Electrophilic Reactions
In the preceding chapters we have seen how new bonds may be formed between nucleophilic reagents and various substrates that have electrophilic centres, the latter typically arising as a result of uneven electron distribution in the molecule. The nucleophile was considered to be the reactive species.
In this chapter, we shall consider reactions in which electrophilic reagents become bonded to electron-rich substrates, especially those that contain multiple bonds, i.e. alkenes, alkynes, and aromatics.
The π electrons in these systems provide regions of high electron density, and electrophilic reactions feature as the principal reactivity in these classes of compounds. We term the reactions electrophilic rather than nucleophilic since it is the electrophile that provides the reactive species.
Electrophilic Addition To Unsaturated Carbon
The characteristic reaction of alkenes is electrophilic addition, in which the carbon-carbon π bond is replaced by two σ bonds.
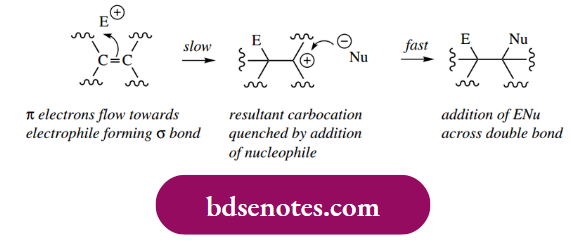
The π bond of an alkene results from the overlapping of p orbitals and provides regions of increased electron density above and below the plane of the molecule.
- These electrons are less tightly bound than those in the σ bonds, so are more polarizable and can interact with a positively charged electrophilic reagent.
- This forms the first part of an electrophilic addition, in which the electrons are used to form a σ bond with the electrophile and leave the other carbon of the double bond electron deficient, i.e. it becomes a carbocation.
- This carbocation is then rapidly captured by a nucleophile, which donates its electrons to form the second new σ bond. This latter step is very much faster than the first step, and thus carbocation formation becomes the rate-determining step in this bimolecular reaction.
Rate = k[alkene][ENu]
where ENu is the electrophilic reagent and k is the rate constant. The carbocation is an intermediate in the reaction sequence and corresponds to a minimum in the energy profile.
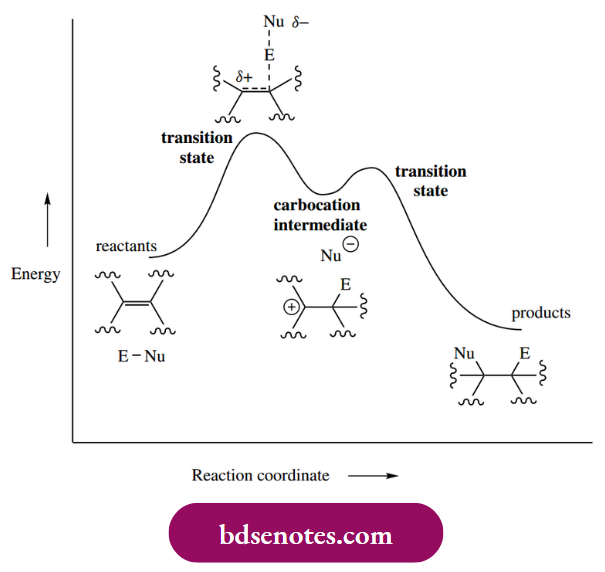
Depends upon producing a high-energy transition state in which there is substantial development of a positive charge on carbon. The reactive carbocation requires a much smaller activation energy for reaction with the nucleophile.
Addition Of Hydrogen Halides To Alkenes
The addition of a hydrogen halide, for example. HCl, to an alkene, is a simple example of an electrophilic addition.
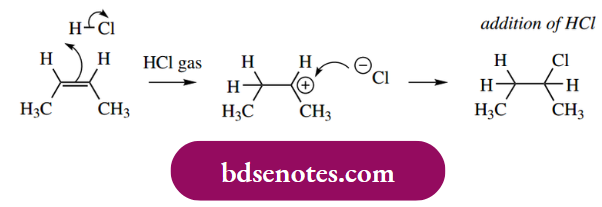
This type of synthetic reaction requires the use of gaseous hydrogen halide. If aqueous acid were employed, then, although water is not such a good nucleophile as halide, it is going to be the most abundant available nucleophile, and the predominant product will be the corresponding alcohol.
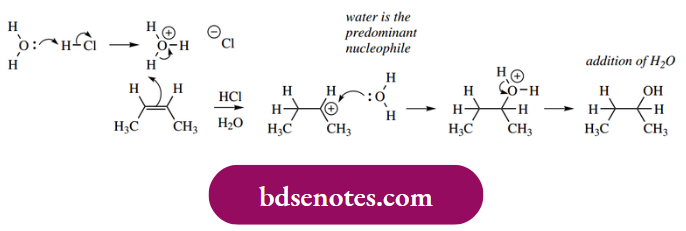
Since HCl will be completely dissociated in water, the electrophile in this case will be the hydronium ion, although the same carbocation will be produced. The reaction is completed by a nucleophilic attack of water, followed by the loss of a proton, thus regenerating the acid catalyst.
- The overall conversion thus becomes the hydration of the alkene. This is an important industrial process, typically employing sulfuric acid, but it is seldom used in the laboratory because the yields are very dependent upon the conditions used, and better routes to alcohols are available.
- Attack of the nucleophile onto a planar carbocation may take place from either face with equal probability, so that it is easy to see that, when a new chiral centre results, a racemic product will be formed, a similarity with SN1 processes.
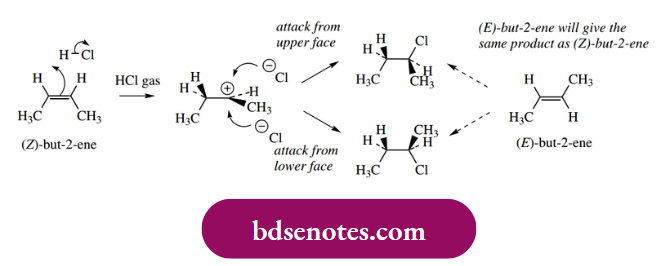
Also, since the same carbocation intermediate will be formed, it can be deduced that the nature of the product will not be dependent upon the configuration of the double bond.
In discussing simple electrophilic additions, the example chosen was but-2-ene. This choice was deliberate, in that it is a symmetrical substrate and it makes no difference which carbon becomes bonded to the electrophile or nucleophile. When the substrate is not symmetrical, we must then consider the relative stabilities of the two carbocations that might be involved in the addition reaction.
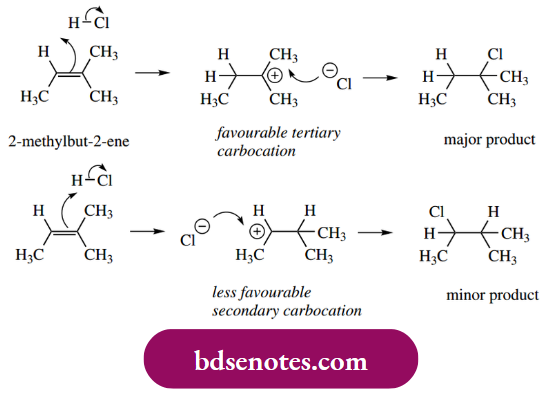
If we consider the protonation of 2-methylbut-2-ene, then two different carbocations might be formed. One of these is tertiary, and thus favourable because three electron-donating alkyl groups help to stabilize the cation by dispersing the charge.
- The alternative carbocation intermediate is less favourable, in that it is secondary, with just two alkyl groups helping to stabilize the carbocation. It follows that the tertiary carbocation is more likely to be formed and that the predominant product will be the result of nucleophilic attack on this intermediate.
- Some of the alternative products will likely be produced since secondary carbocations are reasonably stabilized and frequently produced in reactions.
- Naturally, if the protonation step could lead to either a tertiary or a very unfavourable primary carbocation, then we would expect the product to be almost entirely the result of tertiary carbocation involvement.
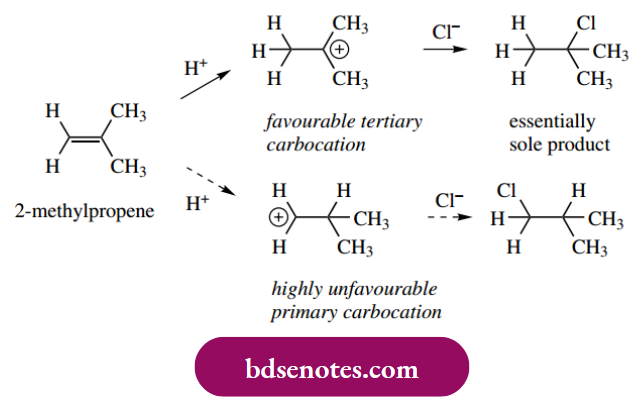
- Long before any reaction mechanism had been deduced, Markovnikov’s rule had been utilized to predict the regiochemistry for the addition of HX to an unsymmetrical alkene. Markovnikov’s rule states that the addition of HX across a carbon-carbon multiple bond proceeds in such a way that the proton adds to the less-substituted carbon atom, i.e. that already bears the greater number of hydrogen atoms.
- Since we now know that carbocation stability controls the regiochemistry of electrophilic addition, it is recommended that the more favoured product be predicted simply from an inspection of the possible carbocation intermediates.
- Alternatively, Markovnikov’s rule should be restated in mechanistic terms, in that the electrophile adds to the double bond to form the more stable carbocation. In some circumstances, this generalization has appeared incorrect, and the so-called anti-Markovnikov addition has been observed. Careful analysis of the reagents has shown that abnormal anti-Markovnikov addition of HX is the result of a radical reaction brought about by the presence of peroxides as radical initiators. This will be discussed further.
- The relative ease with which hydrogen halides react with alkenes is in the order HI > HBr > HCl > HF. This is the same as their relative acidities and indicates that protonation of the alkene is the rate-limiting step for the addition reaction.
Addition Of Halogens To Alkenes
Halogens such as chlorine (Cl2) and bromine (Br2) react readily with alkenes to produce 1,2-dihalogen derivatives. Although the halogen–halogen bond of Cl2 and Br2 is non-polar, it becomes polarized as it approaches the π electrons of the double bond. The electrons in the halogen–halogen σ bond become unequally shared and are disturbed towards the atom furthest away from the polarizing double bond. As a result, the halogen functions as an electrophile, in much the same way as HX.
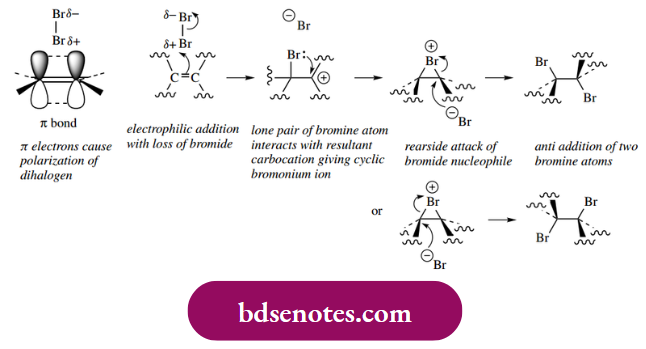
As the reactants get closer, there is a flow of electrons from the π bond to the nearer halogen, followed by the departure of the further halogen as halide. This results in the formation of a carbocation.
- In the next step, we see a significant difference in mechanism when compared with the addition of HX. Instead of the carbocation being quenched by the attack of nucleophiles, there is the formation of a cyclic halonium ion.
- This is achieved by bonding a lone pair of electrons from the large halogen atom to the carbocation, and it helps stabilize the cation by transferring the charge to the halogen. However, the bridging halogen atom now blocks any further attack on the halogen-bonded face of the original double bond, so that when a nucleophile attacks it has to be from the opposite face.
