridones is conversion into chloropyridines by the use of phosphorus oxychloride POCl3 in the presence of PCl5. This appears to react initially on oxygen, forming a good leaving group, which is subsequently displaced by chloride.
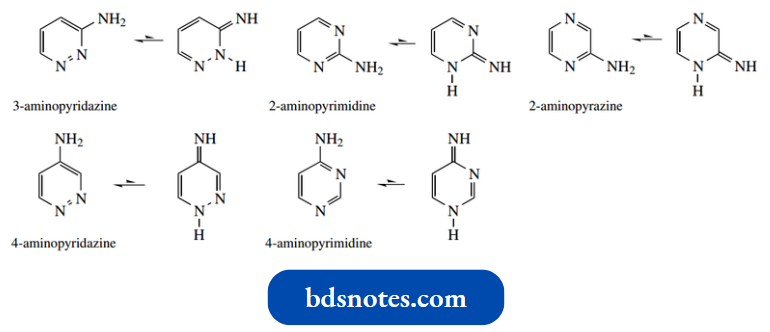
Aminopyridines are also potentially tautomeric with corresponding imino forms.
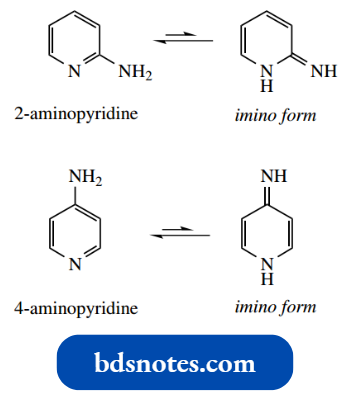
However, 2-aminopyridine and 4-aminopyridine exist almost entirely as amino tautomers – indeed, we have just seen 2-aminopyridine as a product of the Chichibabin reaction.
- Which tautomer is preferred for hydroxy and amino heterocycles is not always easily explained; but, as a generalization, we find that the oxygen derivatives exist as carbonyl tautomers and amino heterocycles favor the amino tautomers.
- At this stage, we should just register the potential for tautomerism in aminopyridines; we shall see important examples with other heterocycles.
Aminopyridines protonate on the ring nitrogen, and 4-aminopyridine is a stronger base than 2-aminopyridine.
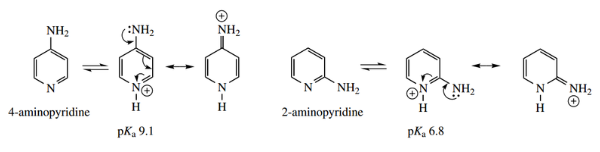
This may be rationalized by a consideration of resonance in the conjugate acids.
- The conjugate acids from ring protonation benefit from charge delocalization, which is greater in 4-amino pyridinium than in 2-amino pyridinium.
- This type of delocalization is not possible in 3-aminopyridinium; 3-aminopyridine (pKa 6.0) is the weakest base of the three aminopy ridines and has basicity more comparable to that of pyridine (pKa 5.2).
Pyrylium Cation And Pyrones
Pyrones are oxygen analogs of pyridones and are potentially aromatic. However, there is little evidence that the dipolar resonance forms of either 2-pyrone or 4-pyrone make any significant contribution.
- Their chemical behavior suggests they should be viewed more as conjugated lactones (2-pyrones) or vinylogous lactones (4-pyrones) rather than aromatic systems since many reactions lead to ring opening.
The pyrylium cation is isoelectronic with pyridine: it has the same number of electrons and, therefore, we also have aromaticity. Oxygen is normally divalent and carries two lone pairs.
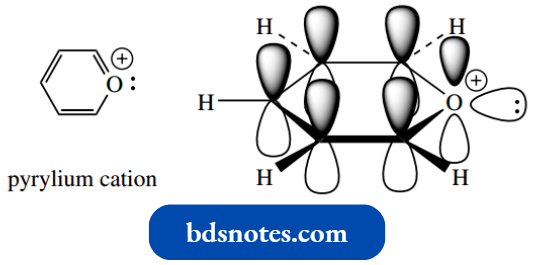
If we insert oxygen into the benzene ring structure, then it follows that, by having one electron in a p orbital contributing to the aromatic sextet, there is a lone pair in an sp2 orbital, and the remaining electron needs to be removed, hence the pyrylium cation.
However, oxygen tolerates a positive charge less readily than nitrogen and aromatic stabilization is less than with pyridine.
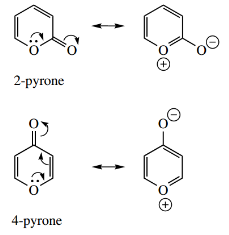
Flavonoids are natural phenolic systems containing pyrylium and pyrone rings and provide the most prominent examples.
We have met some of these systems under antioxidants. Coumarins contain a 2-pyrone system. Note that all of these compounds are fused to a benzene ring and are strictly benzopyran or benzopyrylium systems.
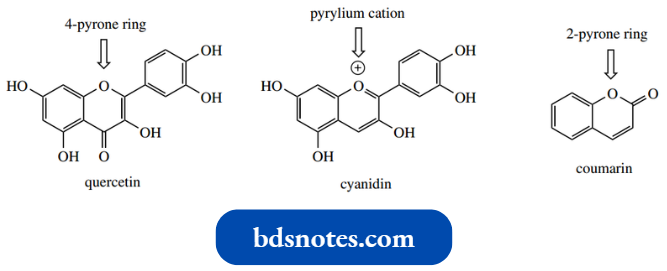
Dicoumarol And Warfarin:
Warfarin provides us with a slightly incongruous state of affairs: it is used as a drug and also as a rat poison.
- It was developed from a natural product, dicoumarol, and provides us with a nice example of how pyrone chemistry resembles that of conjugated lactones rather than aromatic systems.
- Many plants produce coumarins; coumarin itself is found in sweet clover and contributes to the smell of new-mown hay.
- However, if sweet clover is allowed to ferment, oxidative processes initiated by the microorganisms lead to the formation of 4-hydroxycoumarin rather than coumarin.
4-Hydroxycoumarin then reacts with formaldehyde, also produced via the microbial degradative reactions, and provides dicoumarol.
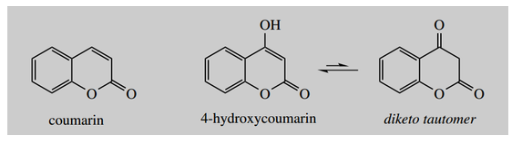
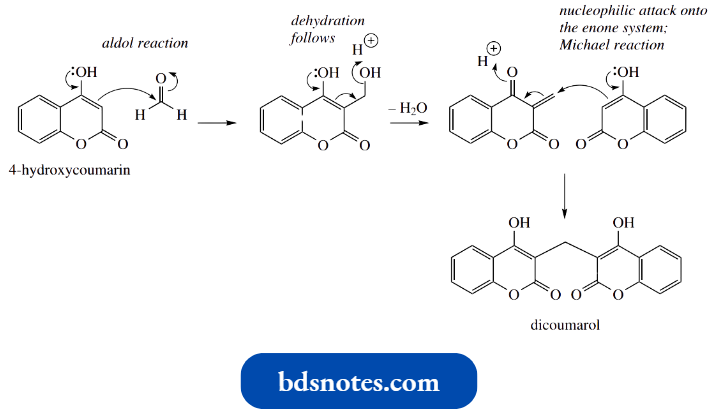
4-Hydroxycoumarin can be considered an enol tautomer of a 1,3-dicarbonyl compound; conjugation with the aromatic ring favors the enol tautomer. This now exposes its potential as a nucleophile.
- Whilst we may begin to consider enolate anion chemistry, no strong base is required and we may formulate a mechanism in which the enol acts as the nucleophile, in a simple aldol reaction with formaldehyde.
Dehydration follows and produces an unsaturated ketone, which then becomes the electrophile in a Michael reaction. The nucleophile is a second molecule of 4-hydroxycoumarin.
Animals fed spoiled sweet clover were prone to fatal hemorrhages. The cause was traced to the presence of dicoumarol.
- This compound interferes with the effects of vitamin K in blood coagulation, the blood loses its ability to clot, and minor injuries can lead to severe internal bleeding.
- Synthetic dicoumarol has been used as an oral blood anticoagulant in the treatment of thrombosis, where the risk of blood clots becomes life-threatening.
It has since been superseded by warfarin, a synthetic development based on the natural product.
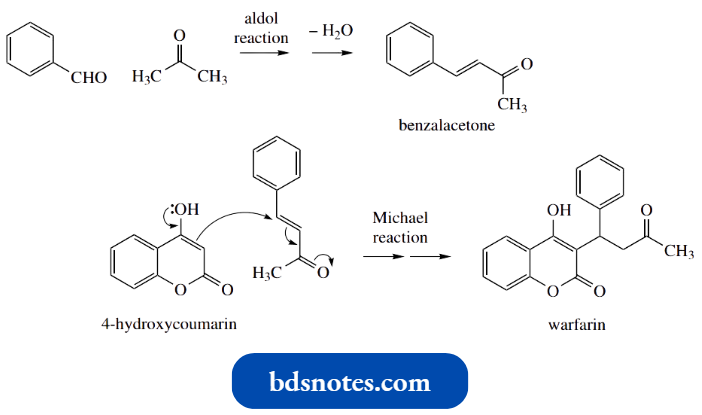
Warfarin was initially developed as a rodenticide and has been widely employed for many years as the first-choice agent, particularly for the destruction of rats.
- After consumption of warfarin-treated bait, rats die from internal hemorrhage.
- Warfarin is synthesized from 4-hydroxycoumarin by a Michael reaction on benzalacetone, again exploiting the nucleophilicity of the hydroxypyrone.
Benzalacetone is the product of an aldol reaction between benzaldehyde and acetone.
Five-Membered Aromatic Heterocycles: Pyrrole
Pyrrole (azacyclopentadiene) is the ring system obtained if we replace the CH2 group of cyclopenta diene with NH.
- Although cyclopentadiene is certainly not aromatic, pyrrole has an aromatic character because nitrogen contributes two electrons, its lone pair, to the π-electron system.
- We have also noted from resonance forms that nitrogen carries a partial positive charge, and the carbons are electron-rich.
This is stronger than the opposing inductive effect.
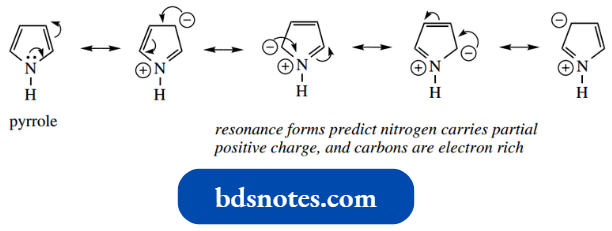
This is reflected in the basicity of pyrrole. Pyrrole is a particularly weak base, with pKa of the conjugate acid −3.8.
First, we should realize that protonation of pyrrole will not occur on nitrogen: nitrogen has already used up its lone pair by contributing to the aromatic sextet, so protonation would necessarily destroy aromaticity.
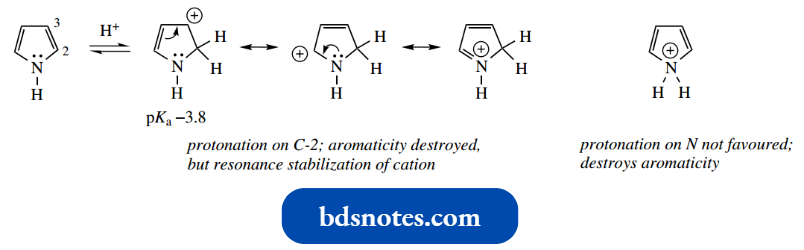
It is possible to protonate pyrrole using a strong acid, but even then the protonation occurs on C-2 and not on the nitrogen.
- Although this still destroys aromaticity, there is some favorable resonance stabilization in the conjugate acid.
- Protonation on C-3 is not as favorable, in that there is less resonance stabilization in the conjugate acid.
- It turns out that, as opposed to acting as a base, pyrrole is potentially an acid (pKa 17.5); it is not a particularly strong acid, but stronger than we might expect for a secondary amine system (pKa about 36).
- This is because the anion formed by losing the proton from nitrogen has a negative charge on the relatively electronegative nitrogen, but maintains its aromaticity.
Unlike in pyrrole, the anion resonance structures do not involve charge separation.
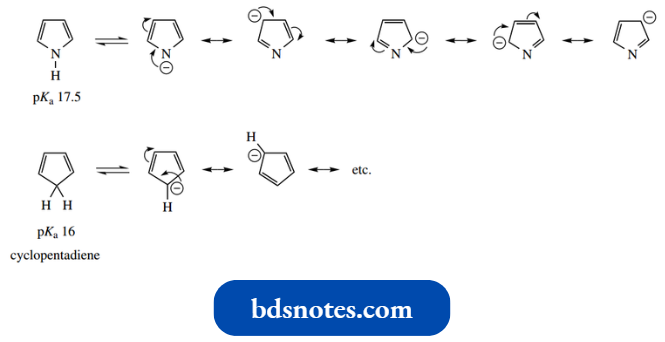
It is appropriate here to compare the acidity of cyclopentadiene, which has pKa 16, is considerably more acidic than most hydrocarbon systems and comparable to water and alcohols.
- Removal of one of the CH2 protons from the non-aromatic cyclopentadiene generates the cyclopentadienyl anion.
- This anion has an aromatic sextet of electrons, two electrons being contributed by the negatively charged carbon.
- The charge distribution in pyrrole leads us to predict that it will react readily with electrophiles; or, put another way, pyrrole will behave as a nucleophile.
- This is indeed the case, and the ease of elec trophilic substitution contrasts with the behavior of pyridine above, where charge distribution favored nucleophilic attack onto the heterocycle.
Although our resonance description of pyrrole shows negative charge can be dispersed to any ring carbon, pyrrole reacts with electrophiles preferentially at C-2 rather than C-3, unless the 2-position is already substituted.
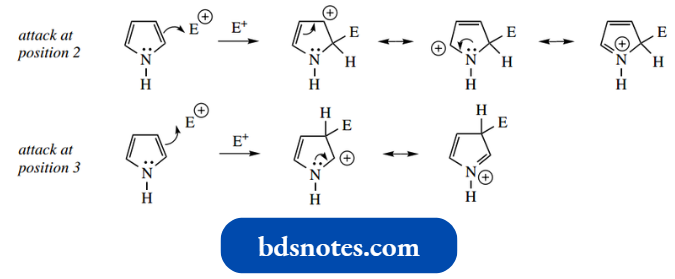
This may reflect that there is more charge dispersion in the addition cation from attack at C-2 than there is from attack at C-3.
- This is, of course, the same argument as used above for C-protonation; protonation (pyrrole acting as a base) also occurs at C-2.
- As with protonation, electrophiles do not react at the nitrogen center. Pyrrole is very reactive towards electrophiles.
For example, treatment with bromine leads to the substitution of all four positions.
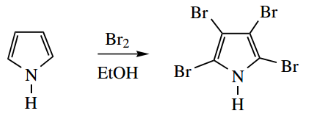
Indeed, it is often difficult to control electrophilic attack so that monosubstitution occurs.
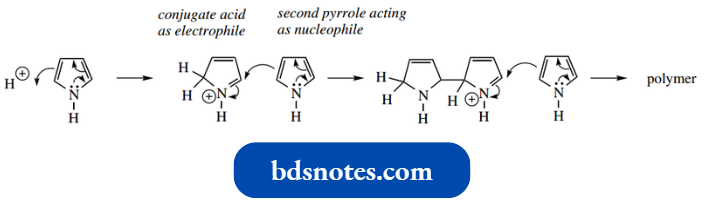
A further problem is that pyrrole polymerizes in the presence of strong acids and Lewis acids, so that typical electrophilic reagents, for example., HNO3 –H2SO4 and RCOCl–AlCl3, cannot be used. Polymerization involves the conjugate acid functioning as the elec trophile.
To achieve useful monosubstitution it is necessary to employ relatively mild conditions, often without a catalyst.
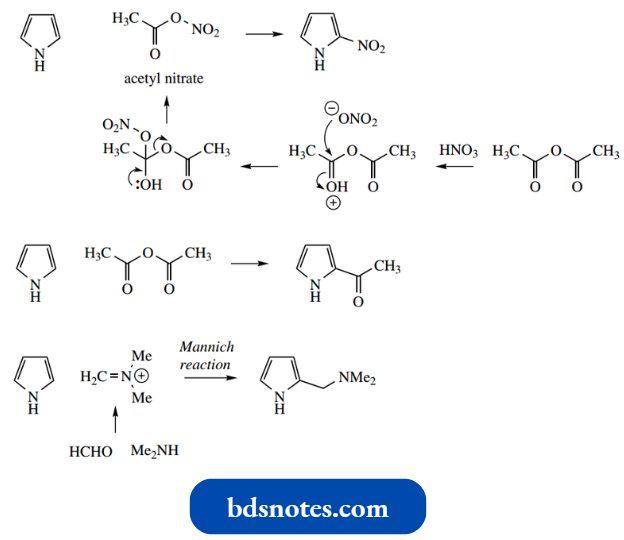
- Nitration may be accomplished with the reagent acetyl nitrate, giving mainly 2-nitropyrrole.