- This means that there now has to be a rear-side attack to open the cyclic halonium ion, in a process resembling an SN2 mechanism. Of course, either carbon might be attacked by the nucleophile, but the consequences are the same.
- The net result is the formation of an 11,2-dialogue system, and, stereochemically, the halogen atoms have been inserted onto opposite faces of the double bond. This is described as anti-addition (Greek: anti = against). A mechanism in which groups become attached to the same face of the double bond would be termed
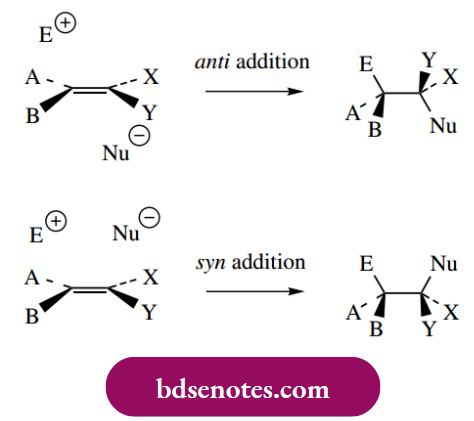
syn addition (Greek: syn = with). The observed addition of halogen with anti-stereochemistry is thus different from the simpler addition of HX, where the initially formed carbocation may be attacked from either face by the nucleophile.
Bromine and chlorine both react via cyclic halonium cations, which we term bromonium and chloronium cations respectively. Fluorine and iodine are hardly ever used for halogenations; iodine is a rather unreactive halogenating agent, whereas, at the other extreme, fluorine is too vigorous to give controllable reactions.
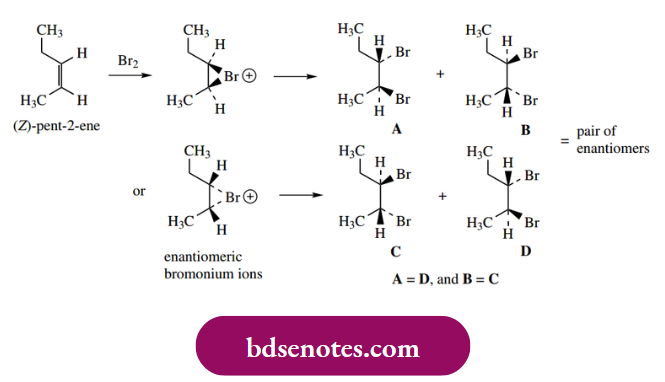
The stereochemical consequences of the electrophilic addition of, say, bromine to certain alkenes can be predicted as follows:
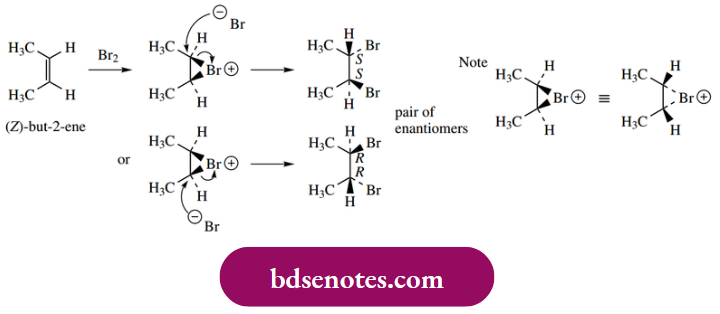
Thus, ( Z)-but-2-ene will react to give 2,3-dibromo butane as a pair of enantiomers, R, R and S, S, a result of the anti-addition A racemic product will thus be formed because there is an equal probability of nucleophilic attack at the two possible centres. Because of the symmetry in the molecules, it is only necessary to consider one bromonium ion, since the mirror image version is identical.
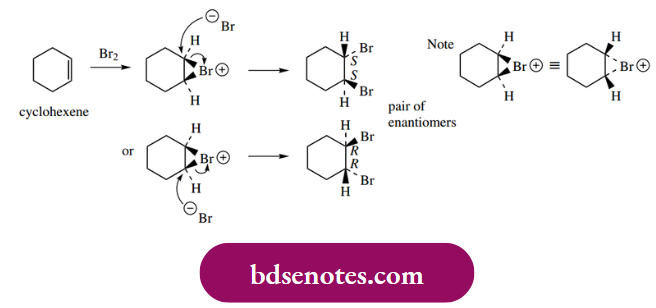
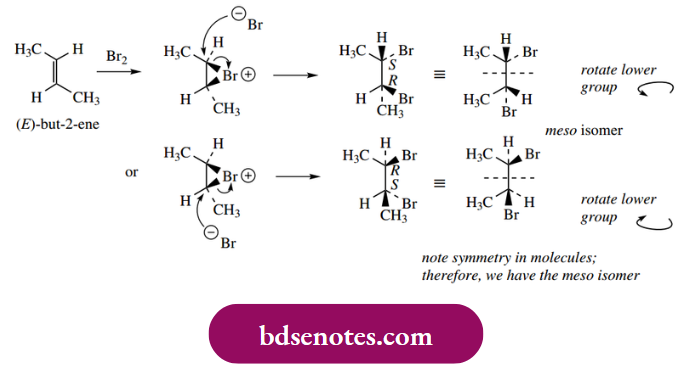
Similarly, cyclohexene will form 1,2-dibromo cyclohexane as a racemic product, again R, R and S, S. Note that the three-membered ring of the bromonium ion must be planar and can only be connected to the cyclohexane ring. When one considers the bromination of (E)-but-2-ene, the product turns out to be the meso R, S isomer, i.e. a single product.
Intriguingly, although there are going to be two different and enantiomeric bromonium ions for an unsymmetrical substrate such as (Z)-pent-2-ene, a pair of enantiomeric products results, due to the two types of nucleophilic attack – try it!
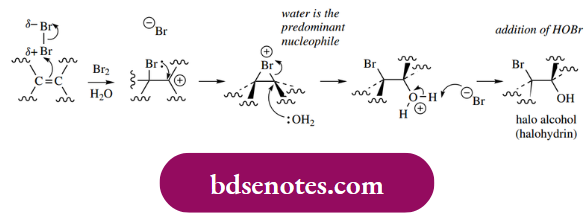
For the HX additions above, we noted that, in aqueous solution, water would be the most abundant nucleophile, and the predominant product would thus be an alcohol derivative. A similar situation holds if we use aqueous bromine or chlorine, for example.
The product is going to be a halo alcohol (halohydrin), and overall we are seeing the electrophilic anti-addition of X+ and HO–. This is sometimes considered as the addition of a hypohalous acid HOX but is much more easily rationalized in terms of halogenation in the presence of water as the predominant nucleophile.
One of the properties of halo alcohols formed in this way is that they can be used to make epoxides, three-membered oxygen heterocycles. This is achieved by treatment with base, and an intramolecular SN2 mechanism is involved.

Note that the bromination–hydroxylation sequence is going to produce anti-addition, and the groups are ideally set up for the SN2 reaction with the necessary rear-side attack.
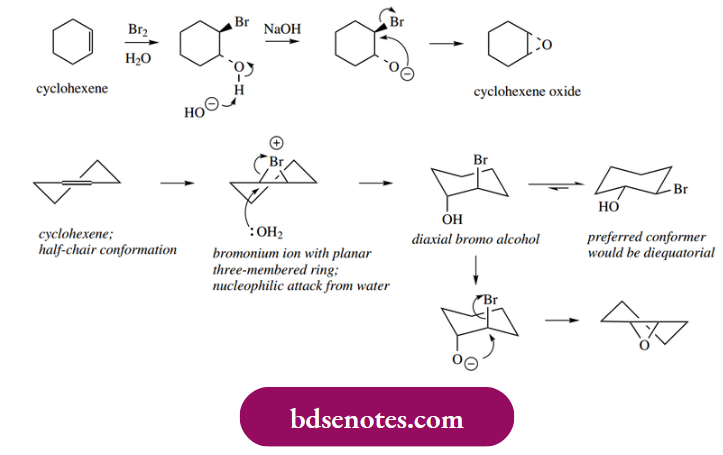
In the synthesis of cyclohexene oxide from cyclohexene shown, this does implicate the less favourable diaxial conformer in the epoxide-forming step. Cyclohexene oxide contains a cis-fused ring system, the only arrangement possible since the three-membered ring is necessarily planar.
Another method of making epoxides is the electrophilic reaction of alkenes with a peroxy acid such as peroxyacetic acid (sometimes simply peracetic acid). Thus, cyclohexene may be converted into the epoxide in a single reaction. Mechanistically, this is an electrophilic attack involving the π electron system of the alkene and the
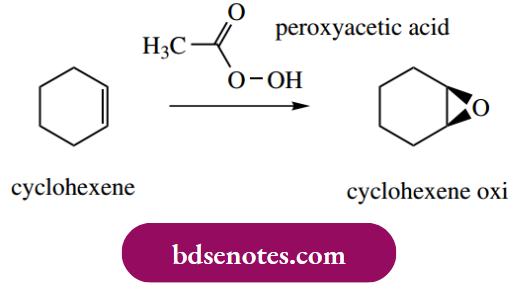
Polarized O–O bond in the peroxy acid. One could realistically suggest a potential carbocation intermediate, followed by a nucleophilic attack of the hydroxyl oxygen, using as precedent the examples seen above.
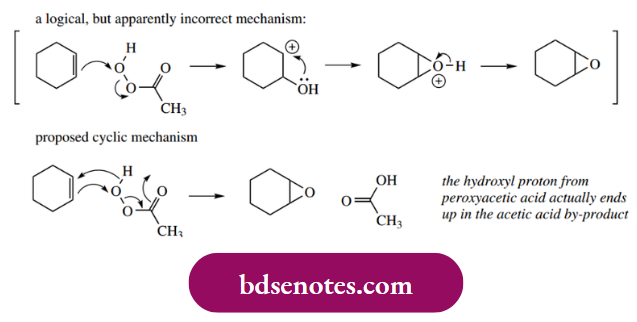
However, since it is found that the hydroxyl proton from peroxyacetic acid ends up in the acetic acid by-product, a messy-looking cyclic mechanism has been proposed.
- This starts with the nucleophilic π bond attacking the peroxy acid oxygen, breaking the O–O bond to form a new carbonyl, with the original carbonyl picking up the hydroxyl’s hydrogen.
- The remaining electrons from the hydroxyl are then used to bond to the electrophilic carbon from the original double bond.
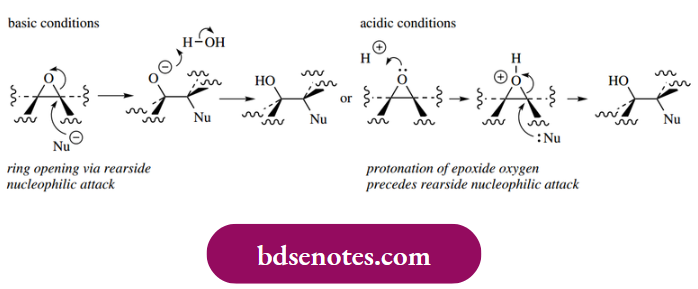
- Epoxides, like cyclic halonium ions, undergo ring opening through the rear side attack of nucleophiles. Two mechanisms are shown, for both basic and acidic conditions.
- Under acidic conditions, protonation of the epoxide oxygen occurs first. The epoxidation–nucleophilic attack sequence also adds substituents to the double bond in an anti-sense.
Basic Conditions:
Halohydrins Biological Activity Of Semi – Synthetic Corticosteroids
Corticosteroids are produced by the adrenal glands and display two main types of biological activity. Glucocorticoids are concerned with the synthesis of carbohydrates from protein and the deposition of glycogen in the liver. They also play an important role in inflammatory processes.
- Mineralocorticoids are concerned with the control of electrolyte balance, promoting the retention of Na+ and Cl–, and the excretion of K+. Synthetic and semi-synthetic corticosteroid drugs are widely used in medicine.
- Glucocorticoids are primarily used for their antirheumatic and anti-inflammatory activities, and mineralocorticoids are used to maintain electrolyte balance where there is adrenal insufficiency.
- The two groups of steroids share a considerable structural similarity, and it is difficult to separate the two types of activity in one molecule. Extensive synthetic effort has been applied to optimize anti-inflammatory activity, whilst minimizing the mineralocorticoid activity, which tends to produce undesirable side-effects.
- One modification that has proved particularly successful has been to create 9 α-halo-11 β-hydroxy compounds. The 11 β- βhydroxyl is present in all glucocorticoids and is known to be essential for activity; the introduction of the halogen atom at position 9 was a major development in this group of drugs. Halogenation was achieved as shown below.
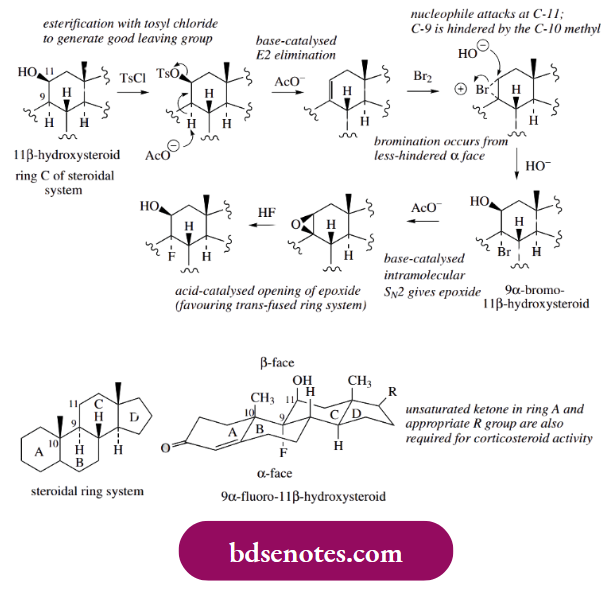
Treatment of the 11 β-hydroxysteroid with tosyl chloride produces a tosylate ester, providing a good leaving group for a base-catalysed E2 elimination. The favoured product is the more substituted 9, 11-alkene. A consideration of the steroid shape (see Box 3.19) shows that the 9 α-proton and the 11 β-tosylate are both axial and, therefore, anti to each other; they are thus ideally positioned for an elimination reaction.
- Bromination of the alkene under aqueous alkaline conditions then leads to the formation of the bromohydrin. Interestingly, this reaction is quite stereospecific. Only one bromonium cation is formed, because the upper face of the steroid is sterically hindered by the methyl groups and the approach of the large bromine molecule occurs from the lower less-hindered α-face. RRing-opening by nucleophilic attack of hydroxide occurs from the upper β-face, and at C-11, since the methyl groups, particularly that at C-10, again hinder attack at C-9.
- The natural glucocorticoid is hydrocortisone (cortisol). Semi-synthetic 9 α-bromohydro cortisone 21-acetate was found to be less active as an anti-inflammatory agent than hydrocortisone 21-acetate by a factor of three, and 9 α-iodohydrocortisone 21-acetate was also less active by a factor of 10.
- However, 9 α-fluorohydrocortisone 21-acetate (fludrocortisone acetate) was discovered to be about 11 times more active than hydrocortisone acetate. Although the bromination sequence shown is equally applicable to chlorine and iodine compounds, fluorine must be introduced indirectly by the β-epoxide formed by base treatment of the 9 α-bromo-11 β-hydroxy analogue.
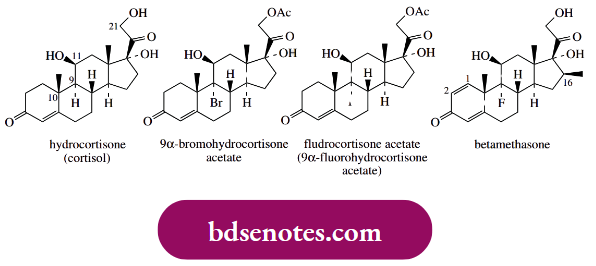
The introduction of a 9 α-fluoro substituent increases anti-inflammatory activity, but it increases mineralocorticoid activity even more (300× ). Fludrocortisone acetate is of little value as an anti-inflammatory, but it is employed as a mineralocorticoid.
- On the other hand, additional modifications may be employed. The introduction of a 1,2-double bond increases glucocorticoid activity over mineralocorticoid activity, and a 16-methyl group reduces mineralocorticoid activity without affecting glucocorticoid activity.
- A combination of these three structural modifications gives valuable anti-inflammatory drugs, for Example. betamethasone, with hardly any mineralocorticoid activity.
Electrophilic Additions To Alkynes
Electrophilic reactions of alkynes can readily be predicted, based on the mechanisms outlined above for alkenes. Of course, the main extension is that addition will initially produce an alkene, which will then undergo further addition.
- Protonation of the alkyne is less favourable than protonation of an alkene, because the resulting vinyl cation is sp hybridized, having σ bonds to just two substituents, a π bond, and a vacant p orbital.
- A vinyl cation is thus less stable than a comparable trigonal sp 2-hybridized carbocation since sp-hybridization brings bonding electrons closer to carbon; it thus becomes less tolerant of positive charge. Protonation, when it occurs, will be on the less-substituted carbon, a secondary vinyl cation being preferred over a primary vinyl cation. Thus, the electrophilic addition of HX follows Markovnikov orientation.
- The vinyl halide product is then able to react with a further mole of HX, and the halide atom already present influences the orientation of addition in this step. The second halide adds to the carbon that already carries a halide.
- In the case of the second edition of HX to RC≡CH, we can see that we are now considering the relative stabilities of tertiary and primary carbocations. The halide’s inductive effect destabilizes the tertiary carbocation.
Nevertheless, this is outweighed by a favourable stabilization from the halide by the overlap of lone pair electrons, helping to disperse the positive charge.

In the case of the electrophilic addition of HX to RC≡CR, it is possible to see even more the role of the first halide atom. After the addition of the first mole of HX, further protonation would give either a secondary carbocation or the tertiary carbocation with a destabilizing inductive effect. The resonance contribution from the first halide atom still defines the position for the second protonation, and thus the nature of the major product, the gem-dihalide.
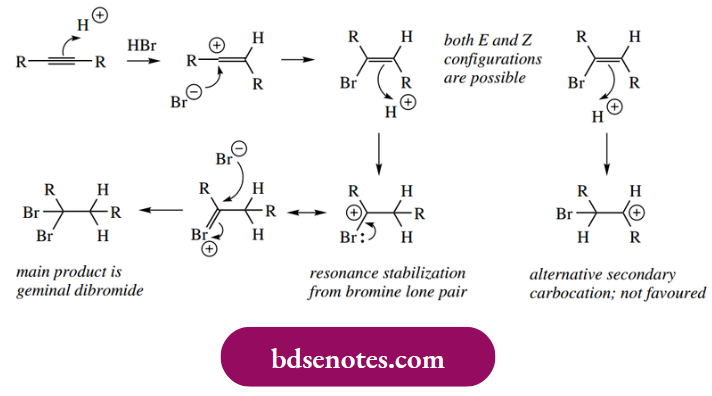
Note also that, if 2 mol of HX is added to an alkyne, it is of no consequence whether the first addition produces an alkene with E or Z stereochemistry since the orientation of addition means the final product has no potential chiral centres.
Predicting the outcome of electrophilic additions to alkynes from an extension of alkene reactivity usually works well and can be applied to halogenations and hydrations. Hydration of an alkyne has a subtle twist, however; the product is a ketone! This can still be rationalized quite readily, though.
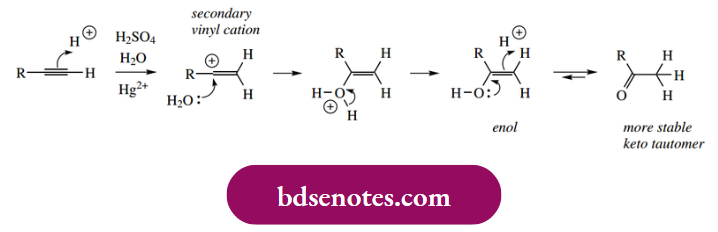
- Protonation of the alkyne produces the more favourable secondary vinyl cation, which is then attacked by water since water is the predominant nucleophile available. The loss of a proton from this produces an enol, which is transformed into a more stable isomeric form, the ketone.
- This transformation is termed tautomerism, and we shall meet it later (see Section 10.1) as an important consideration in the reactivity of many organic compounds.
- This reaction involves only one electrophilic addition, and, although it does not occur readily using simply aqueous acid, it can be achieved by the use of mercuric salts as catalyst. The mercuric ion may function as a Lewis acid to facilitate the formation of the vinyl cation.
Electrophilic Alkylation In Steroids De-Chainb Losynthesis
Polyene macrolide drugs such as amphotericin and nystatin have useful antifungal activity but no antibacterial action (see Box 7.14). Their activity is a result of binding to sterols in the eukaryotic cell membrane; they display no antibacterial activity because bacterial cells do not contain sterol components.
- Binding to sterols modifies cell wall permeability and leads to pores in the membrane and loss of essential cell components. Fungal cells are also attacked in preference to mammalian cells, since the antibiotics bind much more strongly to ergosterol, the major fungal sterol, than to cholesterol, the main animal sterol component.
- This selective action allows these compounds to be used as drugs, though a limited amount of binding to cholesterol is responsible for the side effects of the drugs.
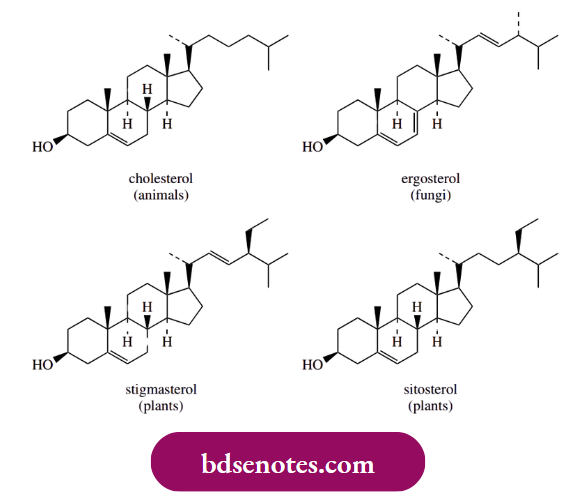
A principal structural difference between ergosterol and cholesterol that affects binding to polyene drugs is the extra methyl group on the side chain in ergosterol. This extra methyl is known to be introduced in nature by an electrophilic alkylation of a double-bond system, and it employs S-adenosylmethionine (SAM) as the electrophilic agent. We have already met SAM as a biochemical alkylating agent through SN2 reactions.
The role of SAM in these electrophilic reactions is similar. It possesses a good leaving group in the neutral molecule S-adenosylhomocysteine, and the methyl group can be donated to the alkene nucleophile in an electrophilic addition.
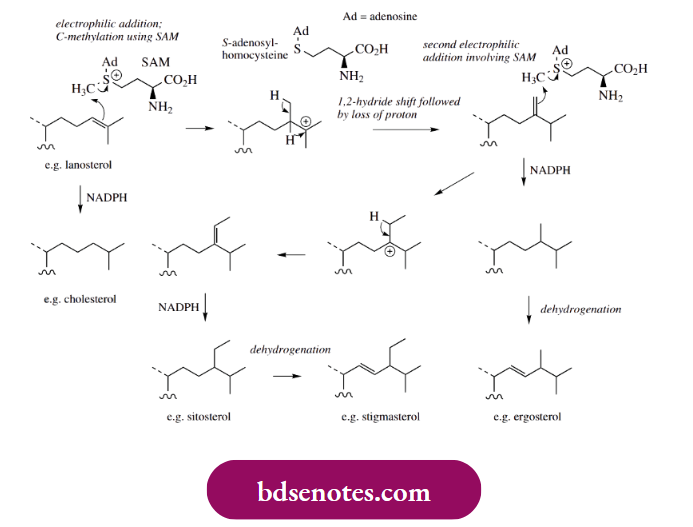
Cholesterol and ergosterol share a common biosynthetic pathway from squalene oxide as far as lanosterol, but then subsequent modifications vary. Part of the route to cholesterol involves the reduction of the side-chain double bond, an enzymic process utilizing the hydride donor NADPH as a reducing agent.