- Acetyl nitrate is formed by reacting acetic anhydride with fuming nitric acid. Since the other product is acetic acid, there is no strong mineral acid present to cause polymerization.
It is also possible to synthesize 2-acetyl pyrrole simply by using acetic anhydride, and pyrrole can act as the nucleophile in the Mannich reaction.
Although pyrrole is a weak acid, it can be deprotonated by using a strong base, for example., sodium hydride, and the anion can be used in typical nucleophilic reactions.
This allows simple transformations such as N-alkylation, N-acylation, and N-sulfonation. Note particularly that whereas pyrrole reacts with elec trophiles at carbon, usually C-2, the pyrrole anion reacts at the nitrogen atom.
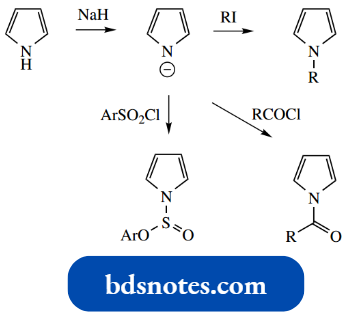
Porphyrins And Corrins:
Pyrrole reacts with aldehydes and ketones under acidic conditions to form polymeric compounds. In many cases these are intractable resin-like materials; however, with appropriate carbonyl compounds, interesting cyclic tetramers can be formed in very good yields.
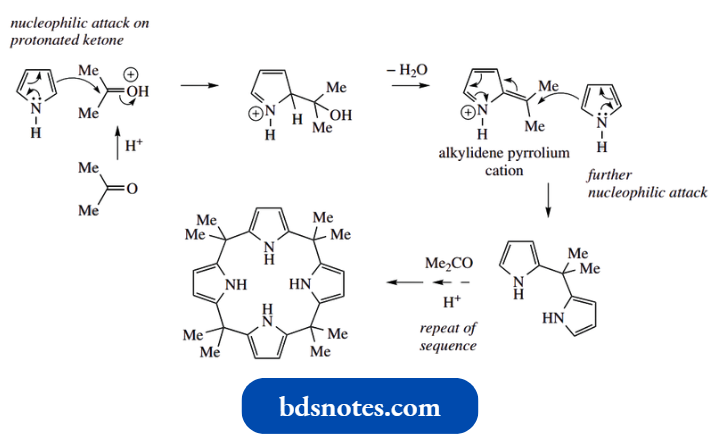
Thus, pyrrole and acetone react as shown above. This involves pyrrole acting as the nucleophile to attack the protonated ketone in an aldol-like reaction.
- This is followed by the elimination of water, facilitated by the acidic conditions. This gives an intermediate alkylidene pyrrolium cation, a highly reactive electrophile that reacts with another molecule of nucleophilic pyrrole.
- We then have a repeat sequence of reactions, in which further acetone and pyrrole molecules are incorporated.
- The presence of the two methyl substituents from acetone forces the growing polymer to adopt a planar array, and this eventually leads to a cyclic tetramer, the terminal pyrrole attacking the alkylidene pyrrolium cation at the other end of the chain.
- The cyclic tetramer shown is structurally related to the porphyrins. The basic ring system in porphyrins is porphin, which is more oxidized than the tetramer from the pyrrole–acetone reaction, and has four pyrrole rings linked together by methine (–CH=) bridges.
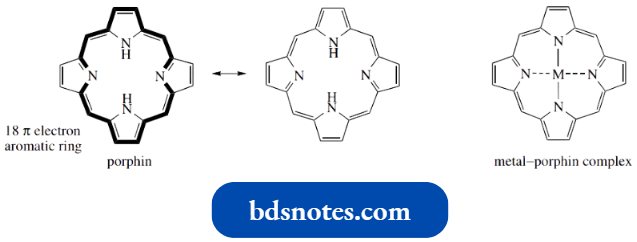
- One of the features of porphin is that it is aromatic. It contains an aromatic 18 π-electron system, which conforms to Huckel’s rule, 4n + 2 with n = 4.
- The aromatic ring weaves around the porphin structure, and is composed entirely of double-bond electrons; it does not incorporate any nitrogen lone pairs.
Note that we can draw Kekule-like resonance structures for porphin. Do not be confused by seeing alternative structures with the double bonds arranged differently.
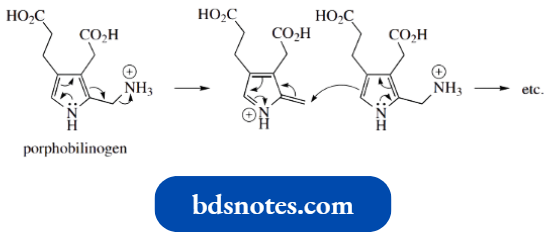
Porphyrin rings are formed in nature by a process that is remarkably similar to that shown above. Though the sequence contains some rather unusual features, the coupling process also involves nucleophilic attack onto an alkylidene pyrrolium cation. This may be generated from the precursor porphobilinogen by the elimination of ammonia.
One of the important properties of porphyrins is that they are complex with divalent metals, the pyrrole nitrogens being ideally spaced to allow this.
Of vital importance to life processes are the porphyrin derivatives chlorophyll and haem. Chlorophyll (a mixture of structurally similar porphyrins; chlorophyll a is shown) contains magnesium, and is, of course, the light-gathering pigment in plants that permits photosynthesis.
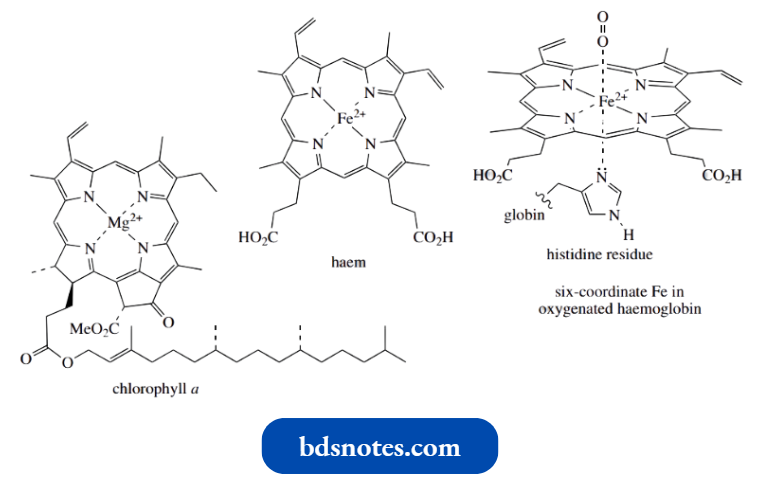
Plants and a few microorganisms use photosynthesis to produce organic compounds from inorganic materials found in the environment, whereas other organisms, such as animals and most microorganisms, rely on obtaining their raw materials in their diet, for example., by consuming plants.
- Haemoglobin, the red pigment in blood, serves to carry oxygen from the lungs to other parts of the body tissue.
- This material is made up of the porphyrin haem and the water-soluble protein globin.
- The haem component shares many structural features with chlorophyll, one of the main differences being the use of Fe2+ as the metal rather than Mg2+ as in chlorophyll.
- The oxygen-carrying ability of hemoglobin involves a six-coordinate iron, with an imidazole ring from the protein (a histidine residue) occupying the sixth position.
- Porphyrin rings containing iron are also a feature of the cytochromes. Several cytochromes are responsible for the latter part of the electron transport chain of oxidative phosphorylation that provides the principal source of ATP for an aerobic cell.
- Their function involves alternate oxidation-reduction of the iron between Fe2+ (reduced form) and Fe3+ (oxidized form).
The individual cytochromes vary structurally, and their classification (a, b, c, etc.) is related to their absorption maxima in the visible spectrum. They contain a haem system that is covalently bound to protein through thiol groups.
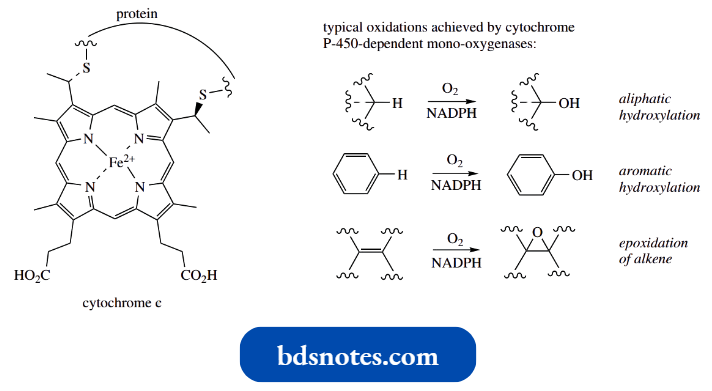
An especially important example is cytochrome P-450, a coenzyme of the so-called cytochrome P-450-dependent mono-oxygenases.
- These enzymes are frequently involved in biological hydroxylations, either in biosynthesis or in the mammalian detoxification and metabolism of foreign compounds such as drugs.
- Cytochrome P-450 is named after its intense absorption band at 450 nm when exposed to CO, which is a powerful inhibitor of these enzymes.
- A redox change involving the Fe atom allows binding and the cleavage of molecular oxygen to oxygen atoms, with subsequent transfer of one atom to the substrate.
- In most cases, NADPH features as a hydrogen donor, reducing the other oxygen atom to water. Many such systems have been identified, capable of hydroxylating aliphatic or aromatic systems, as well as producing epoxides from alkenes.
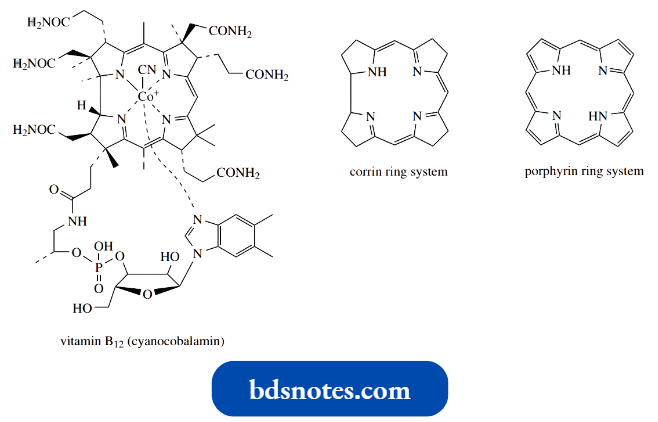
- A related ring system containing four pyrroles is seen in vitamin B12, but this has two pyrroles directly bonded and is termed a corrin ring.
- Vitamin B12 is extremely complex and features six-coordinate Co2+ as the metal component. Four of the six coordination are provided by the corrin ring nitrogens and a fifth by a dimethyl benzimidazole moiety.
- The sixth is variable, being cyano in cyanocobalamin (vitamin B12), but other anions may feature in vitamin B12 analogs.
Vitamin B12 appears to be entirely of microbial origin, with intestinal flora contributing to human dietary needs. Insufficient vitamin B12 leads to pernicious anemia, a disease that results in nervous disturbances and low production of red blood cells.
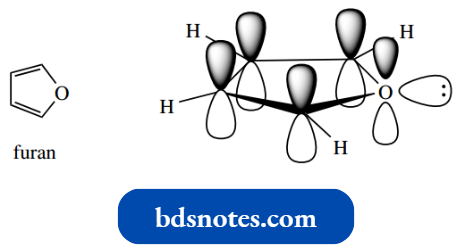
Furan And Thiophene:
Furan and thiophene are the oxygen and sulfur analogs respectively of pyrrole. Oxygen and sulfur contribute two electrons to the aromatic sextet but still retain lone pair electrons.
- There is one significant difference, however, in that oxygen uses electrons from a 2p orbital, whereas the electrons that sulfur contributes originate from a 3p orbital.
- In the case of furan H of thiophene, this reduces orbital overlap with the carbon 2p orbitals.
- Both compounds are thus aromatic, and their chemical reactivity reflects what we have learned about pyrrole.
- The most typical reaction is electrophilic substitution. However, we find that pyrrole is more reactive than furan towards electrophiles and thiophene is the least reactive; all are more reactive than benzene
- This relates to the relative stability of positive charges located on nitrogen, oxygen, and sulfur. We used similar electronegativity reasoning to explain the relative basic strengths of nitrogen, oxygen, and sulfur derivatives.
Furan is also the ‘least aromatic’ of the three, i.e. it has the least resonance stabilization, and undergoes many reactions in which the aromatic character is lost, for example., addition reactions or ring opening.
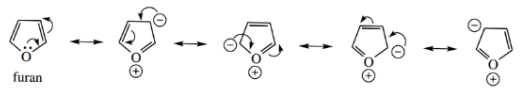
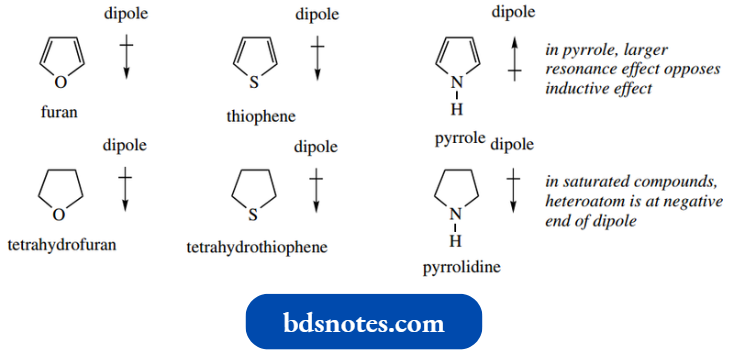
Note that the dipoles of furan and thiophene are opposite in direction to those in pyrrole.
- In furan and thiophene, there is a greater inductive effect opposing the resonance effect, whereas in pyrrole the resonance contribution was greater.
In the non-aromatic analogs, the heteroatom is at the negative end of the dipole in all cases.
Nevertheless, we can interpret the reactions of furan and thiophene by logical consideration as we did for pyrrole.
- In electrophilic substitutions, there is again a preference for 2- rather than 3-substitution, and typical electrophilic reactions carried out under acidic conditions are difficult to control.
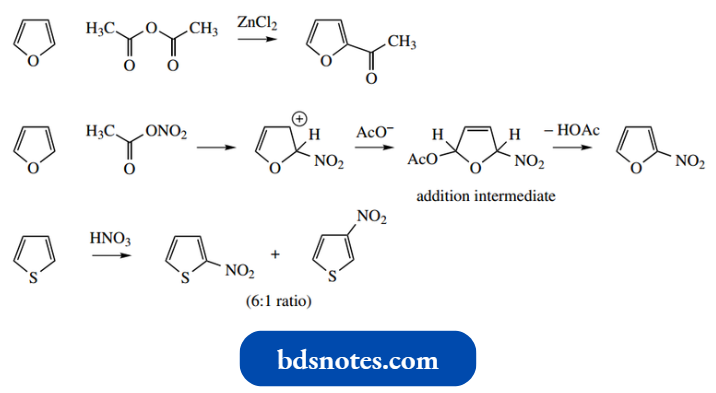
- However, because of lower reactivity compared with pyrrole, it is possible to exploit Friedel-Crafts acylations, though using less-reactive anhydrides rather than acyl chlorides, and weaker Lewis acids than AlCl3.
- Nitration can be achieved with acetyl nitrate rather than nitric acid. In the case of furan, this is slightly anomalous, in that it involves an addition intermediate by combination of the carbocation with acetate.
This subsequently aromatizes by loss of acetic acid. The less-reactive thiophene can even be nitrated with concentrated nitric acid when it yields a mixture of 2- and 3-nitrothiophene.
Six-membered rings with two heteroatoms
Diazines:
A diazabenzene, i.e. a benzene ring in which two of the CH functions have been replaced with nitrogen, is termed a diazine.
- Three isomeric variants are possible; these are called pyridazine, pyrimidine, and pyrazine.
- These structures are all aromatic, the nitrogen atoms functioning in the same way as the pyridine nitrogen, each contributing one p electron to the aromatic sextet, with a lone pair in an sp2 orbital.
The diazines are much weaker bases than pyridine (pKa 5.2).
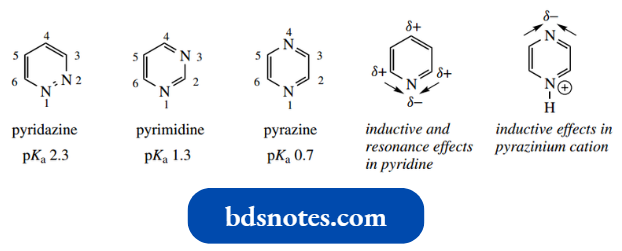
If we consider the inductive and resonance effects in pyridine, we have seen that these both draw electrons toward the nitrogen.