- During ergosterol biosynthesis, the side-chain double bond is involved in an electrophilic reaction with SAM, in addition to yielding the anticipated tertiary carbocation. This carbocation then undergoes a Wagner–Meerwein 1,2-hydride shift, an unexpected change, and subsequently loses a proton from the SAM-derived methyl group to generate a new alkene.
- NADPH reduction of this double bond leads to the C-methylated sidechain, as found in ergosterol, though further unsaturation needs to be introduced via an enzymic dehydrogenation reaction.
- Plant sterols such as stigmasterol typically contain an extra ethyl group when compared with cholesterol. Now this is not introduced by an electrophilic ethylation process; instead, two successive electrophilic methylation processes occur, both involving SAM as a methyl donor.
- Indeed, it is a methylene derivative like that just seen in ergosterol formation that can act as the alkene for further electrophilic alkylation. After proton loss, the product has a side-chain with an ethylidene substituent; the side-chains of the common plant sterols stigmasterol and sitosterol are then related by repeats of the reduction and dehydrogenation processes already seen in ergosterol formation.
Carbocation Rearrangements
Carbocations are highly reactive intermediates and are notorious for their ability to rearrange into more stable variants. We have already met this concept when we considered carbocation rearrangements as competing reactions during SN1 nucleophilic substitutions.
- Since carbocations are also involved in electrophilic reactions, we must expect that analogous rearrangements might occur in these. This is indeed the case. In Section 6.4.2, we included examples of rearrangements in carbocations formed during electrophilic additions, because identical processes are involved, and it was more appropriate to consider these topics together, rather than separately.
- As a simple example, note that the major products obtained as a result of the addition of HBr to the alkenes shown below are not always those initially expected. For the first alkene, protonation produces a particularly favourable carbocation that is both tertiary and benzylic (see Section 6.2.1); this then accepts the bromide nucleophile.
- In the second alkene, protonation produces a secondary alkene, but hydride migration then leads to a more favourable benzylic carbocation. As a result, the nucleophile becomes attached to a carbon that was not part of the original double bond. Further examples of carbocation rearrangements will be met under electrophilic aromatic substitution.
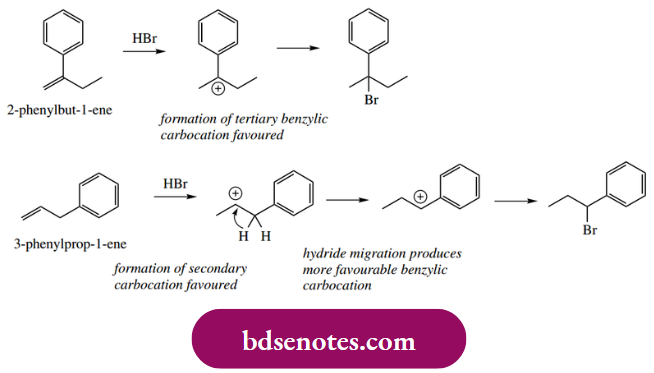
Rearrangements are an unexpected complication, and it is sometimes difficult to predict when they might occur. We need to look carefully at the structure of any proposed carbocation intermediate and consider whether any such rearrangements are probable. In most cases, we shall only need to rationalize such transformations, and will not be trying to predict their possible occurrence.
Electrophilic Addition To Conjugated Systems
The first step in the reaction of HX with an alkene is protonation to yield a more stable cation. If we extend this principle to a conjugated diene, for Example. but-1,3-diene, then we can see that the preferred carbocation will be produced if protonation occurs on a terminal carbon atom; protonation of either C-2 or C-3 would produce an unfavourable primary carbocation.
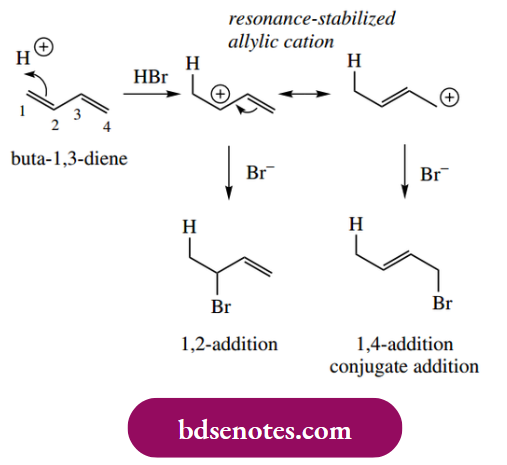
At first glance, this appears to be a secondary carbocation, but on further examination, one can see that it is also an allylic cation. Allylic carbocations are stabilized by resonance, resulting in the dispersal of the positive charge (see Section 6.2.1). From these two resonance forms, we can predict that both carbons 2 and 4 will be electron deficient.
- Now this has particular consequences when we consider subsequent attack of the nucleophile X− on to the carbocation. Two possible centres may be attacked, resulting in two different products. The products are the result of either 1,2-addition or 1,4-addition. The addition across the four-carbon system as in the 1,4-adduct is termed conjugate addition.
- Now the allylic cation has two limiting structures, one of which is a secondary carbocation and the other a primary carbocation. We would expect the secondary carbocation to contribute more than the primary carbocation, and this is usually reflected in the proportions of the two products obtained from the reaction carried out at low temperatures. Why is the rider about low temperatures? Well, the product ratio is different if the same reaction is carried out at higher temperatures, and typically the 1,2- adduct now predominates.
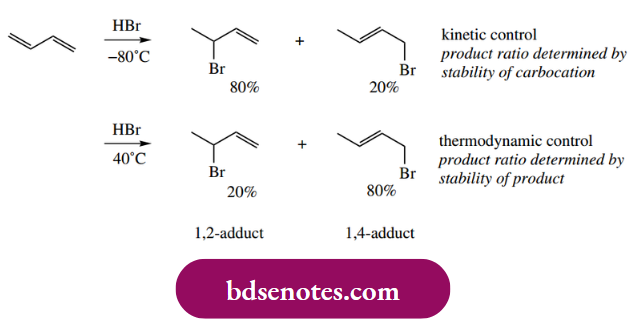
At the higher temperature, the thermodynamic stability of the product is an important consideration, with the 1,4-adduct, a disubstituted alkene, being more stable than the 1,2-adduct, which is a monosubstituted alkene.
An essential part of the reasoning is that the reaction is reversible so that the product can lose halide to regenerate the allylic cation. Thus, the product mixture from the lower temperature reaction is converted upon heating into the product mixture corresponding to the higher temperature.
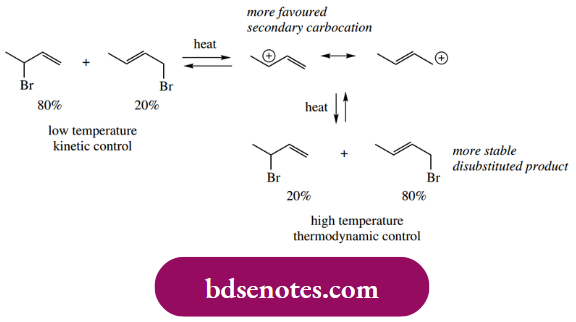
- These concepts are termed kinetic control and thermodynamic control. At the lower temperature, the product ratio is determined by the relative importance of the carbocations, with the predominant one reacting faster. At the higher temperature, the product ratio is determined by the stability of the product, with the more stable one predominating.
- These products are, of course, the result of the addition of just 1 mol of HX to the conjugated system. The second double bond could also react if further HX were available, with regiochemistry now following the principles already established in Section 8.1.
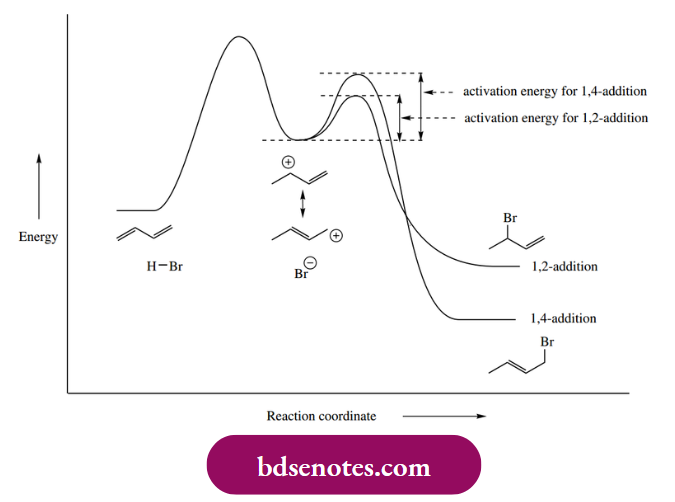
- The energy diagram for kinetic versus thermodynamic control is shown in Figure 8.2. This may be interpreted as follows. The 1,4-addition product is of lower energy, i.e. more stable, than the 1,2-addition product. The critical step, however, is the interaction of the bromide nucleophile with the allylic cation.
The activation energy leading to the 1,2-addition product is lower than that leading to the 1,4-addition product. Therefore, at lower temperatures, the 1,2-adduct is formed faster and becomes the dominant product.
- At the lower temperature, though, there is insufficient energy available to overcome the much larger energy barrier for the reverse reactions, so neither reaction is reversible. Both products are formed but do not revert to the allylic cation. Therefore, we have kinetic control: the product ratio depends upon which product is formed faster.
- At higher temperatures there is now sufficient energy available to overcome both activation energies with ease, and, more importantly, the reverse reactions become feasible. We can also see that the less stable 1,2-addition product will revert to the allylic cation faster than the 1,4-addition product simply because the energy barrier is that much less.
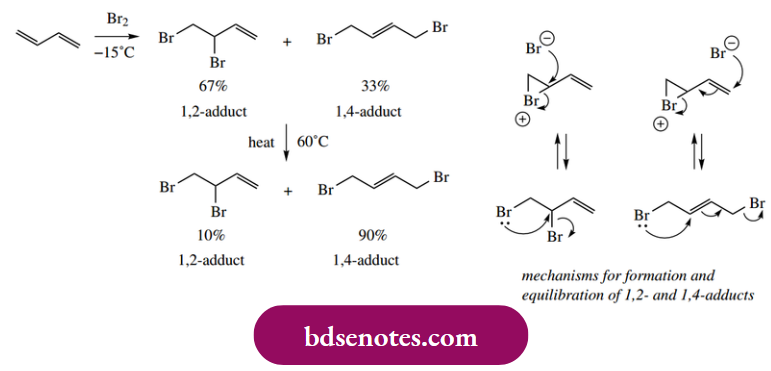
- The dominant equilibrium product will thus become the more stable material, i.e. the 1,4-addition product; we now have thermodynamic control. Similar observations emerge from the addition of halogens to butadiene. Thus, low-temperature bromination gives predominantly the 1,2-adduct.
- At higher temperatures, the 1,4-adduct is the main product, and the mixture from the lower temperature reaction equilibrates to the same product ratio. The 1,4-product is the thermodynamically more stable; it has the more substituted double bond, and the two large bromine atoms are further apart in this isomer.
- Mechanisms for the formation and equilibration of the products can be written as shown, using bromonium cation intermediates. It is perhaps less easy to see why the 1,2-adduct should be the kinetically controlled product until
we consider that the bromonium cation is a stabilized carbocation (see Section 8.1.2). We can then use the same type of rationalization as with the carbocation intermediates in HBr addition.
Carbocations As Electrophiles
As we have seen in Section 8.1, the reaction of an alkene with an electrophile produces a carbocation that is subsequently attacked by a nucleophile. However, the carbocation is itself an electrophilic species and is vulnerable to attack by another alkene molecule, provided that the alkene concentration is sufficiently high. For example, protonation of styrene leads to a secondary carbocation that is favoured by being benzylic.
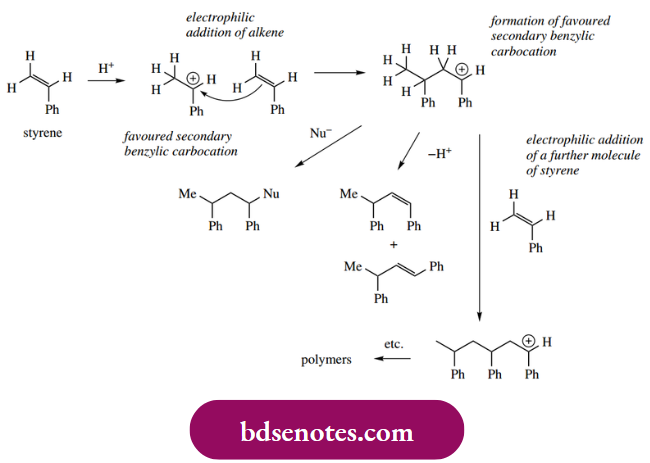
- This carbocation now becomes the electrophile and may be attacked by the π electrons of a second styrene molecule, the regiochemistry of attack being the same as with the original protonation, giving the secondary benzylic carbocation. Now this carbocation may suffer several fates. It may be attacked by a nucleophilic species or, more likely, it may lose a proton to yield an alkene.
- Alternatively, it may act as the electrophile for reaction with a further styrene molecule, generating yet another carbocation. It can be seen that this type of process may then continue, giving polymeric products: polystyrene. The final carbocation will be discharged most probably by loss of a proton. The process is termed cationic polymerization. In practice, the process is more useful for generating dimers and trimers than polymers, and industrial polymers are usually produced by radical processes.
- Cationic polymerization is, of course, an intermolecular electrophilic addition process. Intramolecular electrophilic addition involving two double bonds in the same molecule may be used to generate a cyclic system. Thus, the trienone shown is converted into a mixture of cyclic products when treated with sulfuric acid.
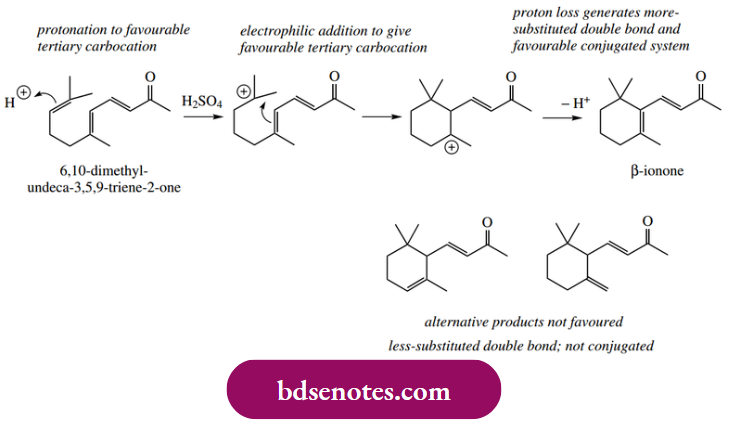
This is easily rationalized by protonation of the terminal alkene, yielding the preferred tertiary carbocation. The carbocation is then attacked by π electrons from the neighbouring double bond, creating a new σ bond and a ring system. Note that this results in a favourable tertiary carbocation and a favourable strain-free six-membered ring.
The products are then formed by the loss of a proton from this carbocation, with a choice of protons that may be lost, so that a mixture of products in varying proportions results. β-Ionone is the predominant product. This is the most substituted alkene and has the added stability conferred by extending conjugation with the unsaturated ketone.
Electrophilic Additions To Carbocations In Terpenoid And Steroid Biosynthesis
Terpenoids and steroids account for a huge group of natural products and provide us with many useful materials, including flavouring and perfumery agents, aromatherapy oils, some vitamins, steroidal hormones and a range of drugs. Although spanning a vast range of chemical structures, these compounds all derive from two simple precursors, dimethylallyl diphosphate and isopentenyl diphosphate.
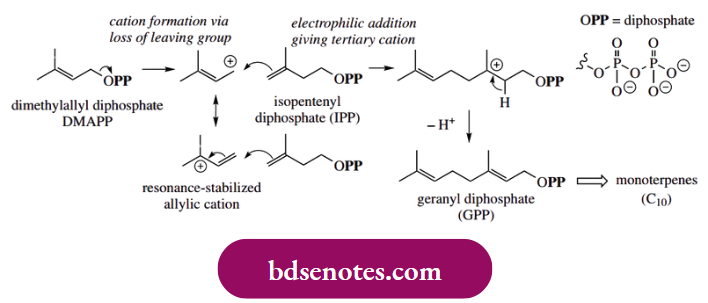
- Dimethylallyl diphosphate is responsible for generating the carbocation. Loss of diphosphate as the leaving group produces a resonance-stabilized allylic cation. An intermolecular electrophilic addition follows, with isopentenyl diphosphate as the source of π electrons.
- In addition, the cationic species takes place at the terminal carbon that is sterically less congested – at first glance, we appear to be invoking the less favourable resonance form, but an alternative addition through the double bond onto the tertiary cation could be drawn.
- At this stage, it is important to appreciate that these reactions are enzyme-controlled so that we can have two different species reacting in a highly specific manner. The carbocation product is the more stable tertiary carbocation, as we might predict, and this loses a proton to form the more substituted alkene product, geranyl diphosphate.
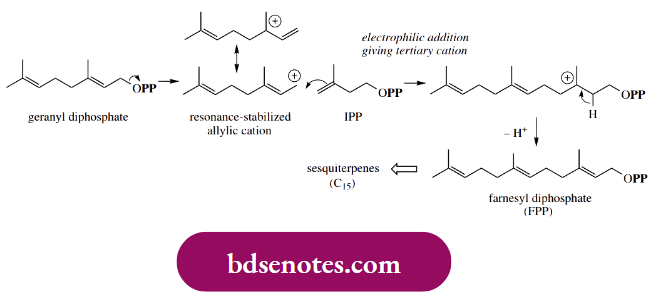
An exactly analogous process can then occur, in which geranyl diphosphate provides the allylic cation, and a further molecule of isopentenyl diphosphate adds on, giving farnesyl diphosphate; this can subsequently yield geranylgeranyl diphosphate.
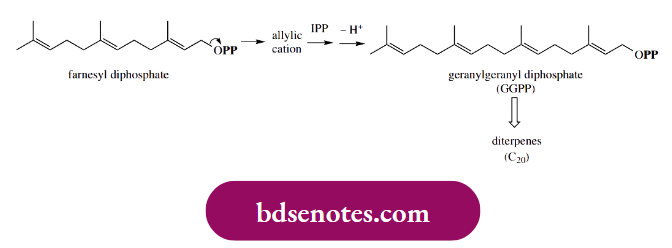
- The compounds geranyl diphosphate, farnesyl diphosphate, and geranylgeranyl diphosphate are biochemical precursors of monoterpenes, sesquiterpenes, and diterpenes respectively, and virtually all subsequent modifications of these precursors involve the initial formation of an allylic cation through loss of diphosphate as the leaving group.
- The formation of cyclic terpenoids involves intramolecular electrophilic addition, and this can be exemplified by the following monoterpene structures, again with all reactions being enzyme-controlled.
- Before cyclization can occur, however, there has to be a change in stereochemistry at the 2,3-double bond, from E in geranyl diphosphate to Z, as in neryl diphosphate. It should be reasonably clear that geranyl diphosphate cannot possibly cyclize to a six-membered ring, since the carbon atoms that need to bond are not close enough to each other.
- The change in stereochemistry is achieved through allylic cations and linalyl diphosphate.
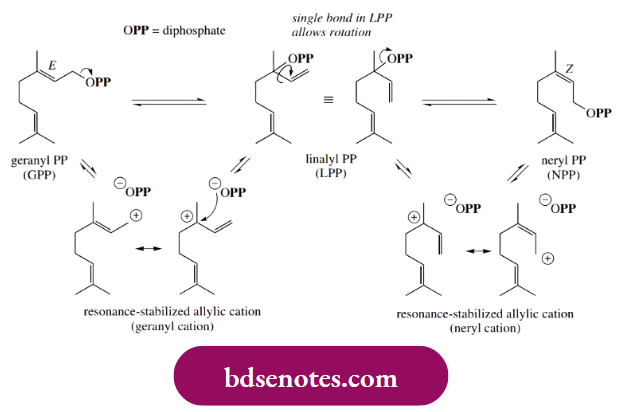
- Geranyl diphosphate ionizes to the resonance-stabilized geranyl carbocation, which, in nature, can recombine with the diphosphate anion in two ways, either reverting to geranyl diphosphate or forming linalyl diphosphate. In linalyl diphosphate, the original double bond from geranyl diphosphate has now become a single bond, and free rotation is possible.
- Ionization of linalyl diphosphate then occurs, giving a resonance-stabilized neryl carbocation, one form of which now has a Z double bond. Recombination of this with diphosphate leads to neryl diphosphate, a geometric configurational isomer of geranyl diphosphate. It is normally very difficult to change the configuration of a double bond.
- Nature achieves it easily in this allylic system via carbocation chemistry, and, in metabolic processes, geranyl diphosphate can be isomerized through linalyl diphosphate to neryl diphosphate.
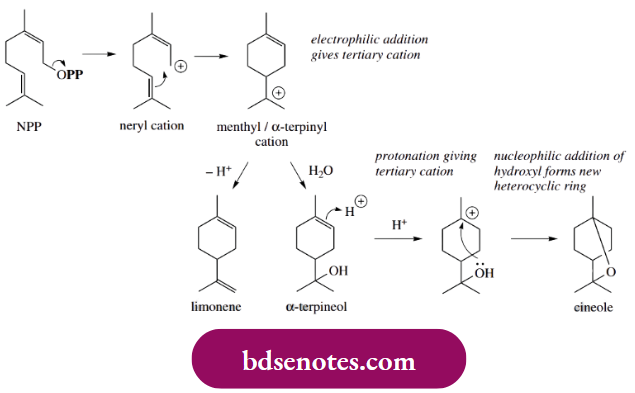
Carbocations As Electrop Hiles:
Cyclization involves the neryl cation with electrophilic attack from the double bond giving the favoured tertiary carbocation and a favourable six-membered ring. Loss of a proton from this action results in the formation of limonene, actually the less-substituted alkene, so this is where enzyme control takes over.
- Alternatively, discharge of the cation by addition of water as a nucleophile leads to α-terpineol. By a similar sequence, α-terpineol may be transformed into cineole. This requires the generation of a carbocation by protonation of the double bond; the proton is added so that the favoured tertiary cation is formed.
- Cineole formation then involves nucleophilic attack from the alcohol group with the generation of a further ring system, this time a heterocyclic ring. Limonene is a major constituent of lemon oil, α-terpineol is found in pine oil, and cineole is the principal component of eucalyptus oil.
- By far the most impressive example of electrophilic addition in natural product formation is in the biosynthesis of steroids. The substrate squalene oxide is cyclized to lanosterol in a process catalysed by a single enzyme. Lanosterol is then converted into the primary animal-steroid cholesterol. Squalene oxide comes from squalene, which is itself formed through a combination of two molecules of farnesyl diphosphate.