- Therefore, a second nitrogen will have destabilizing effects on the conjugate acid formed by the protonation of the first nitrogen.
- The order of basicity in pyridazine, pyrimidine, and pyrazine is influenced by secondary effects, which will not be considered here.
- Deprotonation is very difficult and would require extremely strong acids; in the case of pyridazine, it is essentially impossible because of the need to establish positive charges on adjacent atoms.
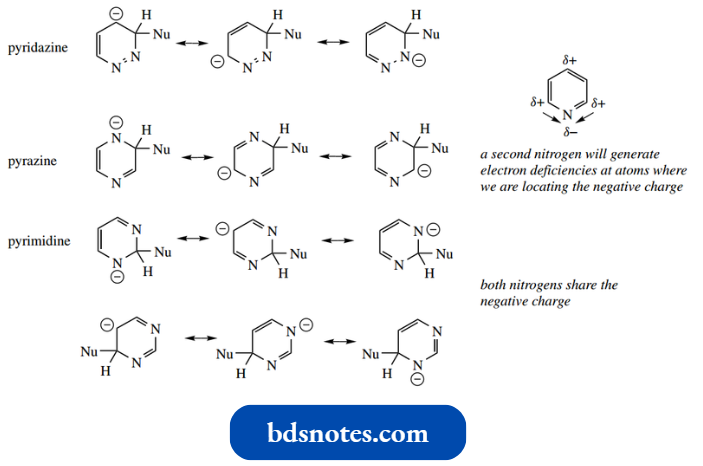
- In general, we can consider that the extra nitrogen, through its combined effects, makes the other ring atoms more electron deficient than they would be in pyridine; as a result, the diazines are more susceptible to nucleophilic attack than pyridine.
- In pyrazines and pyridazines, the second nitrogen helps by withdrawing electrons from atoms that would carry a negative charge in the addition anion.
In pyrimidines, the two nitrogens share the negative charge of the addition anion, and pyrimidines are the more reactive towards nucleophiles.
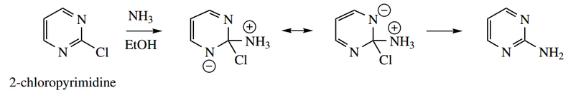
Halodiazines react readily with nucleophiles with a displacement of the halide-leaving group.
- This follows what we have seen with halopyridines, but the halodiazines are more reactive because of the influence of the extra nitrogen.
- Thus, 2-chloropyrazine and 3-chloropyridazine easily yield the corresponding amino derivatives on heating with ammonia in an alcohol solution.
- The 2- and 4-halo pyrimidines are even more reactive and substitute at room temperature. This is because of the improved delocalization of negative charge in the addition anion.
5-Halopyrimidines are the least susceptible to nucleophilic displacement: the halogen is neither α nor γ to nitrogen, and cannot benefit from any favorable charge localization on nitrogen.
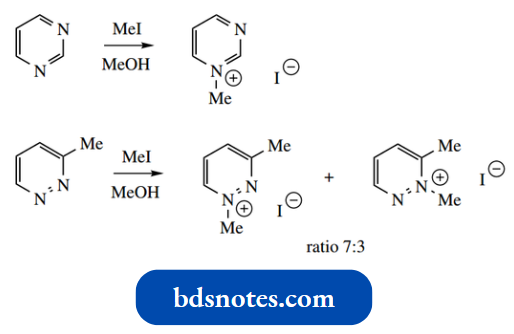
Diazines are generally resistant to electrophilic attack on carbon, and, as for pyridine, addition to nitrogen is observed.
- Alkyl halides give mono-quaternary salts; di-quaternary salts are not formed under normal conditions.
- Of course, if the diazine ring carries a substituent that makes the starting material non-symmetric, then the product will almost always be a mixture of two isomeric quaternary salts.
Steric and inductive effects rather than resonance effects appear to influence the reaction and formation of the major product.
Tautomerism In Hydroxy- And Amino-Diazines:
We have seen that 2- and 4-hydroxypyridines exist primarily in their tautomeric ‘amide-like’ pyridone forms.
- This preference over the ‘phenolic’ tautomer was related to these compounds still retaining their aromatic character, with further stabilization from the carbonyl group.
- 3- Hydroxypyridine cannot benefit from this additional stabilization. In contrast, 2-aminopyridine and 4-aminopyridine exist almost entirely as amino tautomers, although they are potentially tautomeric with imino forms.
- We also encounter tautomerism in hydroxy- and amino-diazines, and the preference for one tautomeric form over the other follows what we have seen with the pyridine derivatives.
- Thus, except for 5-hydroxy pyrimidine, all the mono-oxygenated diazines exist predominantly in the carbonyl tautomeric form.
We term these ‘amide-like’ tautomers diazinones. 5-Hydroxypyrimidine is analogous to 3- hydroxypyridine, in that the hydroxyl is wrongly positioned for tautomerism.
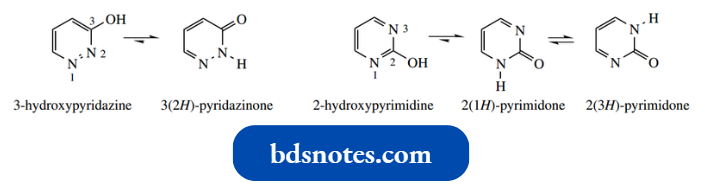
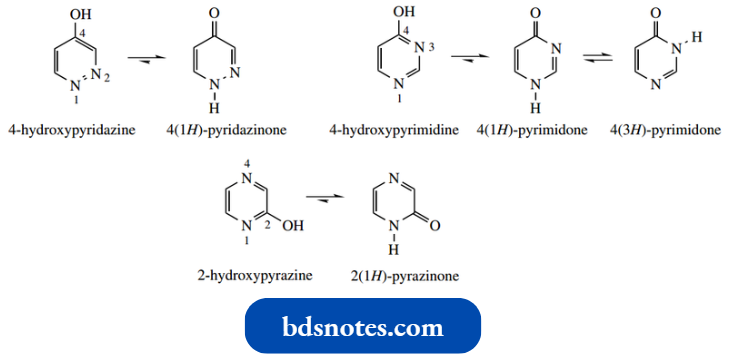
The diazinon tautomers are identified by using terminology such as 3(2H)-pyridazine for the car bonyl tautomer of 3-hydroxypyrazine.
- The 3(2H) prefix signifies the position of the oxygen (3- pyridazine) and specifies the NH is at position 2.
- Note that, in addition to the diazine–diazinone tau H tomerism, when the nitrogens have a 1,3-relationship there is further tautomerism possible, for example., 4(1H)- pyrimidone 4(3H)-pyrimidone.
Diazinones may be converted into chlorodiazines by the use of phosphorus oxychloride, just as pyridones yield chloropyridines.
Aminodiazines exist in the amino form. These compounds contain two-ring nitrogens and a primary amino group.
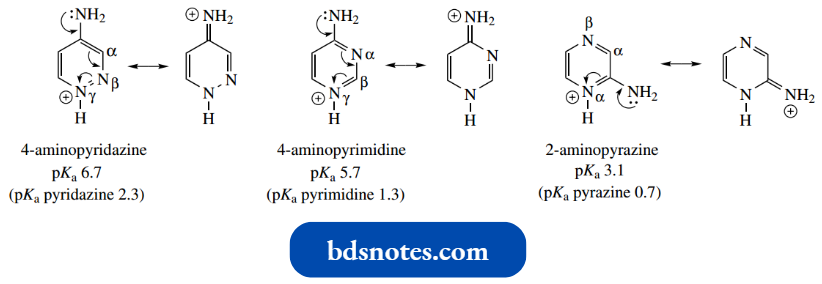
Interestingly, they are more basic than the unsubstituted diazine and always protonate on a ring of nitrogen.
- This allows resonance stabilization of the conjugate acid utilizing the lone pair of the amino substituent.
- It has been found that one can predict which nitrogen is protonated from the ring nitrogen–amino substituent relationship, which follows the preference sequence γ > α > β, as in the examples shown.
This can be related to achieving maximum charge distribution over the molecule.
Pyrimidines And Nucleic Acids:
The storage of genetic information and the transcription and translation of this information are functions of the nucleic acids deoxyribonucleic acid (DNA) and ribonucleic acid (RNA).
- They are polymers whose building blocks are nucleotides, which are themselves combinations of three parts, i.e. a heterocyclic base, a sugar, and phosphate.
- The bases are either monocyclic pyrimidines or bicyclic purines (see Section 14.1). Three pyrimidine bases are encountered in DNA and RNA, cytosine (C), thymine (T), and uracil (U).
- Cytosine is common to both DNA and RNA, but uracil is found only in RNA and thymine is found only in DNA.
In nucleic acid, the bases are linked through an N-glycoside bond to a sugar, either ribose or deoxyribose; the combination base plus sugar is termed a nucleoside. The nitrogen bonded to the sugar is shown.
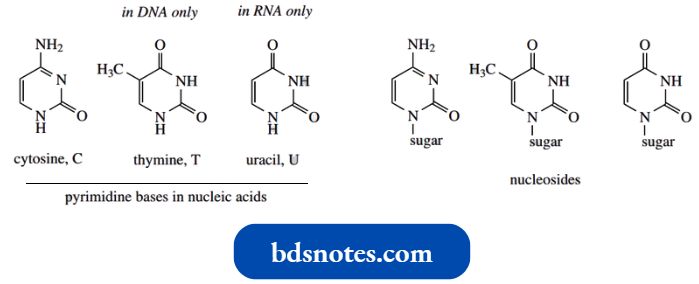
We should note particularly that uracil and thymine are dioxypyrimidines, whereas cytosine is an amino oxypyrimidine. All three pyrimidines are thus capable of existing in several tautomeric forms.
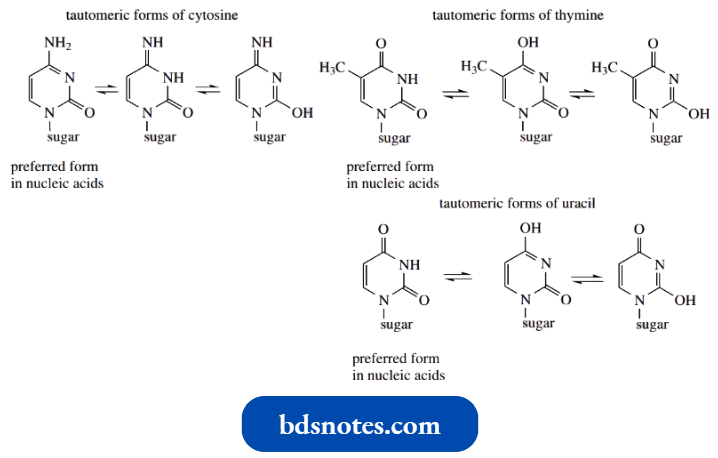
The number of possible forms is reduced somewhat by the fact that one of the nitrogens is bonded to the sugar in the nucleic acid; it no longer carries hydrogen to participate in tautomerism.
- The tautomeric forms indicated are found to predominate in nucleic acids. The oxygen substituents exist almost entirely as carbonyl groups, whereas
- the amino group is preferred over possible imino forms. Although we are accustomed to thinking of nucleic acids containing ‘pyrimidine’ bases, this is not strictly correct. Cytosine exists as an aminopyrimidone, and thymine and uracil are pyrimidines. Further, they are not particularly basic.
- Cytosine is the most basic of the three (pKa 4.6), in that the amino group by a resonance effect can stabilize the conjugate acid (compare 4-aminopyrimidine pKa 5.7 above). Thymine and uracil are very weak bases, in that they are ‘amide-like’.
- The most far-reaching feature of nucleic acids is the ability of the bases to hydrogen bond to other bases.
- This property is fundamental to the double helix arrangement of the DNA molecule, and the translation and transcription via RNA of the genetic information present in the DNA molecule.
- Hydrogen bonding occurs between complementary purine and pyrimidine bases and involves either two or three hydrogen bonds.
- In DNA, the base pairs are adenine–thymine and guanine-cytosine. In RNA, base pairing involves guanine-cytosine and adenine–uracil.
- This property will be discussed in detail in Section 14.2, but it is worth noting at this stage that hydrogen bonding is achieved between amino substituents (N–H) and the oxygen of carbonyl groups.
- These functions in the pyrimidine bases arise directly from the tautomeric preferences.
Five-Membered Rings With Two Heteroatoms:
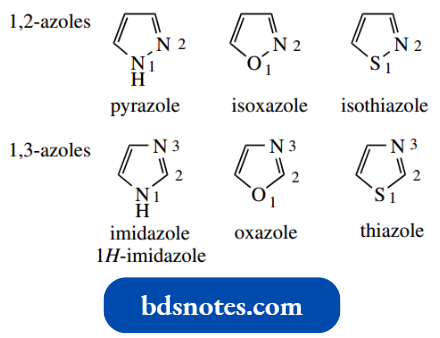
We have looked at the five-membered aromatic heterocycles pyrrole, furan, and thiophene. The introduction of a second heteroatom creates azoles. This name immediately suggests that nitrogen is one of the heteroatoms.
- As soon as we consider valencies, we discover that to draw a five-membered aromatic heterocycle with two heteroatoms, it must contain nitrogen.
- A neutral oxygen or sulfur atom can have only two bonds, and we cannot, therefore, have more than one of these atoms in any aromatic heterocycle. On the other hand, there is potential for having as many nitrogens as we like in an aromatic ring.
- Thus, in five-membered aromatic heterocycles with two heteroatoms, we can have two nitrogens, one nitrogen plus one oxygen, or one nitrogen plus one sulfur.
The heteroatoms can be positioned only 1,2 or 1,3. The numbering of the ring system starts from the heteroatom with the higher atomic number; nitrogen will always be the higher of the two numbers in the oxazole and thiazole systems. In imidazole, numbering begins at the NH.
We can visualize these heterocycles as similar to the simpler aromatic systems pyrrole, furan, and thiophene.
- For example, in imidazole, each carbon and nitrogen will be sp2 hybridized, with p orbitals contributing to the aromatic π system. The carbon atoms will each donate one electron to the π system.
- Then, as in pyrrole, the NH nitrogen supplies two electrons, and, as in pyridine, the =N– supplies one electron and retains a lone pair.
Oxygen or sulfur would also supply two electrons, as we saw in furan and thiophene.
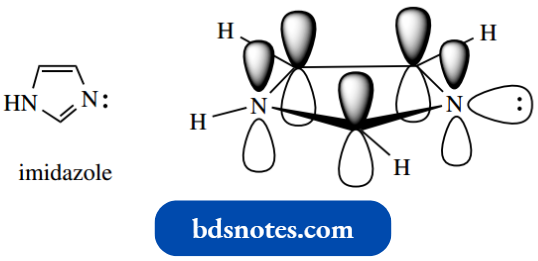
It also follows that a compound like imidazole has one pyridine-like nitrogen and one pyrrolelike nitrogen.
- We may thus expect to see imidazole having properties resembling a combination of either pyridine- or pyrrole-like reactivity.
- The availability and location of lone pair electrons are crucial to our understanding of imidazole chemistry, and it often helps to include these in the structure.
1,3-Azoles: Imi Dazole, Oxazole, And Thiazole:
Imidazole (pKa 7.0) is a stronger base than either pyridine (pKa 5.2) or pyrrole (pKa − 3.8).
- When we compared the basicity of pyridine with that of the aliphatic amine piperidine (pKa 11.1), we implicated the higher s character of the pyridine lone pair (sp2) compared with that in piperidine (sp3) to account for pyridine’s lower basicity.
- Even so, imidazole seems abnormally basic for a compound with sp2-hybridized nitrogen.
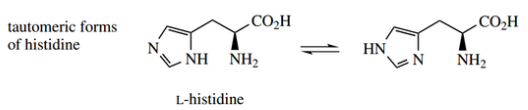
- Therefore, when we meet structures for the imidazole-containing amino acid histidine, we may encounter either of the tautomeric forms shown.
- Though there imidazole appears to stem from the symmetry of the conjugate acid, and the resonance stability conferred by this.
The 1,3-relationship allows the two nitrogen atoms to share the charge equally. Note that pKa 7.0 means imidazole is 50% protonated in water.
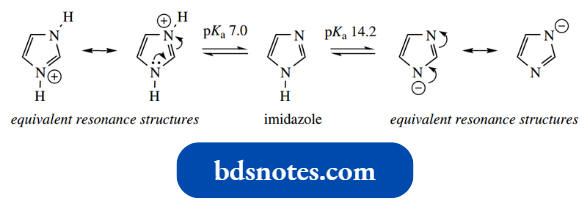
Imidazole (pKa 14.2) is also more acidic than pyrrole (pKa 17.5); this, again, is a feature conferred by symmetry and enhanced resonance stabilization in the conjugate base.