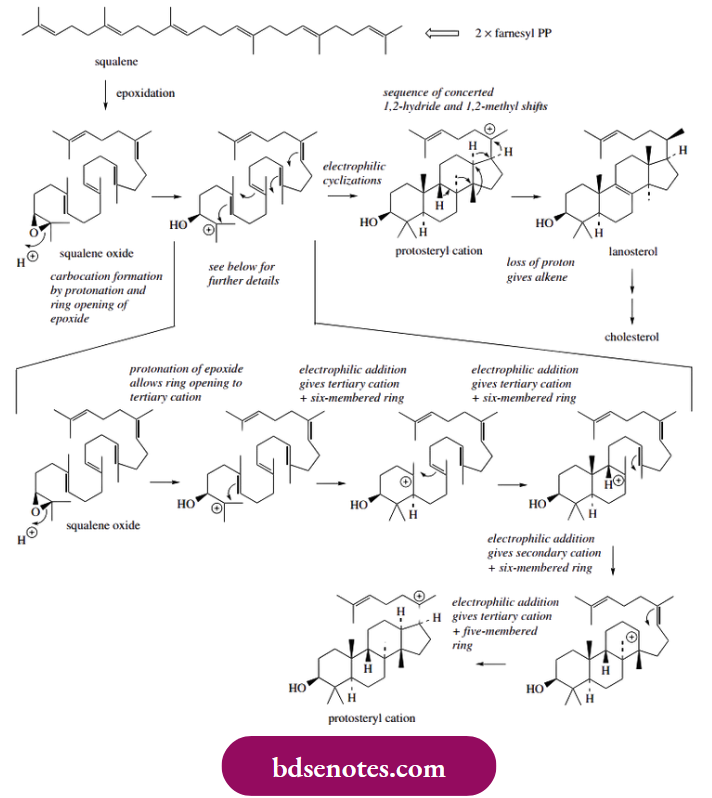
squalene oxide suitably positioned and folded onto the enzyme surface, a series of electrophilic cyclizations can be used to rationalize the formation of the polycyclic structure. The cyclizations are carbocation-mediated and proceed in a stepwise sequence.
- Thus, protonation of the epoxide group will allow the opening of this ring and the generation of the preferred tertiary carbocation. This is suitably placed to allow electrophilic addition to a double bond, formation of a six-membered ring and production of a new tertiary carbocation.
- This process continues twice more, generating a new carbocation, until the protosteryl cation is formed. This is then followed by a sequence of concerted Wagner–Meerwein migrations of methyls and hydrides leading to lanosterol. It is not appropriate to discuss these migrations in this chapter, but this aspect is studied further.
- Note that the preferred tertiary carbocation (Markovnikov addition) is produced in all of the cyclizations, except in one case, the third ring, which appears to be formed in an anti-Markovnikov sense. The latest studies now show that the reaction as illustrated above is not quite correct.
- The third ring is first produced as a five-membered one, reassuringly by Markovnikov addition via the predicted tertiary carbocation, and it is subsequently expanded to a six-membered ring through a Wagner–Meerwein 1,2-alkyl shift. This should not be thought of as a complication; simply note the formation of a biochemically important polycyclic ring system through a series of electrophilic additions.
Electrophilic Aromatic Substitution
Electrophilic reactions with aromatic substrates tend to result in substitution. This should not be viewed as markedly different behaviour from alkenes, but merely as an obvious consequence of aromatic stabilization dictating the fate of the initial carbocation.
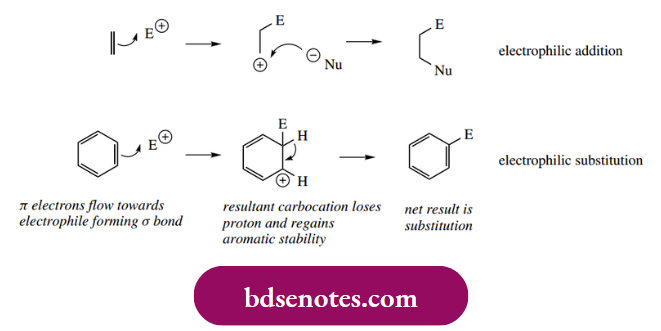
- However, there are differences, in that an electrophilic attack on the n aromatic ring is energetically less favourable than an attack on the n alkene. This is because the initial addition reaction leading to carbocation formation uses up one of the p orbitals that normally contribute to the π electron system, and thereby creates an sp3-hybridized centre.
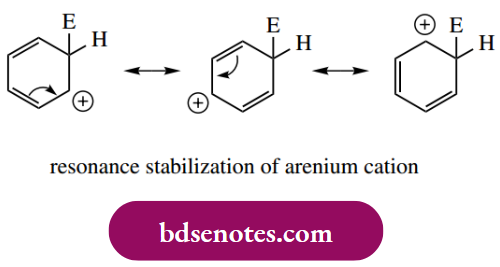
- This means that the π electron delocalization characteristic of an aromatic system is destroyed. However, there is also some good news: the carbocation generated (uranium cation) is resonance stabilized and is considerably more favourable than the corresponding simple trigonal cation from an alkene.
- Accordingly, the electrophilic addition can occur; but, rather than reacting with a nucleophile, the cation loses a proton and this leads to restoration of the aromatic π electron system. The overall reaction is thus substitution.
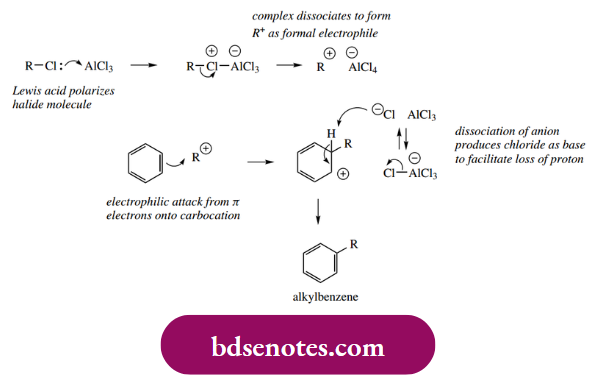
Because the initial electrophilic attack and carbocation formation results in loss of aromatic stabilization, the electrophiles necessary for electrophilic aromatic substitution must be more reactive than those that typically react with alkenes. Thus, chlorination or bromination generally occurs only in the presence of a Lewis acid, which allows a greater fraction of the positive charge to develop on the electrophilic atom. The role of the Lewis acid AlCl3 in the chlorination of benzene is illustrated below; we can consider the electrophilic species as Cl+.
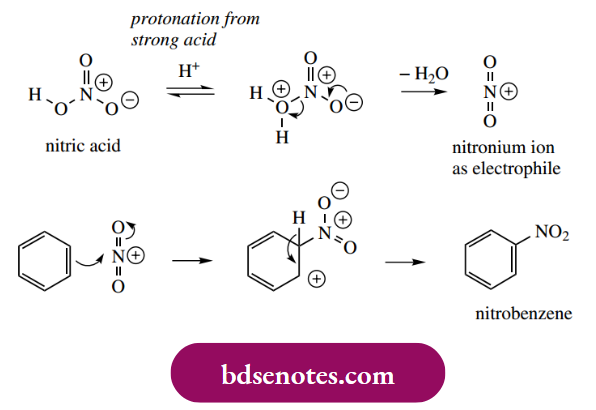
Nitration of an aromatic ring using nitric acid also requires the presence of sulfuric acid. Nitric acid is protonated by the stronger sulfuric acid, leading to loss of water and production of the nitronium ion as electrophile.
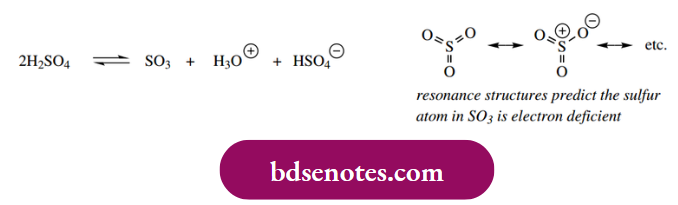
Aromatic sulfonation occurs with fuming sulfuric acid, where the electrophile is sulfur trioxide. This is present in concentrated sulfuric acid as a result of the equilibrium shown.
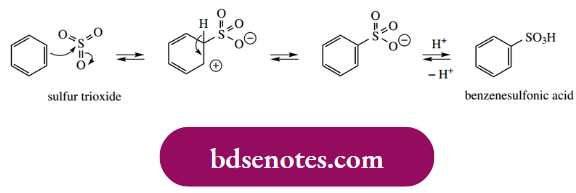
The product of electrophilic aromatic substitution is a sulfonic acid. Unusually, sulfonation is found to be reversible; it is possible to replace an SO3H group attached to an aromatic ring with hydrogen by heating the sulfonic acid with steam.
Electrophilic Alkylations: Friedel–Crafts Reactions
Particularly useful reactions result from Friedel–Crafts alkylations and acylations, in which the electrophile is developed from either an alkyl halide or an acyl halide in the presence of a Lewis acid. The alkylation reaction is mechanistically similar to the halogenation process above, with the Lewis acid increasing polarization in the alkyl halide.
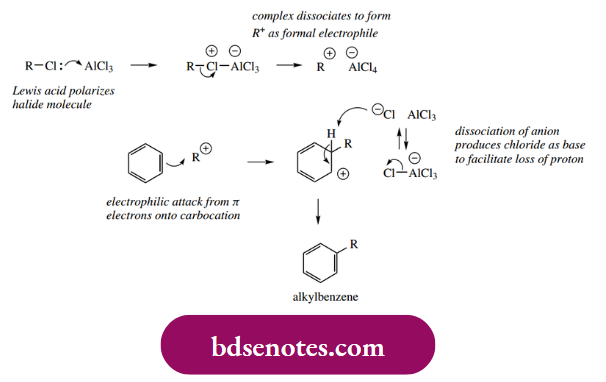
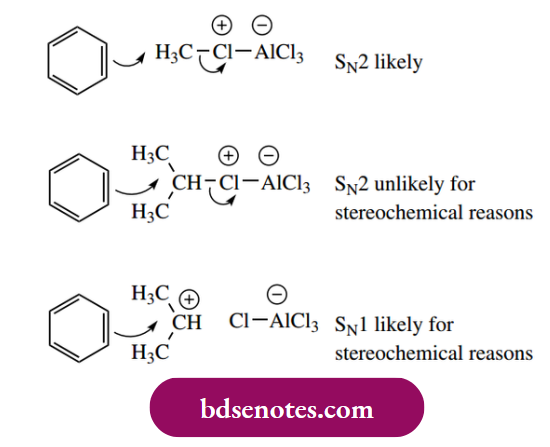
- However, although we invoked a Lewis acid complex to provide the halonium electrophile, there is considerable evidence that, where appropriate, the electrophile in Friedel–Crafts alkylations is the dissociated carbocation itself.
- Of course, a simple methyl or ethyl cation is unlikely to be formed, so there we should assume a Lewis acid complex as the electrophilic species. On the other hand, if we can get a secondary or tertiary carbocation, then this is probably what happens.
- There are good stereochemical reasons why a secondary or tertiary complex cannot be attacked. Just as we saw with SN2 reactions, if there is too much steric hindrance, then the reaction becomes SN1 type.
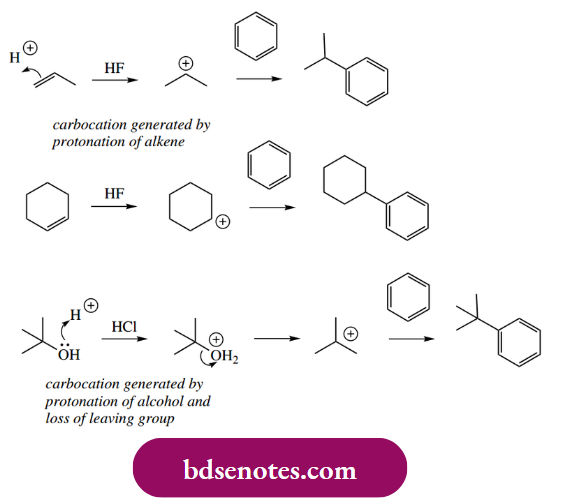
Indeed, we can also achieve alkylation of an aromatic ring by using any system that generates a carbocation. In effect, we are paralleling the concept of carbocations as electrophiles as in Section 8.3, but using an aromatic substrate. Thus, an alkene in strongly acidic conditions, or an appropriate alcohol in acid, may be used to generate a carbocation and achieve electrophilic substitution.
This involvement of carbocations limits the utility of Friedel–Crafts alkylations, because, as we have already noted with carbocations, rearrangement reactions complicate the anticipated outcome. For instance, when a Lewis acid is used to generate what would be a primary carbocation, rearrangement to a secondary carbocation is likely to occur. Both types of cationic species can then bond to the aromatic ring, and a mixture of isomeric products is formed.
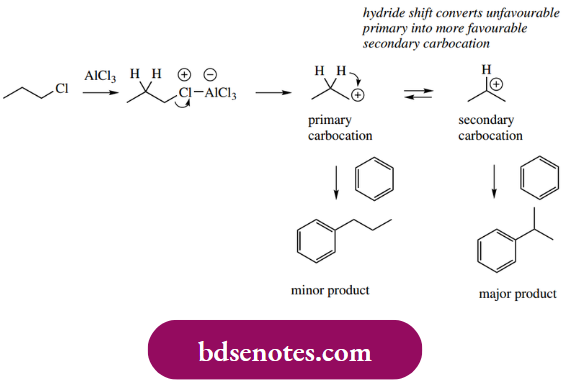
- To be satisfactory, a Friedel–Crafts alkylation requires one relatively stable secondary or tertiary carbocation to be formed from the alkyl halide by interaction with the Lewis acid, i.e. cases where there is not going to be any chance of rearrangement.
- Note also that we are unable to generate carbocations from an aryl halide – aryl cations are unfavourable – so we cannot use the Friedel–Crafts reaction to join aromatic groups. There is also one further difficulty, as we shall see below.
- This is the fact that the introduction of an alkyl substituent onto an aromatic ring activates the ring towards further electrophilic substitution. The result is that the initial product from Friedel–Crafts alkylations is more reactive than the starting material, with the consequence that di-, tri-, and poly-alkylated products also tend to be formed. It may be possible to minimize this if the starting material is readily available, for Example. benzene, and can thus be used in large excess.
Electrophilic Acylations Friedel – Crafts Reactions
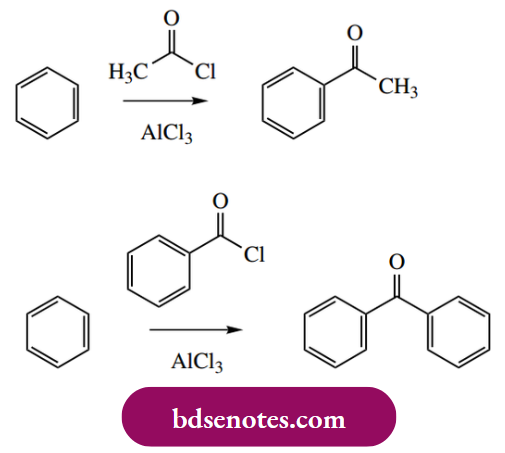
In Friedel–Crafts acylations, an acyl halide, almost always the chloride, in the presence of a Lewis acid is employed to acylate an aromatic ring. The process is initiated by the polarization of the carbon–chlorine bond of the acyl chloride, resulting in the formation of a resonance-stabilized acylium ion.
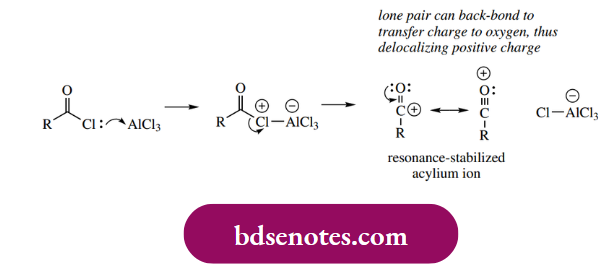
The acylium ion is now our electrophile, and aromatic substitution proceeds in the predicted manner. The intermediate cation is subsequently deprotonated to yield the acylated product. However, this acyl derivative is a ketone, which can also be complex with the Lewis acid.
Accordingly, the final complex must be decomposed by treatment with water, and the significant consequence is that the Lewis acid has to be supplied in stoichiometric amounts, in sharp contrast to Friedel–Crafts alkylations, where only catalytic amounts need to be used.
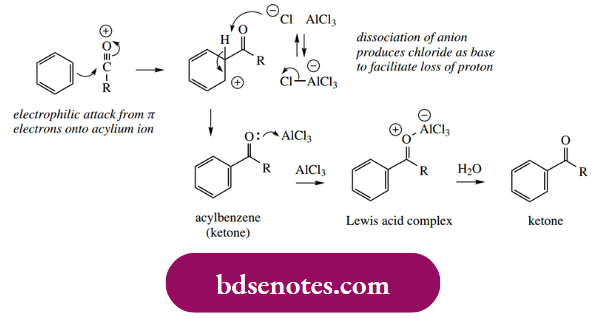
- A similar problem of complex formation may be encountered if either amino or phenol groups are present in the substrate, and the reaction may fail. Under such circumstances, these groups need to be blocked (protected) by making a suitable derivative.
- Nevertheless, Friedel–Crafts acylations tend to work very well and with good yields, uncomplicated by multiple acylations, since the acyl group introduced deactivates the ring towards further electrophilic substitution. This contrasts with Friedel–Crafts alkylations, where the alkyl substituents introduced activate the ring towards further substitution.
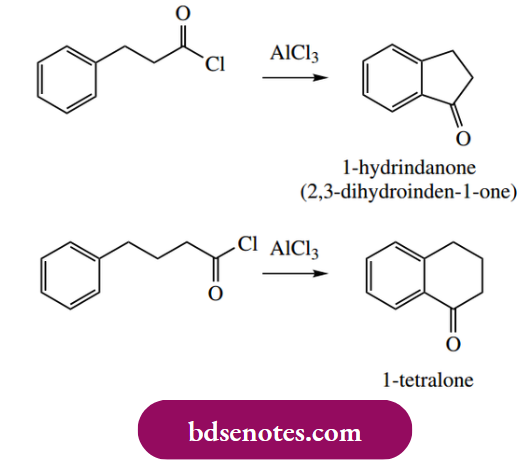
A useful extension of Friedel–Crafts acylation is an intramolecular reaction leading to cyclic products. Thus, five- and six-membered rings are readily and efficiently created by use of an appropriate aryl acyl chloride, as shown below.
Effect Of Substituents On Electrophilic Aromatic Substitution
Substituents already bonded to an aromatic ring influence both the rate of electrophilic substitution and the position of any further substitution. The effect of a particular substituent can be predicted by a consideration of the relative stability of the first-formed uranium cation, the formation of which constitutes the rate-limiting step. In general, substituents that are electron-releasing activate the ring to further substitution – they help to stabilize the arenium ion. Substituents that are electron withdrawing destabilize the arenium ion and, therefore, are deactivating and hinder further substitution.
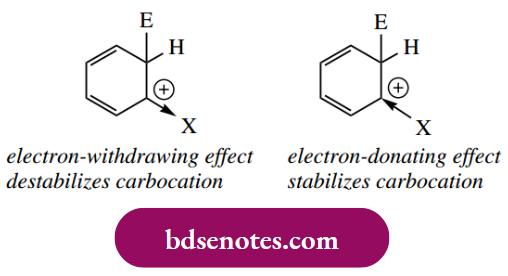
For the position of further substitution, we also need to consider resonance forms of the areniumion.
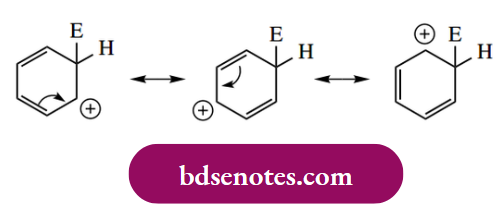
- From these resonance forms, we can deduce that positions ortho and para to the position of attack are electron deficient. This means that any pre-existing substituent will produce maximum effect if it is located in any of these positions. We normally think in terms of the existing substituent directing the attack of the electrophile to a position that optimizes the stability of the arenium ion.
- Electron-releasing substituents are thus ortho and para directing because they help to stabilize the arenium ion; electron-withdrawing substituents destabilize the arenium ion more if they are ortho or para, and, consequently, they are found to be meta directing. The observed electron-releasing and electron-withdrawing properties of various groups are summarized in Table 8.1, though to understand these
Directing Effects Of Substituents In Electrophilic Aromatic Substitution

- fully we need to consider the properties in terms of both inductive effects and delocalization or resonance effects. We must also appreciate that the term ‘directing’ indicates the major product(s) formed and not the exclusive product. Mixtures of products are the norm. Stabilization of the arenium ion through electron-donating effects is typical of alkyl substituents. This is not strictly an inductive effect.
- But Is Derived From Overlap Of Σ Orbitals With The Aromatic π Orbital System. Thus, With Both Ortho And Para Addition, There Is One Resonance Form Of The Arenium Ion That Is Particularly Favourable,
- The Positive Charge Is Adjacent To The Electron-Donating Alkyl Group, Which Helps To Charge, Aith A Carbocation. There Is No Particularly Favourable Resonance Form Resulting From Meta Addition.
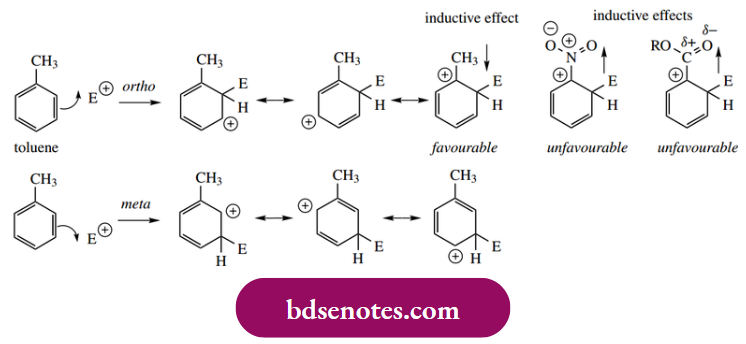
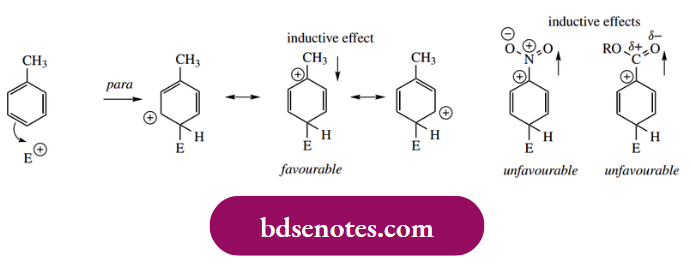
Nitration of toluene gives approximately 59% o-nitrotoluene, 37% p-nitrotoluene, and only 4% m-nitrotoluene. On the other hand, nitration of nitrobenzene gives about 93% m-dinitrobenzene, 6% o-dinitrobenzene, and 1% p-dinitrobenzene.
- Since nitro is an electron-withdrawing group, those resonance forms stabilized by the presence of an alkyl substituent are going to be seriously destabilized when alkyl is replaced by an electron-withdrawing group, and so meta substitution predominates.
- This can be appreciated even more readily when one looks at the representation of the nitro group or, say, an ester group the unfavourable resonance forms have the positive charge positioned adjacent to a full or partial positive charge.