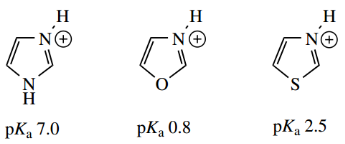
Oxazole (pKa 0.8) and thiazole (pKa 2.5) are weak bases. The basicity of the nitrogen is reduced by the presence of the other heteroatom.
Oxygen and sulfur provide a stronger electron-withdrawing inductive effect, compared with nitrogen, but a much weaker electron-releasing resonance effect.
Tautomerism In Imidazoles:
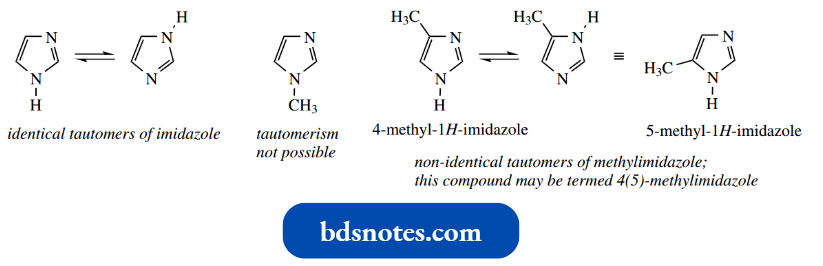
A complicating factor in imidazoles is tautomerism. Imidazole tautomerizes rapidly in solution and consists of two identical tautomers.
- This becomes a problem, though, in an unsymmetrically substituted imidazole, and tautomerism means 4-methylimidazole is in equilibrium with 5-methylimidazole.
- Depending upon substituents, one tautomer may predominate. Tautomerism of this kind cannot occur with N-substituted imidazoles; it is dependent upon the presence of an N–H group.
Tautomerism is also not possible with oxazoles or thiazoles.
Therefore, when we meet structures for the imidazole-containing amino acid histidine, we may encounter either of the tautomeric forms shown. Though there will usually be no indication that tautomers exist, do not think there is a discrepancy in structures.
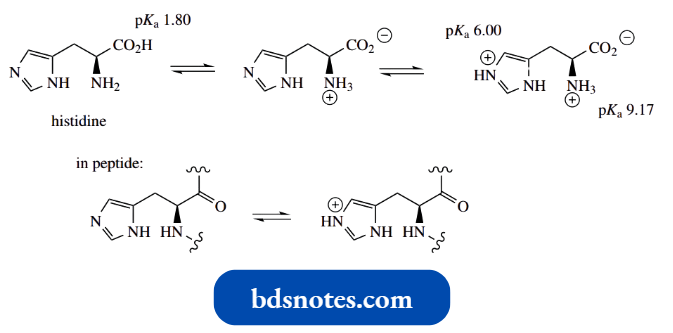
The Imidazole Ring Of Histidine: Acid-Base Properties
The amino acid histidine contains an imidazole ring. We have just seen that unsubstituted imidazole as a base has pKa 7.0. From the Henderson–Hasselbalch equation
⇒ \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\text { [base }]}{\text { [acid }]}\)
we can deduce that in water, at pH 7, the concentrations of acid and conjugate base are equal, i.e. imidazole is 50% protonated.
- The imidazole side-chain of histidine has a pKa value of 6.0, making it a weaker base than the unsubstituted imidazole.
- This reflects the electron-withdrawing inductive effect of the amino group, or, more correctly the ammonium ion, since amino acids at pH values around neutrality exist as doubly charged zwitterionic forms.
- Using the Henderson–Hasselbalch equation, this translates to approximately 9% ionization of the heterocyclic side-chain of histidine at pH 7.
In proteins, pKa values for histidine side chains are estimated to be in the range 6–7, so the level of ionization will, therefore, be somewhere between 9 and 50%, depending upon the protein.
This level of ionization is particularly relevant in some enzymic reactions where histidine residues play an important role.
- This means that the imidazole ring of a histidine residue can act as a base, assisting in the removal of protons, or that the imidazolium cation can act as an acid, donating protons as required.
- The terminology used for such donors and acceptors of protons is a general acid catalyst and general base catalyst respectively.
- A typical role for the histidine imidazole ring is shown below, in the enzyme mechanism for a general base-catalyzed hydrolysis of an ester.
- The imidazole nitrogen acts as a base to remove a proton from water, generating hydroxide that attacks the carbonyl. Subsequently, the alkoxide-leaving group is deprotonated by the imidazolium ion.
General Base-Catalysed Ester Hydrolysis:
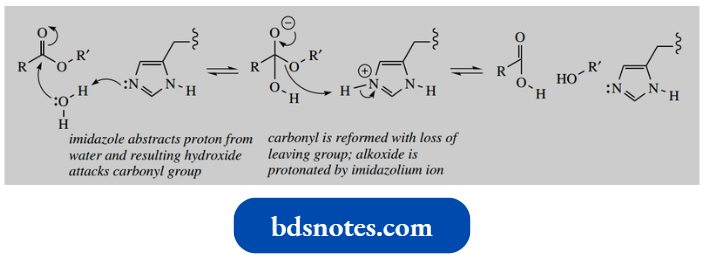
The beauty of this is that we effectively have the same mechanism as in the hydrolysis of an ester using aqueous sodium hydroxide. However, with the enzyme catalyst, this is all taking place at pH 7 or thereabouts.
- Implicit in the above mechanism, though not emphasized, is the pronounced ability of imidazole rings to hydrogen bond.
- Imidazole resembles water, in that it is both a very good donor and a very good acceptor for hydrogen bonding.
Imidazole (and also pyrazole) has a higher-than-expected boiling point, ascribed to intermolecular hydrogen bonding. This leads to polymeric-like structures for imidazole and dimers for pyrazole.
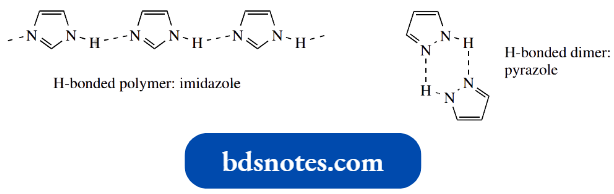
In enzymic mechanisms, we are not usually going to get imidazole–imidazole hydrogen bonding, but the ability of imidazole to hydrogen bond to water, to other small molecules, and carboxylic acid side chains facilitates the enzyme reaction by correctly positioning the reagents.
Histamine And Histamine Receptors:
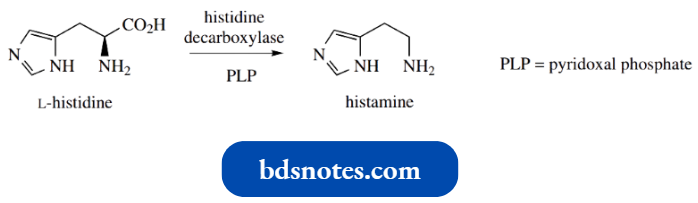
Most people have heard of antihistamines, even if they have little concept of the nature of histamine.
Histamine is the decarboxylation product from histidine and is formed from the amino acid by the action of the enzyme histidine decarboxylase.
The mechanism of this pyridoxal phosphate-dependent reaction will be studied in more detail later.
Histamine is released from mast cells during inflammatory or allergic reactions. It then produces its typical response by interaction with specific histamine receptors, of which there are several types.
- H1 receptors are associated with inflammatory and allergic reactions, and H2 receptors are found in acid-secreting cells in the stomach. Drugs to target both of these types of receptors are widely used.
- The term antihistamine usually relates to H1 receptor antagonists. These drugs are valuable for pain relief from insect stings or for the treatment and prevention of allergies such as hay fever.
- Major effects of histamine include dilation of blood vessels, inflammation and swelling of tissues, and narrowing of airways.
- In serious cases, life-threatening anaphylactic shock may occur, caused by a dramatic fall in blood pressure. Remarkably, current H1 receptor antagonists, for example., diphenhydramine, bear little if any structural similarity to histamine.
- The main clinical use of H2 receptor antagonists is to inhibit gastric secretion in the treatment of stomach ulcers.
These agents all contain features that relate to the histamine structure, in particular the heterocyclic ring. Cimetidine and ranitidine are the most widely used in this class.
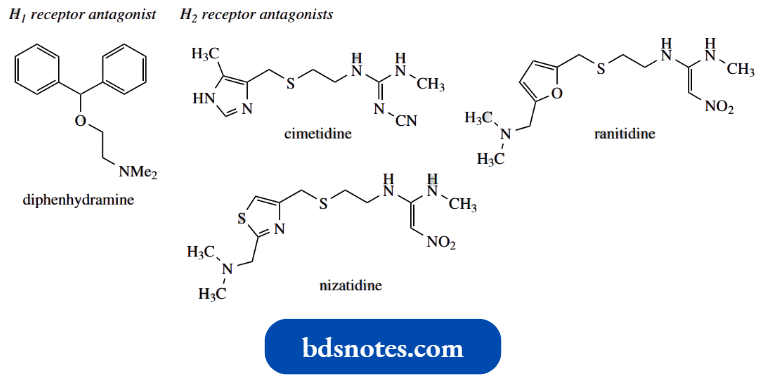
Cimetidine contains an imidazole ring comparable to histamine, a sulfur atom (thioether group) in the side chain, and a terminal functional group based upon a guanidine.
- Ranitidine bears considerable similarity to cimetidine, but there are some important differences.
- The heterocycle is now furan rather than imidazole, and the guanidine has been modified to an amidine. A newer drug, nizatidine, is a variant of ranitidine with a thiazole heterocyclic ring system.
Reactivity Of 1,3-Azoles:
Electrophiles can add to N-3, the azomethine =N–, of 1,3-azoles as they can to the pyridine nitrogen.
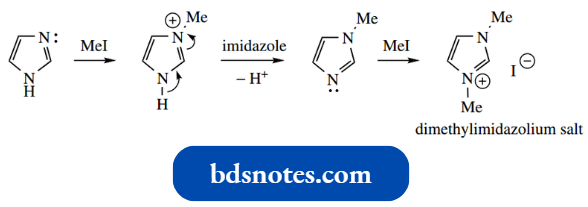
N Alkylation is complicated in the case of imidazole by the possibility of forming a dialkyl imidazolium salt; the first-formed protonated N-alkyl imidazole can be deprotonated by imidazole, then alkylated further.
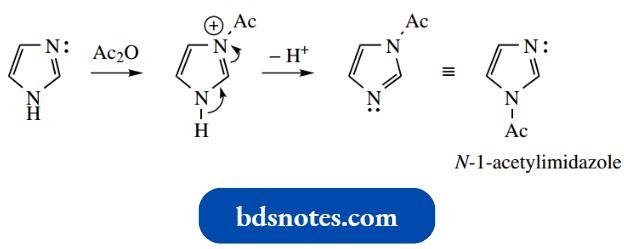
N-acylation is mechanistically similar, and mono-acylation can be accomplished by using two molar equivalents of imidazole to one of the acylating agents, the second mole serving to deprotonate the first-formed N-3-acyl imidazolium salt.
- Note that in Ac dimethylimidazolium salt alkylation and acylation, it is the =N– that acts as the nucleophile; this carries the only lone pair.
However, proton loss occurs from the other nitrogen, giving the impression that the N–H has been alkylated or acylated.
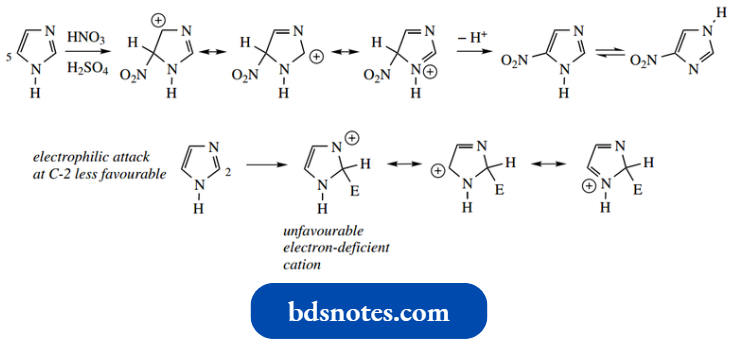
The 1,3-diazoles are much less susceptible to electrophilic substitution than pyrrole, furan, and thiophene, but are more reactive than pyridine. Imidazole is the most reactive and may be nitrated readily.
- Substitution occurs at C-5, but tautomerism then leads to the 4(5) mixture.
- The position of substitution may be predicted from a consideration of resonance structures: attack at C-5 provides maximum delocalization with no particularly unfavorable resonance forms.
There is less delocalization after the attack at C-2; one of the resonance forms has an unfavorable electron-deficient nitrogen.
In general, the 1,3-diazoles do not react by nucleophilic substitution, although imidazole can participate in the Chichibabin reaction with substitution at C-2; the position of substitution is equivalent to that noted with pyridine.
- Nucleophilic species that are strong bases, like sodium amide, are more likely to remove the NH proton (pKa 14).
However, oxazole and thiazole do not have any NH, and the most acidic proton is that at C-2. The electronegative oxygen and sulfur can support an adjacent negative charge.
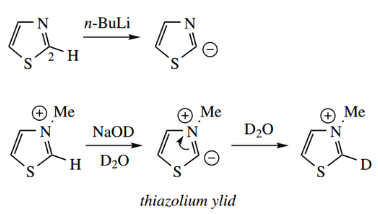
It is found that quaternary salts of 1,3-azoles are deprotonated at C-2 in the same way.
- Rates of deprotonation are considerably faster because of the influence of the quaternary center that provides a favorable inductive effect.
- The conjugate base bearing opposite charges on adjacent atoms is termed a ylid (or ylide; pronounced il-ide). This ylid, with neg negative charge on carbon, is potentially a nucleophilic species.
- Thus, it is found that both oxazoline and thiazolium salts undergo H–D exchange at C-2 remarkably quickly under basic conditions, illustrating very simply this nucleophilic behavior.
The Thiazolium Ring In Thiamine:
Thiamine (vitamin B1), in the form of thiamine diphosphate (TPP), is a coenzyme of considerable importance in carbohydrate metabolism.
- Dietary deficiency leads to the condition beriberi, characterized by neurological disorders, loss of appetite, fatigue, and muscular weakness.
- We shall study several TPP-dependent reactions. At this stage, we should merely examine the structure of thiamine, and correlate its properties with our knowledge of heterocycles.
- Thiamine contains two heterocyclic rings, a pyrimidine, and a thiazole, the latter present as a thiazolium salt.
The pyrimidine portion is unimportant for our understanding of the chemistry of TPP, though it may play a role in some of the enzymic reactions.
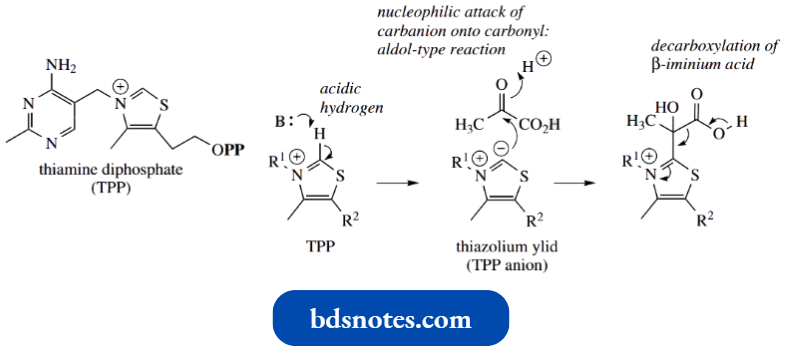
The proton in the thiazolium ring is relatively acidic (pKa about 18) and can be removed by even weak bases to generate the carbanion or ylid; a ylid is a species with positive and negative charges on adjacent atoms.
- This ylid is an ammonium ylid with extra stabilization provided by the sulfur atom. The lid can act as a nucleophile and is also a reasonable leaving group.
- Prominent among TPP-dependent reactions is the oxidative decarboxylation of pyruvic acid to acetyl-CoA; this reaction links the glycolytic pathway to the Krebs cycle.
- The addition of the thiazolium ylid to the carbonyl group of pyruvic acid is the first reaction of this sequence, and this allows the necessary decarboxylation, the positive nitrogen in the ring acting as an electron sink.
In due course, the thiazolium ylid is regenerated as a leaving group. We shall look at this sequence in more detail.
1,2-Azoles: Pyrazole, Isoxazole, And Isothiazole
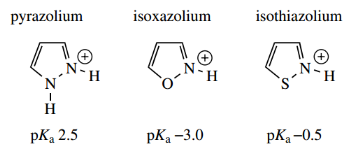
As in the 1,3-azoles, the =N–nitrogen carries a lone pair of electrons and 1,2-azoles are thus potentially basic.
- However, the direct linking of the two heteroatoms has a base-weakening effect.
- Thus, pyrazole has pKa 2.5 and isoxazole pKa − 3.0. The higher basicity in pyrazole is probably related to the symmetry of the contributing resonance structures.
The greater electron-withdrawing effect of oxygen compared with sulfur is reflected in the basicity of isothiazole (pKa−0.5).
Heterocycles Fused To A Benzene Ring:
Many interesting and important heterocyclic compounds contain fused ring systems.
- Some of the common ones are the result of fusing a heterocycle to equivalent resonance structures in a benzene ring, and these have long-established trivial names, for example., indole, quinoline, and isoquinoline.
- Systematic names can be derived by relating to the parent heterocycle and using the prefix benzo to indicate its fusion with benzene.
It is necessary to define which of the bonds in the heterocycle is fused to benzene, and this is accomplished through the use of a bond descriptor, a lowercase italic letter in square brackets.
Thus, indole is benzo[b]pyrrole, quinoline is benzo[b]pyridine, and isoquinoline, an isomer of quinoline with a different type of fusion, becomes benzo[c]pyridine. A few other examples are shown below.
- Note that the bonds of the heterocycle are lettered starting from the heteroatom.
- Where we have two similar heteroatoms, lettering is chosen to produce the lower alternative. Thus, quinazoline is benzo[d]pyrimidine, not benzo[e]pyrimidine.
Where the heteroatoms are different, just as we number from the atom of higher atomic number, we also letter from the same atom. Hence, benzo[d]isoxazole is quite different from benzo[c]isoxazole.
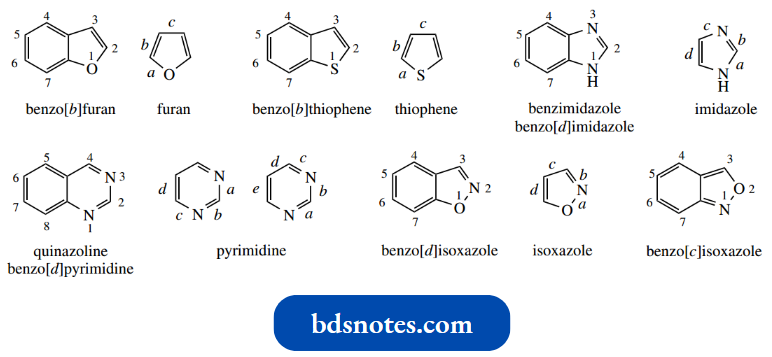
The final fused ring system is then given a completely new numbering system, different from that of the heterocycle.
- Typically, this starts adjacent to the bridgehead atom and then proceeds around the fused ring. The major criterion is to generate the lowest number for the first heteroatom.
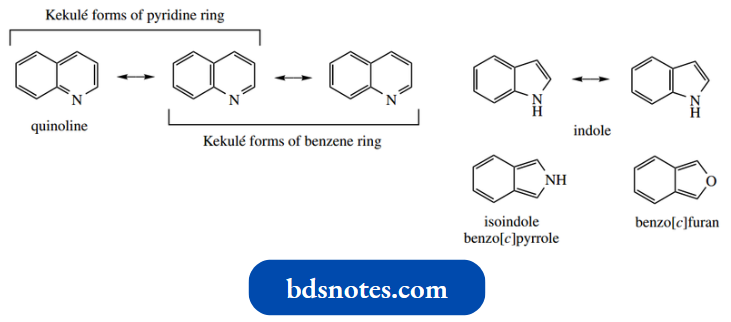
- Note that, in most cases, we have little regard for Kekulé forms of pyridine rings in which the Kekule form of a benzene or pyridine ring is drawn. The three versions of quinoline shown are simply contributing resonance forms.
However, some structures, such as isoindole, benzo[c]furan, or benzo[c]isoxazole above, can only be drawn in one way without invoking charge separation.
Quinoline And Isoquinoline
Quinoline and isoquinoline are benzopyridines. They behave by showing the reactivity associated with either the benzene or the pyridine rings.
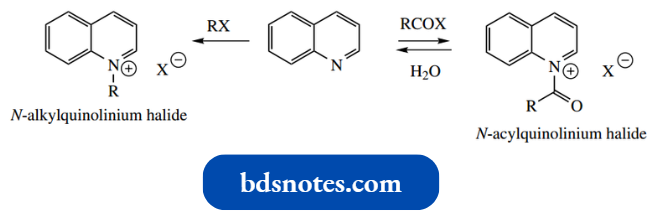
- Quinoline is basic with a pKa of 4.9, similar to that of pyridine (pKa 5.2).
- As with pyridine, the nitrogen carries a lone pair in an sp2 orbital. Alkyl halides and acyl halides also react with nitrogen to give N-alkyl- and N-acyl quinolinium salts.
The N-alkyl salts are stable, but the N-acyl salts hydrolyze rapidly in the presence of water.
Quinoline is much more reactive towards elec trophilic substitution than pyridine, but this is because substitution occurs on the benzene ring, not on the pyridine.
- We have already seen that pyridine carbons are unreactive towards electrophilic reagents, with strongly acidic systems protonating the nitrogen first, further inhibiting the reaction.
- This is again true in quinoline, so that the protonated system is involved in the reaction, and the benzene ring undergoes substitution.
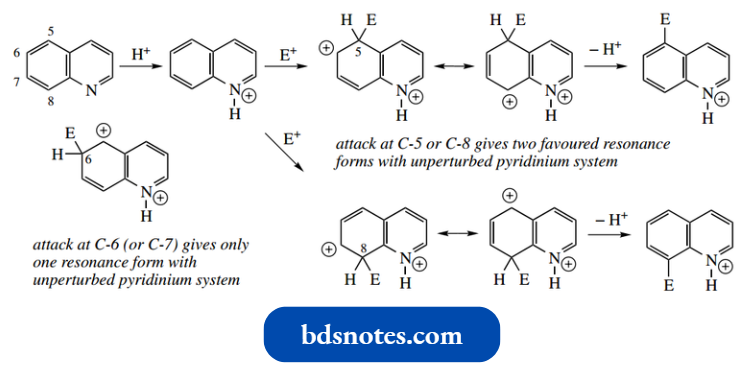
With a nitrating mixture of HNO3 –H2SO4, the products are 5- and 8-nitroquinoline in roughly equivalent amounts.
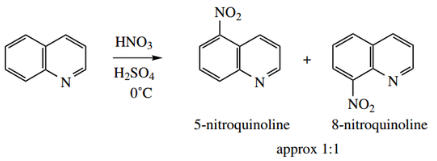
This may be rationalized by considering the stability of intermediate addition cations.
- When the electrophile attacks at C-5 or C-8, the intermediate cation is stabilized by resonance, each having two favorable forms that do not perturb the aromaticity of the pyridinium system.
- In contrast, for attack at C-6 or C-7, there is only one such resonance form. We used similar reasoning to explain why naphthalene undergoes preferential electrophilic substitution at the α-positions.
Whilst we may be a little unhappy about the protonation of a quinolinium cation to an intermediate that carries two positive charges, we find that N-methylquinolinium salts also undergo nitration at a similar rate to quinoline; so this mechanism appears correct.
Nucleophilic substitution occurs at C-2, and to a lesser extent C-4, as might be predicted from similar reactions with pyridine.
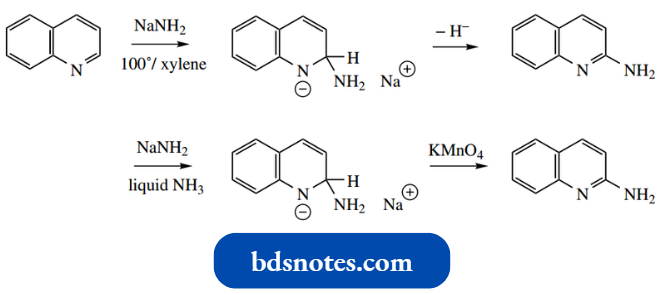
- Chichibabin amination occurs rather more readily than with pyridine, ggiving2-aminoquinoline.
- A typical hydride abstraction process occurs when quinoline is heated with sodium amide.
However, better yields have been achieved by performing the reaction at low temperatures in liquid ammonia solvent and then oxidizing the intermediate dihydroquinoline salt using potassium permanganate.
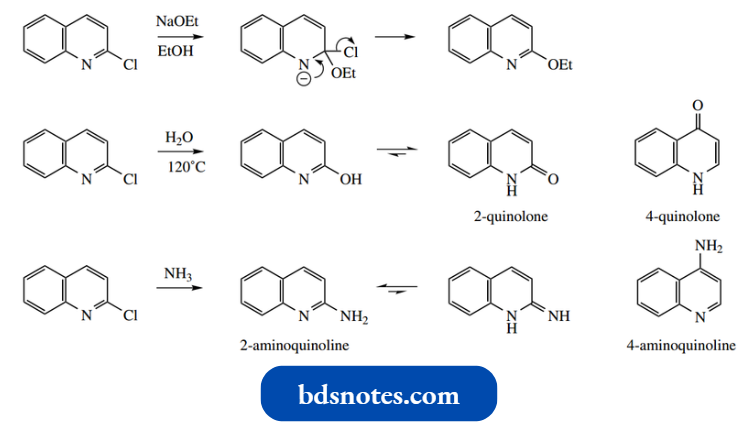
Quinolines carrying 2- or 4-halo substituents undergo nucleophilic substitution readily, in the same manner as 2- and 4-halo pyridines.
- Hydroxyquinolines with the hydroxyl at positions 2 or 4 exist mainly in the carbonyl form, i.e. 2-quinolone and 4-quinolone.
Note, however, that hydroxyls on the benzene ring would be typical phenols. Again, aminoquinolines follow the pyridine precedent, and the tautomeric imino forms are not observed.
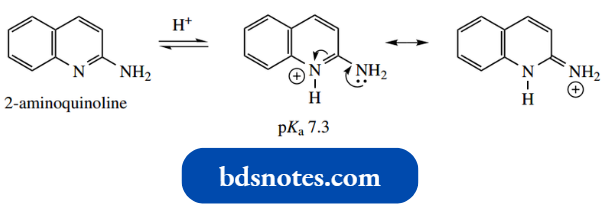
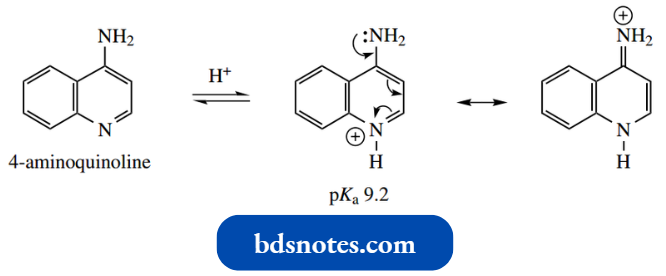
Both 2-aminoquinoline and 4-aminoquinoline pro tonate first on the ring nitrogen, with 4-aminoquin oline being the more basic, the conjugate acid benefiting from increased charge distribution through H+ resonance.
No such resonance structures can be drawn for 3-aminoquinoline, which is much less basic (pKa 4.9).
Quinolone Antibiotics:
The quinolone antibiotics feature as the one main group of antibacterial agents that is synthetic, and not derived from or based upon natural products, as are penicillins, cephalosporins, macrolides, tetracyclines, and aminoglycosides.
The first of these compounds to be employed clinically was nalidixic acid; more recent drugs in current use include ciprofloxacin, norfloxacin, and ofloxacin
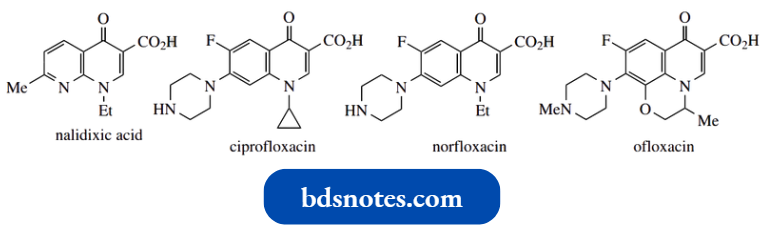
‘Quinolone’ as a descriptor is an oversimplification, since nalidixic acid contains two fused pyridine rings rather than a benzopyridine, and ofloxacin has a morpholine ring fused to the quinolone.
- Nevertheless, the quinolone substructure is generally used when referring to this group of antibiotics.
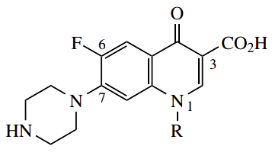
The most important structural features for good antibacterial activity are a carboxylic acid at position 3, a small alkyl group at position 1, a 6-fluorine substituent, and a nitrogen heterocycle, often piperazine, at position 7.
Quinolones are good general antibiotics for systemic infections, and they are particularly useful for urinary tract infections because high concentrations are excreted into the urine.
- The mode of action involves interference with DNA replication by inhibiting DNA gyrase, a bacterial enzyme related to mammalian topoisomerases that breaks and reseals double-stranded DNA during replication.
- Isoquinoline (pKa 5.4) has a similar basicity to quinoline and pyridine and also undergoes N-alkylation and N-acylation.
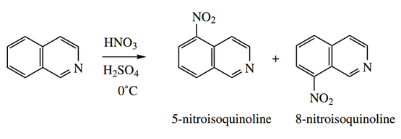
Nitration occurs smoothly to give predominantly 5-nitroisoquinoline; the isoquinolinium cation reacts more readily than the quinolinium cation.
Nucleophilic substitution occurs exclusively at position 1 in isoquinoline; the alternative position C-3 is quite unreactive.
This is explained by the loss of benzene resonance in the intermediate anion. Thus, Chichibabin amination gives 1-aminoisoquinoline.
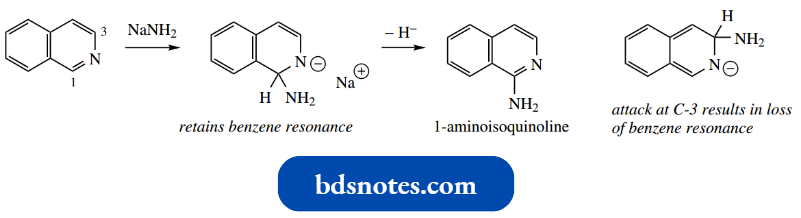
Substitution with displacement of halide occurs readily at C-1 and much less readily at C-3 for the same reasons, i.e. the loss of benzene resonance if C-3 is attacked.
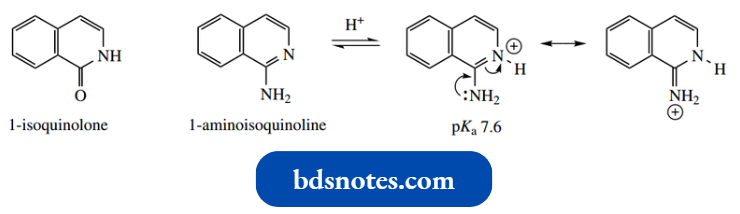
1-Isoquinolone exists completely in the carbonyl form, whereas 1-amino isoquinoline is the normal tautomer.
The basicity of 1-amino isoquinoline (pKa 7.6) is similar to that of 2-aminoquinoline (pKa 7.3).
Indole
Indole is the fusion of a benzene ring with a pyrrole. Like quinoline and isoquinoline, indole behaves as an aromatic compound.
- However, unlike quinoline and isoquinoline, where the reactivity was effectively part benzene and part pyridine, the reactivity in indole is modified by each component of the fusion.
- The closest similarity is between the chemistry of pyrroles and indoles. Indoles, like pyrroles, are very weak bases. The conjugate acid of indole has pKa − 3.5; that of pyr role has pKa − 3.8.
As in the case of pyrrole, nitrogen has already contributed its lone pair to the aromatic sextet, so N-protonation would necessarily destroy aromaticity in the five-membered ring.
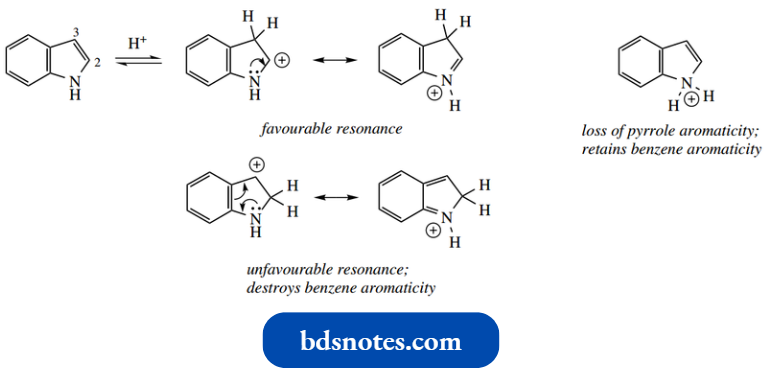
Nevertheless, an equilibrium involving the N-protonated cation is undoubtedly set up, since acid-catalyzed deuterium exchange of the N hydrogen occurs rapidly, even under very mild acidic conditions.
- Protonation eventually occurs preferentially on carbon, as with pyrrole; but there is a difference, in that this occurs on C-3 rather than on C-2. This is the influence of the benzene ring.
- It can be seen that protonation on C-3 allows resonance in the five-membered ring and charge localization on nitrogen.
In contrast, any resonance structure from protonation at C-2 destroys the benzene ring aromaticity.
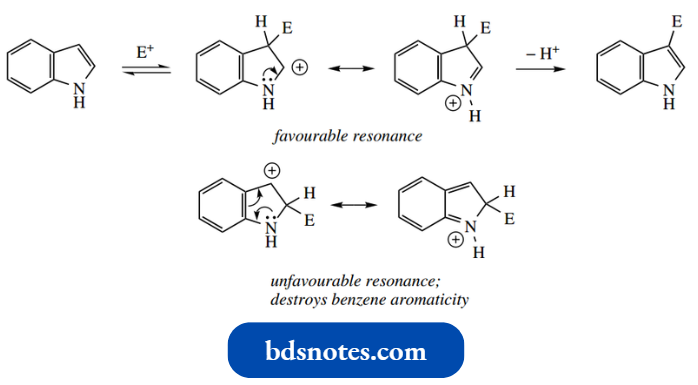
Indole is very reactive towards electrophiles, and it is usually necessary to employ reagents of low reactivity. Nitration with HNO3 –H2SO4 is unsuccessful (compare pyrrole), but can be achieved using benzoyl nitrate.
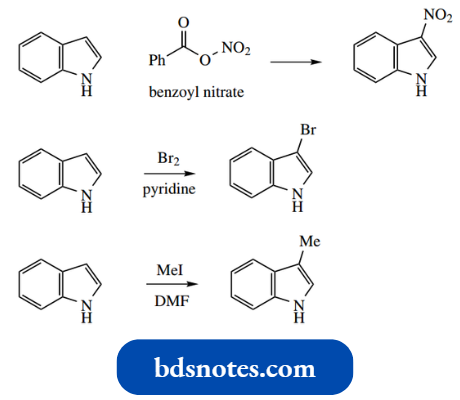
It is also possible to brominate and methylate at C-3; however, conditions must be controlled carefully, since further electrophilic reactions may then occur.
Treatment with acetic anhydride leads to 1,3-diacetyl indole. Indole reacts readily as the nucleophile in Mannich reactions. This provides convenient access to other derivatives, as shown below.
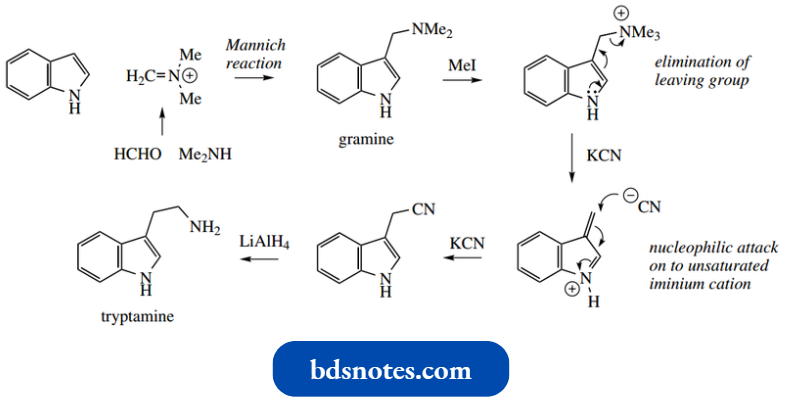
Thus, quaternization at the side-chain nitrogen allows the ready elimination of trimethylamine. This is facilitated by the electron-releasing ability of the indole nitrogen and can be brought about by a mild base.
- By choosing KCN as the mild base, the transient 3-methyleneindoleninium salt can be trapped by cyanide nucleophiles, leading to indole acetonitrile.
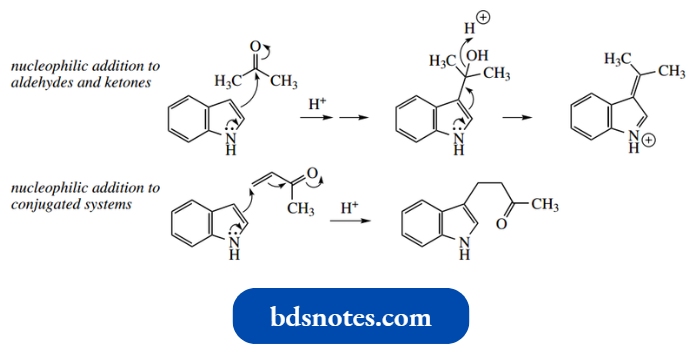
- Reduction of the nitrile group with LAH provides a route to tryptamine. A simple addition to carbonyl compounds occurs under mild acidic conditions.
- Examples are given illus trate reaction with acetone, an aldol-like reaction, and conjugate addition to methyl vinyl ketone, a Michael-like reaction.
The first-formed alcohol products in aldol-like reactions usually dehydrate to give a 3- 3-alkylidene-3H-indolium cation.
We noted above that pyrrole, though a very weak base, is potentially acidic (pKa 17.5).
- This was because the anion formed by losing the proton from nitrogen has a negative charge on the relatively electronegative nitrogen, but maintains its aromaticity.
The indole anion is also formed by the loss of the N–H proton (pKa 16.2) using sodium amide sodium hydride, or even a Grignard reagent as a base.
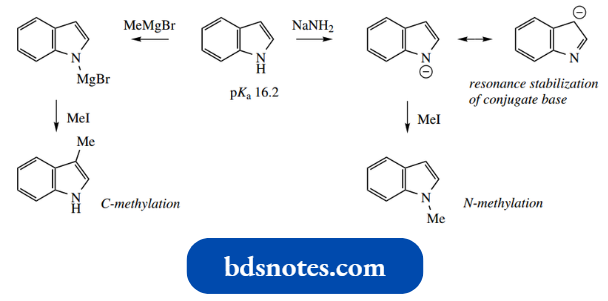
The indole anion is resonance stabilized, with a negative charge localized mainly on nitrogen and C-3.
- It can now participate as a nucleophile, for example., in alkylation reactions. However, this can lead to N-alkylation or C-alkylation at C-3.
- Which is the predominant product depends upon several variables; but, as a general rule, if the associated metal cation is sodium, then the anion is attacked at the site of highest electron density, i.e. the nitrogen.
- Where the cation is magnesium, i.e. the Grignard reagent, then the partial covalent bonding to nitrogen prevents attack there, and the reaction occurs at C-3.
Indoles In Biochemistry:
Some rather important indole derivatives influence our everyday lives. One of the most common ones is tryptophan, an indole-containing amino acid found in proteins.
- Only three of the protein amino acids are aromatic, the other two, phenylalanine and tyrosine are simple benzene systems.
- None of these aromatic amino acids is synthesized by animals and they must be obtained in the diet. Despite this, tryptophan is surprisingly central to animal metabolism.
It is modified in the body by decarboxylation (see Box 15.3) and then hydroxylation to 5-hydroxytryptamine (5-HT, serotonin), which acts as a neurotransmitter in the central nervous system.
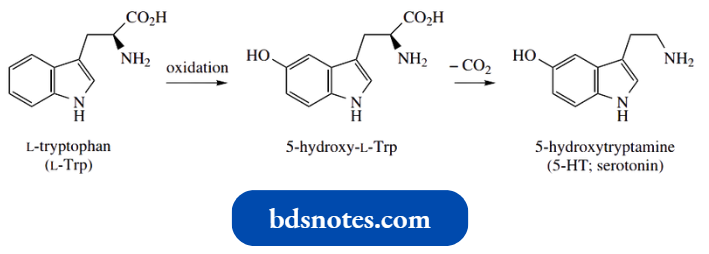
Serotonin mediates many central and peripheral physiological functions, including contraction of smooth muscle, vasoconstriction, food intake, sleep, pain perception, and memory, a consequence of it acting on several distinct receptor types.
- Although 5-HT may be metabolized by monoamine oxidase, platelets and neurons possess a high-affinity mechanism for reuptake of 5-HT.
- This mechanism may be inhibited by the widely prescribed antidepressant drugs termed selective serotonin reuptake inhibitors (SSRI), for example., fluoxetine (Prozac), thereby increasing levels of 5-HT in the central nervous system.
- Migraine headaches that do not respond to analgesics may be relieved by the use of an agonist of the 5-HT1 receptor since these receptors are known to mediate vasoconstriction.
- Though the causes of migraine are not clear, they are characterized by dilation of cerebral blood vessels.
- 5-HT1 agonists based on the 5-HT structure in current use include the sulfonamide derivative sumatriptan, and the more recent agents naratriptan, rizatriptan, and zolmitriptan.
These are of considerable value in treating acute attacks.
Several of the ergot alkaloids also interact with 5-HT receptors. Some are used medicinally, but the most notorious is the semi-synthetic derivative lysergic acid diethylamide (LSD).
- This is itself an indole derivative, though the indole is part of a more complex fused-ring system.
- Nevertheless, from the structural similarities, it is not difficult to see why LSD might trigger 5-HT receptors.
- It has the additional ability to interact with noradrenaline and dopamine receptors, thus generating a complex pharmacological response.
- LSD is probably the most powerful psychotomimetics known, intensifying and distorting perceptions.
Experiences can vary from beautiful visions to living nightmares, and no two ‘trips’ are alike.
Also known to be hallucinogenic are the indole derivatives psilocin and psilocybin found in the so-called magic mushrooms, Psilocybe species.
- Ingestion of these small fungi causes visual hallucinations with rapidly changing shapes and colors.
- Psilocybin is the phosphate of psilocin; although based on 4-hydroxytryptamine, it also acts on 5-HT receptors.
Melatonin is N-acetyl-5-methoxytryptamine, a simple derivative of serotonin. It is a natural hormone secreted by the pineal gland in the brain during the hours of darkness.
- It is involved in controlling the body’s day-night rhythm, the ability to sleep during the night, and to stay awake during the day.
- When given as a drug, melatonin induces sleep and adjusts the internal body clock. It is now used as a means of reducing the effects of jet lag
- Plants also require hormones to trigger their growth patterns. One of these is indole-3-acetic acid, which controls cell elongation and is produced in the growing shoot tips.
- One of the major subdivisions of plant alkaloids is termed the indole alkaloid group. All contain the basic indole heterocycle, and many have valuable pharmacological activity that can be exploited in drug materials.
- The indole portion is very often fused to another heterocycle; we shall see some typical structures, where we shall consider them under fused heterocycles.
Fused Heterocycles
There is ample scope for increasing structural complexity by fusing two or more heterocycles.
- Shown below are a few of the ring systems encountered in natural compounds, many of which have interesting, and potentially useful, biological properties.
- Note that in some cases the rings are fused so that the heteroatom can be at the ring junction and is thus common to both rings.
This gives us even more combinations.
We do not wish to consider these further, but instead, we shall concentrate on just two groups of fused heterocycles of particular importance, the purines and pteridines. Both purine and pteridine are parent heterocycles for nomenclature purposes.
- The systematic procedure for naming fused heterocycles is an extension of what we saw when we considered a benzene ring fused to a heterocycle. The main difference is that we have to identify bonds in two different rings to indicate the fusion.
- We use lettering for bonds in one heterocycle and numbers for bonds in the other. Numbering is used for the ‘sub stituent’ ring and lettering for the ‘root’ ring, and all are put in square brackets between substituent and root.
We do not wish to include a great amount of detail, but we shall use purine and pteridine to illustrate the approach and to provide a modest level of familiarity when such names are encountered.
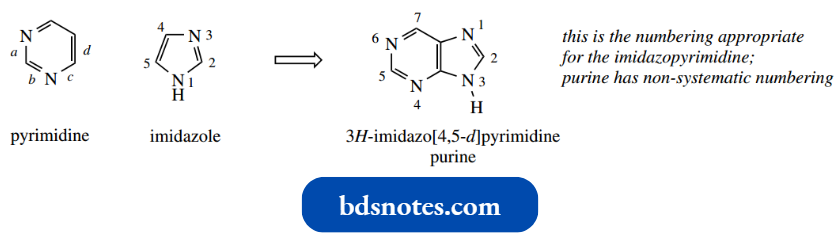
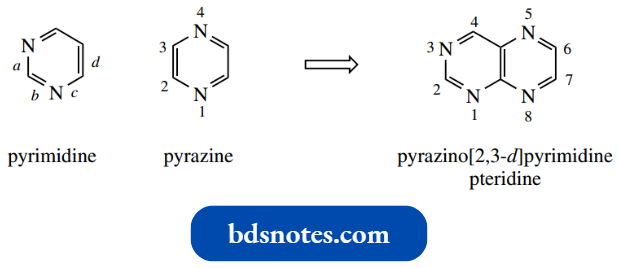
The fused heterocycle is then given its numbering system, starting adjacent to a bridgehead atom to generate the lowest number for the first heteroatom. nucleic acids are bonded to sugar through N-9, the additional potential for tautomerism in the imidazole ring is no longer of concern.
Fused Heterocycles Purines:
Purines, along with pyrimidines, feature as bases in nucleic acids, DNA, and RNA. A purine is the product of fusing a five-membered imidazole ring onto a six-membered pyrimidine ring.
The accepted numbering system unfortunately is non-systematic and treats purine as a pyrimidine derivative, the pyrimidine ring being numbered first and separately from the other ring.
Purine in solution exists as a roughly equimolar mixture of two tautomeric forms, 9H-purine and 7H purine, tautomerism involving the imidazole ring as we have noted earlier.
- The purine systems in nucleic acids, adenine, and guanine, are aminopurines.
- The amino group is on the pyrimidine ring and, as with aminopyrimidines, these compounds exist as their amino tautomers.
Guanine also has an oxygen substituent on the pyrimidine ring, and this adopts the carbonyl form, also following the behavior of oxypyrimidines.
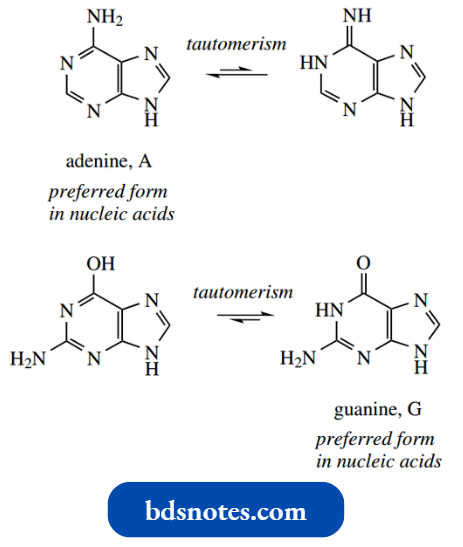
Because adenine and guanine in possible N-protonated forms are produced. This is perhaps unexpected, in that protonation on N-7 would provide a cation that is resonance stabilized in the imidazole ring.
- However, the observed pKa more closely resembles that of pyrimidine (pKa 1.3) rather than that of imidazole (pKa 7.0).
- Amino groups increase basicity (adenine pKa 4.3), though the oxygen substituent in guanine reduces the effect of the amino group (guanine pKa 3.3).
In the aminopurines, the position of protonation appears to be N-1 in adenine, whereas it is N-7 in guanine; this presumably reflects the opposing effects provided by amino groups (electron donating) and carbonyl groups (electron withdrawing).
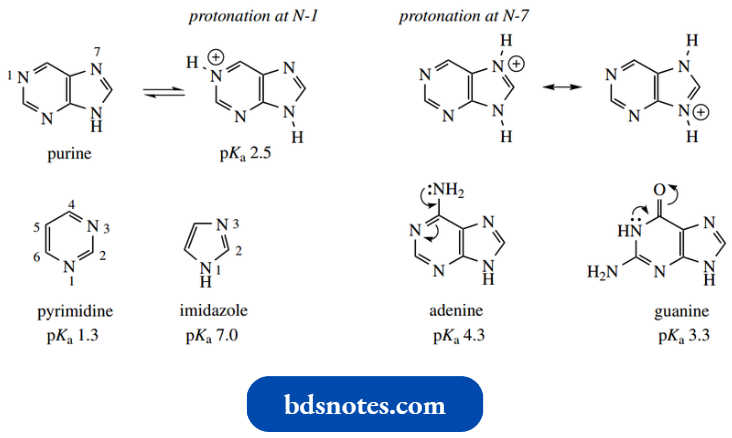
Purine has an acidic pKa of 8.9, making it somewhat more acidic than phenol (pKa 10), and a stronger acid than imidazole (pKa 14.2). The N-9 proton is lost giving an anion with substantial resonance delocalization of charge.
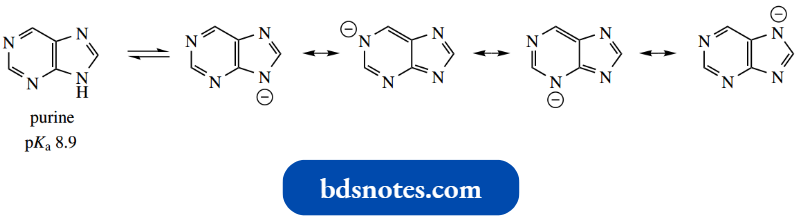
The acidities of adenine (pKa 9.8) and guanine (pKa 9.9) are similar, though different protons are removed.
Adenine loses the N-9 proton, but guanine is ionized at N-1. N-1 is part of an amide-like system, and the charge in the conjugate base can be delocalized to the more favorable electronegative oxygen. The effect is most pronounced in uric acid, a metabolite of purines.
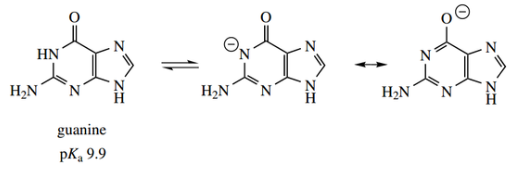
Uric Acid, A Purine Metabolite:
Nucleic acid degradation in humans and many other animals leads to the production of uric acid, which is then excreted.
The process initially involves purine nucleotides, adenosine, and guanosine, which are combinations of adenine or guanine with ribose. The purine bases are subsequently modified as shown.
The amino groups are replaced with oxygen. Although there is a biochemical reaction, the same can be achieved under acid-catalyzed hydrolytic conditions and resembles the nucleophilic substitution on pyrimidines.
- The first-formed hydroxy derivative would then tautomerize to the carbonyl structure. In the case of guanine, the product is xanthine, whereas adenine leads to hypoxanthine.
- The latter compound is also converted into xanthine by an oxidizing enzyme, xanthine oxidase. This enzyme also oxidizes xanthine at C-8, giving uric acid.
- Uric acid is not a carboxylic acid but is a relatively strong acid with pKa 5.8. It has an ‘all-amide’ structure, and there are four potential sites for the loss of a proton.
Deprotonation occurs at N-9. Loss of this proton generates a conjugate base in which the charge can be delocalized to oxygen, giving maximum charge distribution. One resonance form is particularly favorable in having aromaticity in both rings.
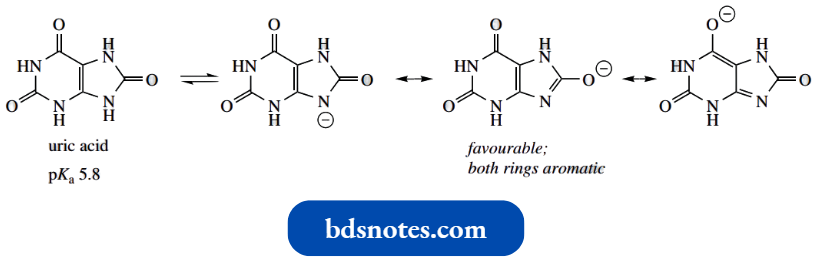
Impaired purine metabolism can lead to a build-up of uric acid, and deposition of salts of uric acid as crystals in the joints.
- This causes the painful condition known as gout. One way of treating gout is to reduce uric acid biosynthesis by specific inhibition of the enzyme xanthine oxidase.
- The hypoxanthine analog allopurinol is a drug that is used for this purpose. Allopurinol resembles hypoxanthine, though it contains a pyrazole ring rather than an imidazole ring.
Allopurinol is oxidized by the enzyme to alloxanthine. This product then acts as an inhibitor of the enzyme, binding to the enzyme, but not being modified further and not being released.
Caffeine, Theobromine, And Theophylline:
After the nucleic acid purines adenine and guanine, the next most prominent purine in our everyday lives is probably caffeine.
- Caffeine, in the form of beverages such as tea, coffee, and cola, is one of the most widely consumed and socially accepted natural stimulants.
- Closely related structurally are theobromine and theophylline. Theobromine is a major constituent of cocoa and related chocolate products.
Caffeine is also used medicinally, but theophylline is much more important as a drug compound because of its muscle relaxant properties, utilized in the relief of bronchial asthma.
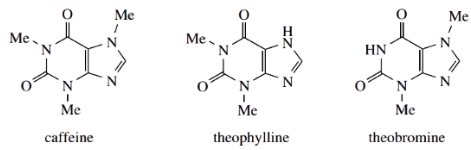
These compounds competitively inhibit phosphodiesterase, resulting in an increase in cyclic AMP and subsequent release of adrenaline.
- This leads to the major effects: a stimulation of the central nervous system (CNS), a relaxation of bronchial smooth muscle, and induction of diuresis.
- These effects vary in the three compounds. Caffeine is the best CNS stimulant and has weak diuretic action. Theobromine has little stimulant action but has more diuretic activity and also muscle relaxant properties.
- Theophylline also has low stimulant action and is an effective diuretic, but it relaxes smooth muscle better than caffeine or theobromine.
- It has been estimated that beverage consumption may provide the following amounts of caffeine per cup or average measure: coffee, 30–150 mg (average 60–80 mg); instant coffee, 20–100 mg (average 40–60 mg); decaffeinated coffee, 2–4 mg; tea, 10–100 mg (average 40 mg); cocoa, 2–50 mg (average 5 mg); cola drink, 25–60 mg.
- The maximal daily intake should not exceed about 1 g to avoid unpleasant side effects, for example., headaches, and restlessness. An acute lethal dose is about 5–10 g.
Caffeine and theobromine may be obtained in large quantities from natural sources, or they may be obtained by total or partial synthesis. Theophylline is usually produced by total synthesis.
Pteridines:
In pteridines, we have a pyrimidine ring fused to a pyrazine ring. There are, of course, several possible ways of combining these two six-membered ring systems; pteridines are pyrazino[2,3- d]pyrimidines.
- We do not want to consider the chemistry of the pteridine ring system here, but instead, we shall look at the structures of two rather important pteridine-based biochemicals, namely folic acid and riboflavin.
- In the latter case, the pteridine is also fused further to a benzene ring, giving an even more complex ring system, a benzo[g]pteridine.
The accompanying diagram shows the derivation of the fused-ring nomenclature. The oxygenated form of the benzopteridine found in riboflavin is also called an isoalloxazine.
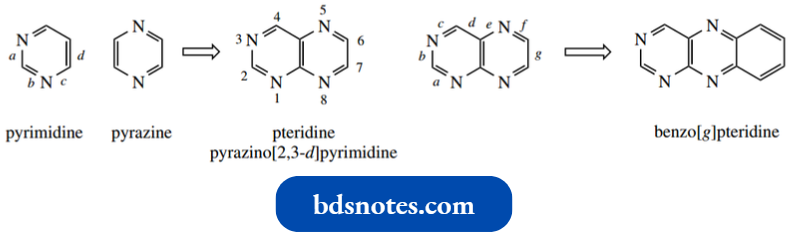
Folic acid (vitamin B9) is a conjugate of a pteridine unit, p-aminobenzoic acid, and glutamic acid. Deficiency of folic acid leads to anemia, and it is also standard practice to provide supplementation during pregnancy to reduce the incidence of spina bifida.
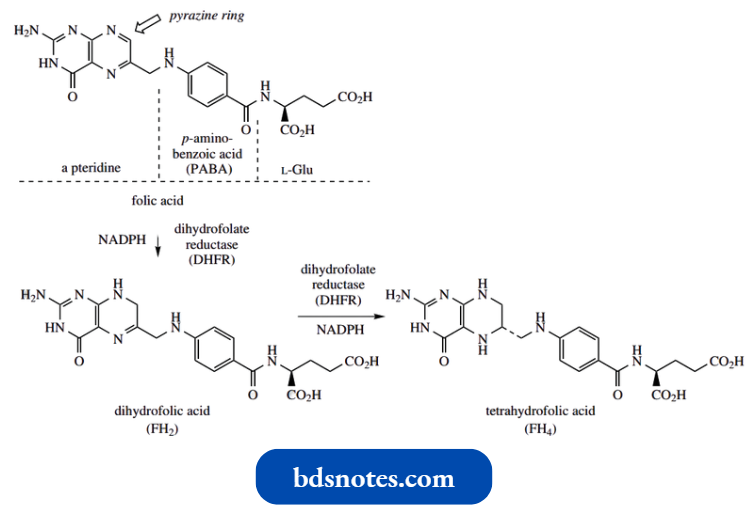
Folic acid becomes sequentially reduced in the body by the enzyme dihydrofolate reductase to give dihydrofolic acid (FH2) and then tetrahydrofolic acid (FH4). Reduction occurs in the pyrazine ring portion.
- Tetrahydrofolic acid then functions as a carrier of one-carbon groups for amino acid and nucleotide metabolism.
- The basic ring system can transfer methyl, methylene, methenyl, or formyl groups, and it utilizes slightly different reagents as appropriate.
- These are shown here; for convenience, we have left out the benzoic acid–glutamic acid portion of the structure.
These compounds are all interrelated, but we are not going to delve any deeper into the actual biochemical relationships.
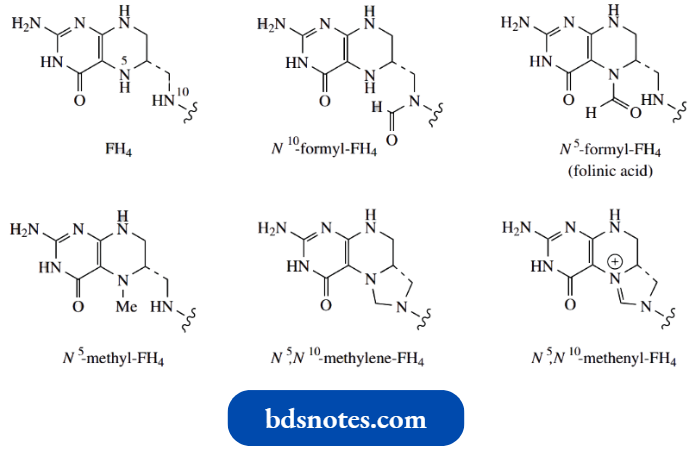
In any case, you might be able to analyze some of the relationships on a purely chemical basis. For example, tetrahydrofolic acid reacts readily and reversibly with formaldehyde to produce N5 and N10-methylene-FH4.
- You could consider N-5 of the reduced pteridine ring reacting with formaldehyde (a one-carbon reagent) to give an iminium cation, which could then cyclize via nucleophilic attack of N-10.
- We might also consider reducing the iminium cation with, say, borohydride to give the N-methyl derivative.
These are not necessarily the same as what is occurring in the enzymic reactions, but they should help to make the structures appear rather more familiar.
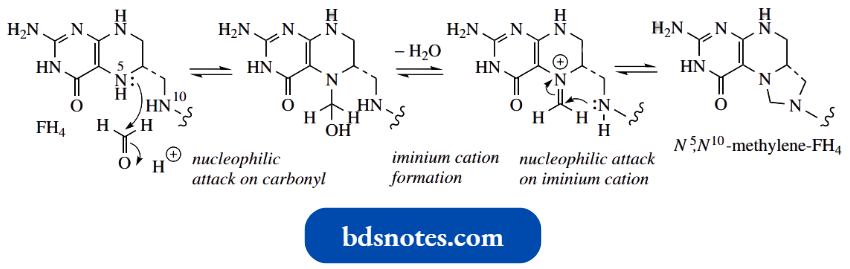
Now we have seen that the usual reagent for biological methylations is S-adenosylmethionine (SAM).
One occasion where SAM is not employed, for fairly obvious reasons, is the regeneration of methionine from homocysteine, after a SAM methylation. For this, N5-methyl-FH4 is the methyl donor, with vitamin B12 also playing a role as a coenzyme.
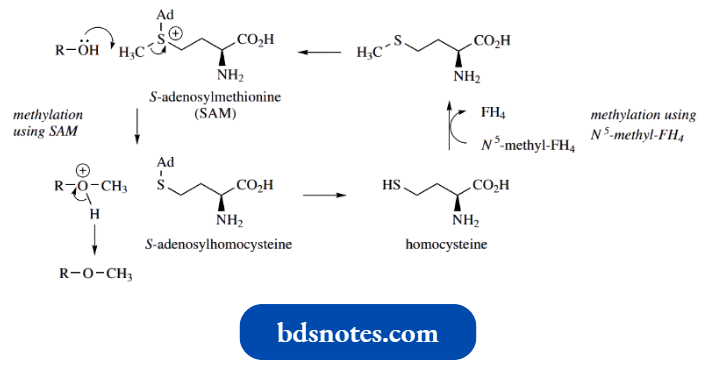
Another vitally important methylation reaction involving folic acid derivatives is the production of the nucleic acid base thymine from uracil.
- Uracil is found in RNA, and thymine is a component of DNA; thymine is the methyl derivative of uracil. For continuing DNA synthesis, it is necessary to methylate uracil.
- In practice, it is the nucleotide deoxyuridylate (dUMP) that is methylated to deoxy thymidylate (dTMP). The methylating agent employed here is N5, N10-methylene-FH4. As a consequence of this reaction, N5, N10– methylene-FH4 is converted into dihydrofolic acid.
- To keep the reaction flowing, this is reduced to FH4, and further N5, N10-methylene-FH4 is produced using a one-carbon reagent.
In this process, the one-carbon reagent comes from the amino acid serine, which is transformed into glycine by the loss of its hydroxymethyl group. The chemistry of the transformations is fairly complex and outside our requirements.
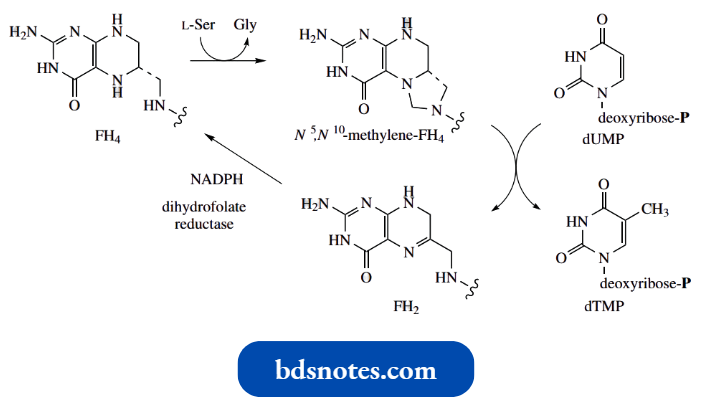
Folic acid derivatives are essential for DNA synthesis, in that they are cofactors for certain reactions in purine and pyrimidine biosynthesis, including the uracil–thymine methylation just described.
- They are also cofactors for several reactions relating to amino acid metabolism.
- The folic acid system thus offers considerable scope for drug action. Mammals must obtain their tetrahydrofolate requirements from their diet, but microorganisms can synthesize this material.
- This offers scope for selective action and led to the use of sulfanilamide and other antibacterial sulfa drugs, compounds that competitively inhibit the biosynthetic enzyme (dihydropteroate synthase) that incorporates p-aminobenzoic acid into the structure.
- Rapidly dividing cells need an abundant supply of dTMP for DNA synthesis, and this creates a need for dihydrofolate reductase activity.
Specific dihydrofolate reductase inhibitors have become especially useful as antibacterials, for example., trimethoprim, and antimalarial drugs, for example., pyrimethamine.
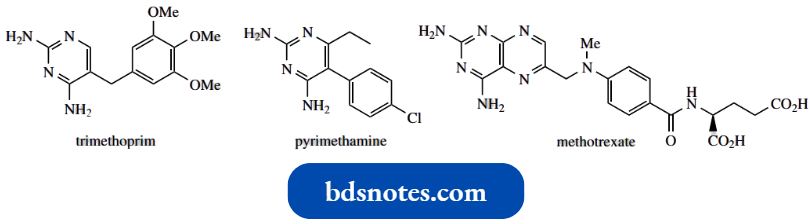
These are pyrimidine derivatives and are effective because of differences in susceptibility between the enzymes in humans and the infective organism.
- Anticancer agents based on folic acid, for example., methotrexate, inhibit dihydrofolate reductase, but they are less selective than the antimicrobial agents and rely on a stronger binding to the enzyme than the natural substrate has.
- They also block pyrimidine biosynthesis. Methotrexate treatment is potentially lethal to the patient and is usually followed by ‘rescue’ with folinic acid (N5-formyl-tetrahydrofolic acid) to counteract the folate-antagonist action.
The rationale is that folinic acid ‘rescues’ normal cells more effectively than it does tumor cells.
Riboflavin:
Riboflavin (vitamin B2) is a component of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), coenzymes that play a major role in oxidation-reduction reactions.
- Many key enzymes involved in metabolic pathways are covalently bound to riboflavin and are thus termed flavoproteins.
- Riboflavin contains an isoalloxazine ring linked to the reduced sugar ribitol.
The sugar unit in riboflavin is the non-cyclic ribitol so FAD and FMN differ somewhat from the nucleotides we encounter in nucleic acids.
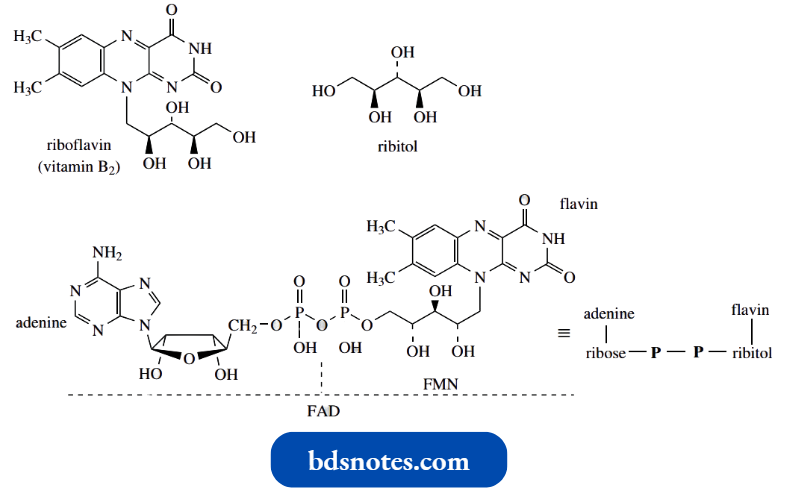
Riboflavin is widely available in foods; dietary deficiency is uncommon, but it manifests itself in skin problems and eye disturbances.
The flavin nucleotides are typically involved in the oxidations creating double bonds from single bonds.
The flavin takes up two hydrogen atoms, represented in the figure as being derived by the transfer of hydride from the substrate and a proton from the medium.
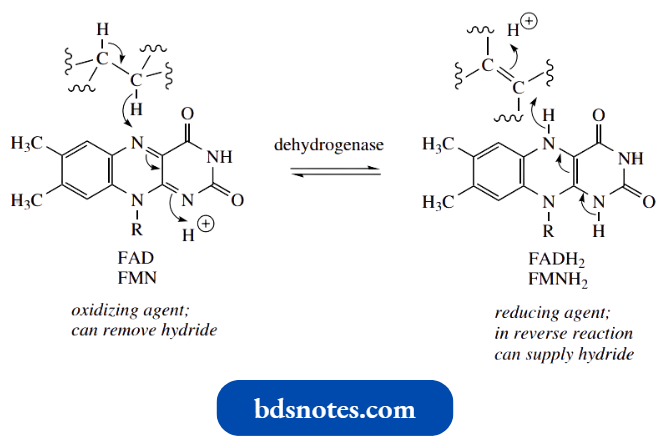
Reductive sequences involving flavoproteins may be represented as the reverse reaction, where hydride is transferred from the coenzyme, and a proton is obtained from the medium.
- The reaction mechanism shown here is in many ways similar to that in NAD+ oxidations, i.e. a combination of hydride and a proton; it is less easy to explain adequately why it occurs, and we do not consider any detailed explanation advantageous to our studies.
- We should register only that the reaction involves the N=C–C=N function that spans both rings of the pteridine system.
Some Classic Aromatic Heterocycle Syntheses:
Our study of heterocyclic compounds is directed primarily to an understanding of their reactivity and importance in biochemistry and medicine.
- The synthesis of aromatic heterocycles is not, therefore, a main theme, but it is useful to consider just a few examples to underline the application of reactions in earlier chapters.
- From the beginning, we should appreciate that the synthesis of substituted heterocycles is probably not best achieved by carrying out substitution reactions on the simple heterocycle.
- It is often much easier and more convenient to design the synthesis so that the heterocycle already carries the required substituents, or has easily modified functions.
We can consider two main approaches for heterocycle synthesis, here using pyridine and pyrrole as targets.
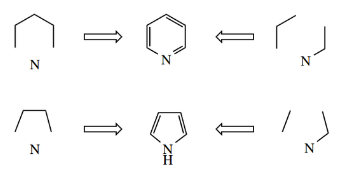
We can insert the heteroatom into the rest of the carbon skeleton, or attempt to join two units, one of which contains the heteroatom, using C–C and C–heteroatom linkages.
- To make the new bonds, two reaction types are most frequently encountered.
- Heteroatom–C bond formation is achieved using the heteroatom as a nucleophile to attack an electrophile such as a carbonyl group.
- Aldol-type reactions may be exploited for C–C bond formation, employing enamines and enols/enolate anions.
- We shall now look at some synthetic procedures that merit the descriptor ‘classic’ because of their general application, and their longevity – some have been around for more than 100 years.
Do not worry about remembering the names: these commemorate the originators, and we should instead concentrate on the chemistry, which we shall see is usually a combination of processes we have already met.
Hantzsch Pyridine Synthesis
In its simplest form, this consists of the condensation of a β-ketoester with an aldehyde and ammonia.
- The product is a 1,4-dihydropyridine, which is subsequently transformed into pyridine by oxidation.
- Several separate reactions occur during this synthesis and the precise sequence of events may not be quite as shown below – they may be in a different order.
- The normal Hantzsch synthesis leads to a symmetrical product. The diesters formed may be hydrolyzed and decarboxylated using the base to give pyridines with less substitution.
Note that we are using the ester groups as activating species to facilitate enolate anion chemistry
Skraup Quinoline Synthesis:
The most general method for synthesizing quinolines employs aniline or a substituted aniline, glycerol, sulfuric acid, and an oxidizing agent such as a ferric salt or nitrobenzene.
The first step is acid-catalyzed dehydration of glycerol to the unsaturated aldehyde acrolein. Variations of the Skraup synthesis use different acroleins instead of glycerols.
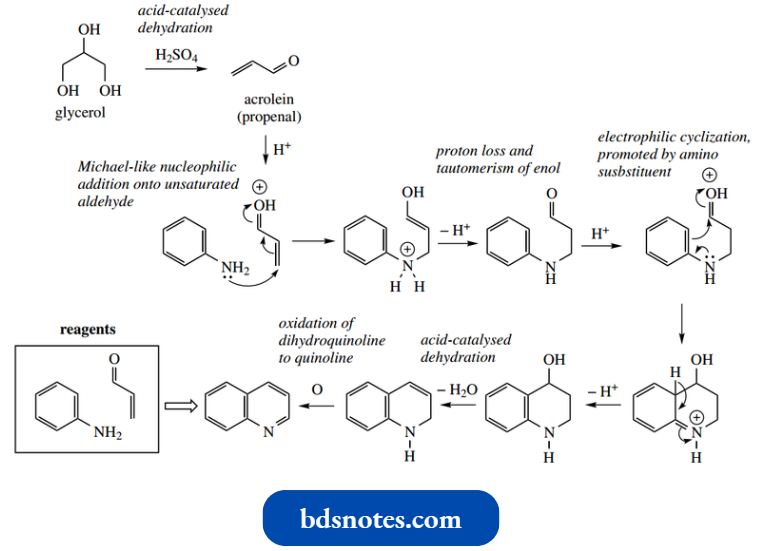
The initial product is a dihydroquinoline; it is formed via a Michael-like addition, then an electrophilic aromatic substitution that is facilitated by the electron-donating amine function.
- A mild oxidizing agent is required to form the aromatic quinoline.
- The Skraup synthesis can be used with substituted anilines, provided these substituents are not strongly electron-withdrawing and are not acid-sensitive.
Bischler–Napieralski Isoquinoline Synthesis:
Isoquinolines are easily prepared by the reaction of an acyl derivative of a β-phenylethylamine with a dehydrating agent, for example., P2O5, then using a catalytic dehydrogenation to aromatize the intermediate 3,4- dihydroisoquinoline.
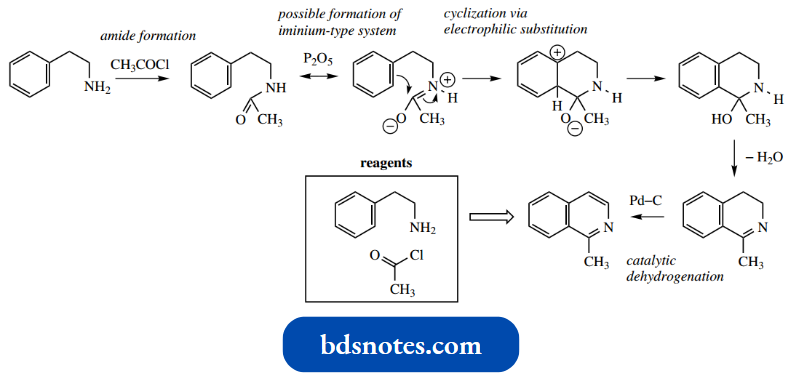
The crucial cyclization step is represented here as dehydrogenation and an iminium-type system, a resonance form of an electrophilic attack involving the aromatic ring of the amide, suitably coordinated with the phosphorus reagent.
- The cyclizing agent P2O5 also dehydrates the intermediate hydroxyamine to a dihydroisoquinoline.
- The isoquinoline is then obtained by heating over a catalyst, effectively reversing a catalytic hydrogenation reaction, facilitated by the generation of aromaticity in the product.
- As in the Skraup synthesis above, electron-withdrawing substituents on the aromatic ring will deactivate it towards electrophilic attack, whereas electron-donating substituents will favor the reaction.
Pictet–Spengler Tetrahydroisoquinoline Synthesis
This approach to the isoquinoline ring, albeit a reduced isoquinoline, is mechanistically similar to the Bischler–Napieralski synthesis, in that it involves an electrophilic attack of an iminium cation onto an aromatic ring. In this case, the imine intermediate is formed by reacting a phenylethylamine with an aldehyde.
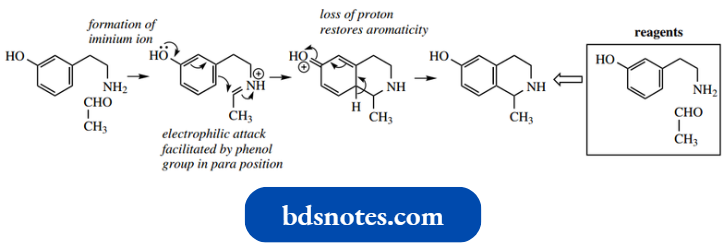
We have already met this reaction as an analog of the Mannich reaction, which we then interpreted as a nucleophilic attack of an electron-rich phenolic ring pontoon iminium cation.
- Is it electrophilic or nucleophilic? It matters little; they are the same, though the descriptor used depends upon which species you consider the more important, the nucleophilic phenol or the electrophilic iminium cation.
- For effective cyclization, we need an electron-donating substituent para to the point of ring closure since the Mannich-type electrophile is less reactive than the phosphorus-linked intermediates in the Bis cheer–Napieralski synthesis.
- It is also found that a similar group in the ortho position does not work, though we could still write an acceptable mechanism With a good electron-donating substituent like hydroxyl, the whole process, imine formation, and cyclization can occur under ‘physiological’ conditions pH 6–7 at room temperature.
- In nature, this is precisely how tetrahydroisoquinoline alkaloids are biosynthesized, though the reactions are enzyme-controlled
Knorr Pyrrole Synthesis:
This approach to the five-membered pyrrole ring reacts an α-aminoketone with a β-ketoester.
The mechanism will probably involve imine formation and then cyclization via an aldol-type reaction using the enamine nucleophile.
Dehydration leads to the pyrrole. Only the key parts of this sequence are shown below.
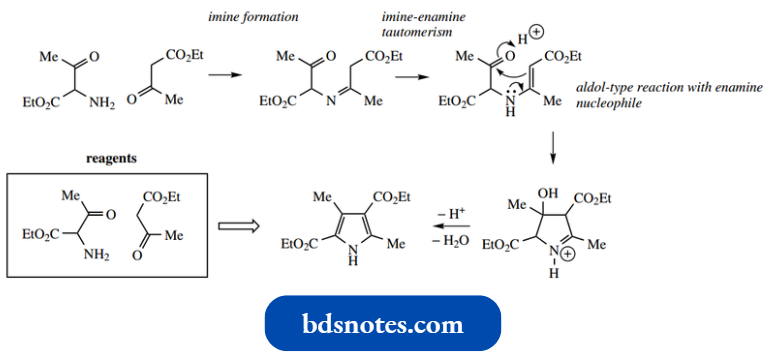
The synthesis works well only with an activated ester like ethyl acetoacetate. Otherwise, self-condensation of the α-aminoketone to a dihydropyrazine occurs more readily than the cyclization.
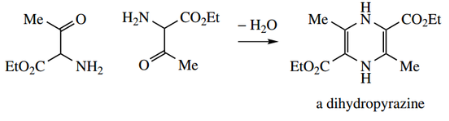
Paal–Knorr Pyrrole Synthesis:
The other major route to pyrroles is the interaction of a 1,4-dicarbonyl compound with ammonia.
The nucleophilic mechanism below shows successive nucleophilic additions of amino groups onto the carbonyls; but, since no intermediates have been isolated, the precise sequence of steps is speculative.
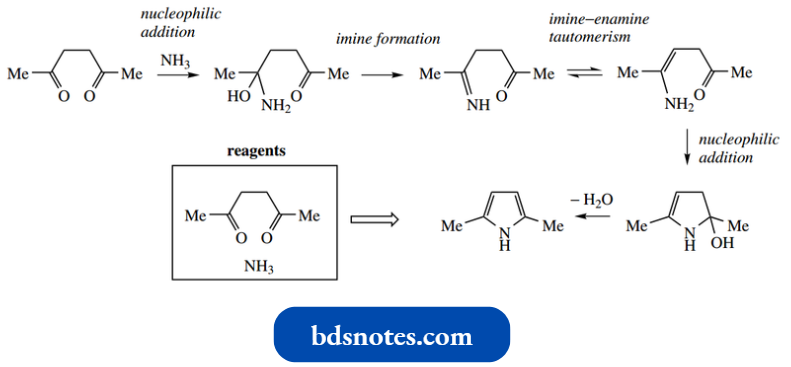
Note, however, that this synthesis gives furans if no ammonia is included. This would involve a nucleophilic attack of an enol tautomer of the substrate onto another carbonyl to give a hemiketal, followed by dehydration. The heteroatom is thus derived from a carbonyl oxygen.
The procedure works well and is usually carried out with acid catalysts under non-aqueous conditions.
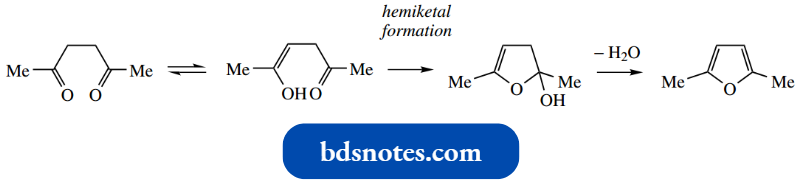
Fischer Indole Synthesis:
The most useful route to indoles is the Fischer indole synthesis, in which an aromatic phenylhydrazone is heated in acid.
- The phenylhydrazone is the condensation product from a phenylhydrazine and an aldehyde or ketone. Ring closure involves a cyclic rearrangement process.
- The hydrazine behaves as an amine towards a carbonyl compound and forms the imine-like product, a hydrazone.
- The cyclic rearrangement involves the enamine tautomer of this hydrazone, and proceeds because the cyclic flow of electrons forms a strong C–C bond whilst cleaving a weak N–N bond.
- This produces what appears to be a di-imine. One of these is involved in rearomatization and creates an aromatic amine.
This then attacks the other imine function, and we get the nitrogen equivalent of a hemiketal. Finally, acid-catalyzed elimination of ammonia gives the aromatic indole system.
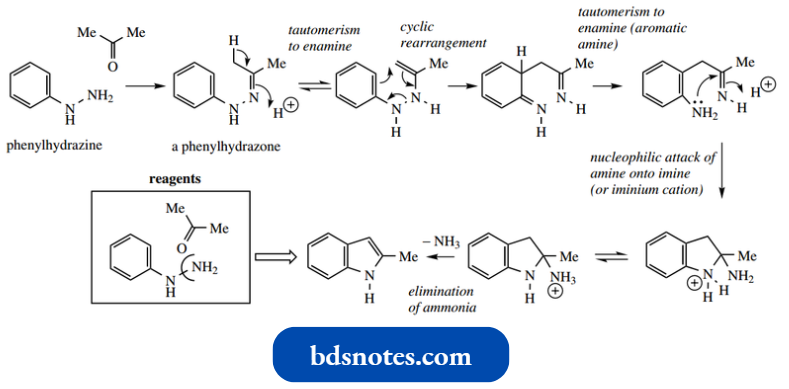
Unfortunately, the reaction fails with acetaldehyde and cannot, therefore, be used to synthesize indole itself. It is possible to use the ketoacid pyruvic acid instead and decarboxylate the product to yield indole.
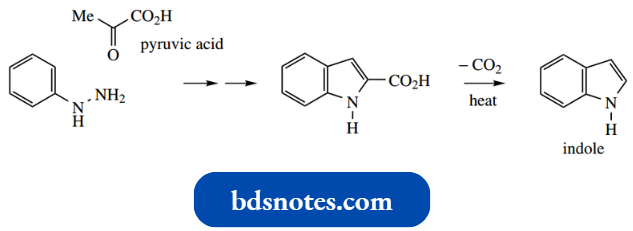
Leave a Reply