- However, we must also realize that toluene undergoes electrophilic substitution some 25 times as readily as benzene because of the beneficial inductive effect, whereas nitrobenzene undergoes nitration only about 10−4 times as readily as benzene because the inductive effect withdraws electrons from the ring.
- Groups that are particularly strong electron releasers do not achieve this by an inductive effect, but they have heteroatoms with lone pair electrons that can stabilize resonance structures by transferring the charge to the heteroatom, i.e. an electron-releasing resonance effect. An amino group is typical of this type of substituent.
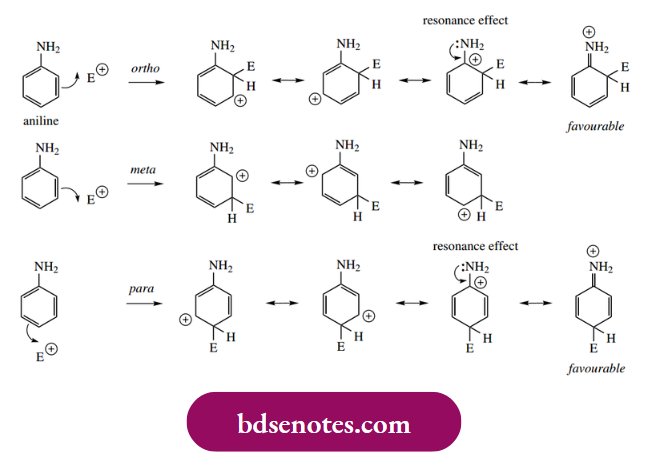
It can be seen that lone pair donation creates another favourable resonance form only when an electrophilic attack is ortho or para to the amino group. There is a small electron-withdrawing inductive effect for an amino group due to the heteroatom, but this effect is vastly overpowered by the resonance effect, and an amino group is strongly activating and gives rise to ortho and para substitution.
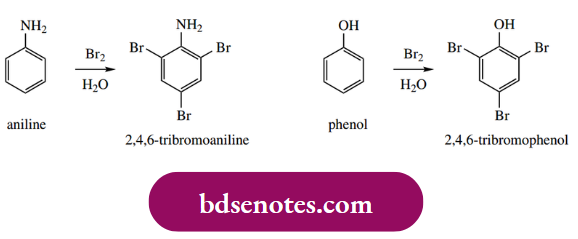
Aniline reacts readily with bromine in water, without any need for a catalyst, giving 2,4,6-tribromoaniline in nearly quantitative yield. The same is true for phenol, which rapidly gives 2,4,6-tribromophenol, because of the powerful activation provided by the phenol group. The activation is so great that all three positions are brominated.
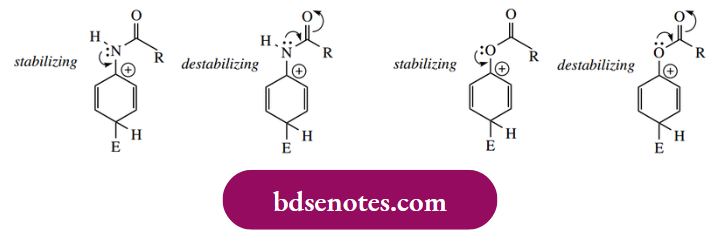
Groups such as amides and esters, where the heteroatom is bonded to the aromatic ring, might be expected to behave similarly; this is true, but the level of activation is markedly less than for amines and phenols.
We can understand this because of two conflicting types of resonance behaviour in such molecules. These groups do activate the ring towards electrophilic attack and are ortho and para-directing, but activation is considerably less than with simple amino and phenolic groups.
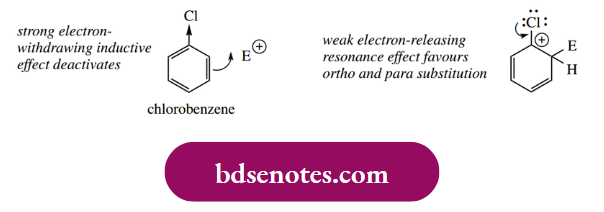
For most substituents, electron-donating ones activate the ring towards electrophilic attack and also direct ortho and para. Conversely, electron-withdrawing substituents are deactivating and direct substitution meta. This appears so straightforward a concept that we must have an exception; this is found with halogen substituents.
- Thus, chlorobenzene is nitrated about 50 times more slowly than benzene, but yields o- and p-nitro products. However, the explanation is simple and does not alter our reasoning. It turns out that, because of the high electronegativity of halogen atoms, we have a very strong electron-withdrawing inductive effect and, consequently, significant deactivation towards electrophilic substitution.
- However, because there is an electron-donating resonance effect we get ortho and para substitution. This lone pair donation is not nearly as effective as with oxygen and nitrogen, however, because, in the larger atom, the orbitals are less able to overlap effectively.
- So we have conflicting trends, deactivation from a strong inductive effect through σ bonds, but ortho and para directing because of a weak resonance effect through the π bond system.
An understanding of electron-donating and electron-withdrawing substituent effects is crucial to designing the synthesis of aromatic derivatives. For example, from the electrophilic substitution reactions we have studied, there are two potential approaches for the synthesis of nitro acetophenone:
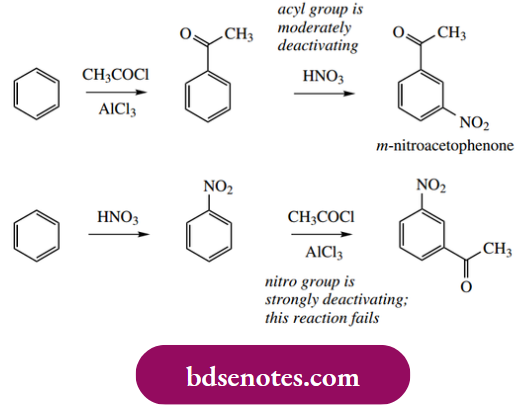
Only the first of these is effective because strongly deactivating groups such as nitro almost completely inhibit Friedel–Crafts acylation (or alkylation), and the alternative sequence shown will fail at the second step.
- Accordingly, the workable route inserts the less effective deactivating group, the acyl group, first, so that the second electrophilic substitution can proceed, even though it tends to be fairly slow.
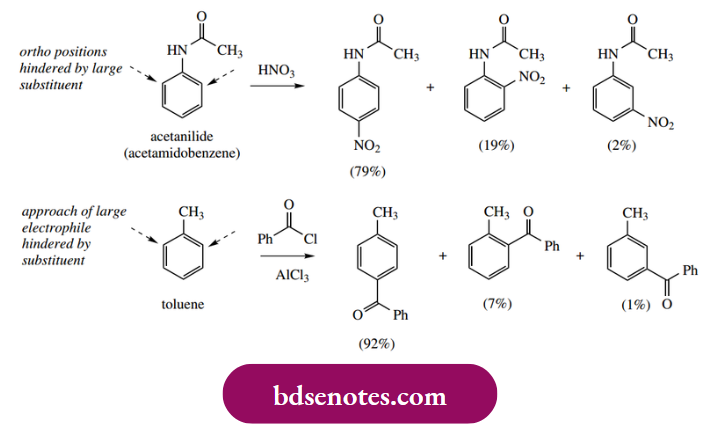
- Although electron-donating substituents activate the ring towards electrophilic attack, they are both ortho and para-directing and an electrophilic substitution reaction can be expected to yield a mixture of products that must be separated. In practice, this problem can be minimal because of steric considerations.
- When the original substituent is large, or the incoming substituent is large, the steric interaction will be considerably less with para substitution than with ortho.
- Thus, both nitration of acetanilide and acylation of toluene give predominantly the para product. Note that small amounts of the meta product are inevitably formed as well as the ortho and para products; these reactions are only regioselective.
This is a good time to have another brief look at Sections 4.3.5 and 4.5.4 and compare how we used similar reasoning to consider the likely stability, or otherwise, of anions and cations to predict the acid-base properties of aromatic amines and phenols.
The rationalizations are essentially identical. The effect of heteroatoms on electrophilic aromatic substitution, Example. the reactions of pyridine will be considered separately.
Synthesis Of Ibuprofen
There are several approaches to the synthesis of the analgesic anti-inflammatory drug ibuprofen. Here is one that employs a relatively simple sequence of reactions, beginning with a Friedel–Crafts acylation of isobutylbenzene.
- The alkyl substituent is weakly electron-releasing and thus activates the ring towards electrophilic substitution. It also directs further substitution to the ortho and para positions.
- As in most Friedel–Crafts acylations, the para product predominates strongly over the ortho, a consequence of the relatively large size of the electrophilic reagent. In this case, we also have a quite large alkyl substituent, again disfavouring the ortho product.
- The subsequent steps are relatively straightforward. Sodium borohydride reduction of the ketone gives an alcohol, and then the alcohol is converted into a nitrile by successive nucleophilic substitution reactions.
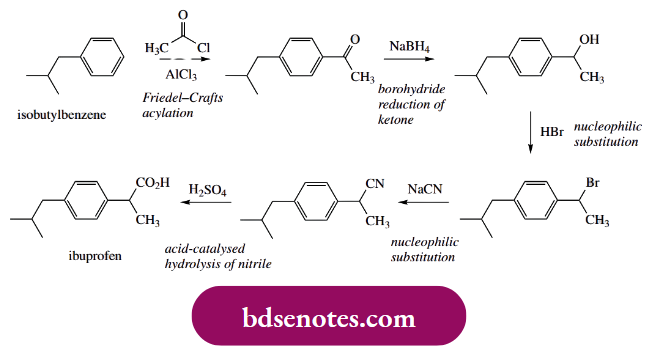
Note that a two-stage process is involved. Since hydroxide is a poor leaving group, nucleophilic substitution requires acidic conditions to protonate the hydroxyl to provide a better-leaving group. We can formulate an SN1 conversation since this would involve a favourable benzylic carbocation.
- HCN is a weak acid (pKa 9.1), so it is not very effective in protonating the hydroxyl group. Thus, the two-stage process is used, with displacement of hydroxyl via bromide, then subsequent displacement of bromide by cyanide, the latter step usually being an SN2 process.
- Lastly, the nitrile group is hydrolysed to a carboxylic acid. The starting material, isobutylbenzene, is readily available but could be synthesized by exploiting another Friedel–Crafts reaction.
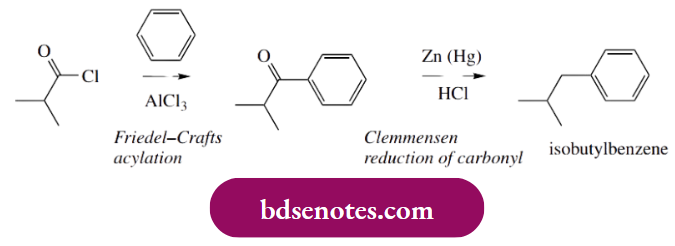
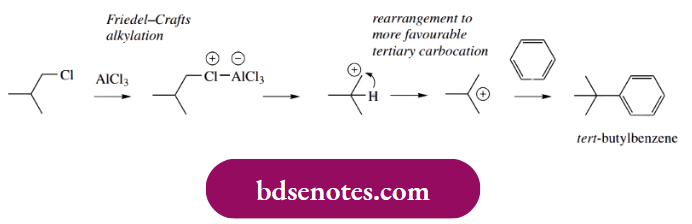
- Note that a Friedel–Crafts alkylation is not a good idea. There is too much chance of rearrangement occurring since we are trying to generate the equivalent of a primary carbocation. We might expect that rearrangement of the primary carbocation to a tertiary carbocation by hydride migration would occur,
- so that the product would turn out to be tert-butylbenzene rather than isobutylbenzene. The approach then is to use Friedel–Crafts acylation, then reduce the carbonyl group by an appropriate method, here a Clemmensen reduction (see below).
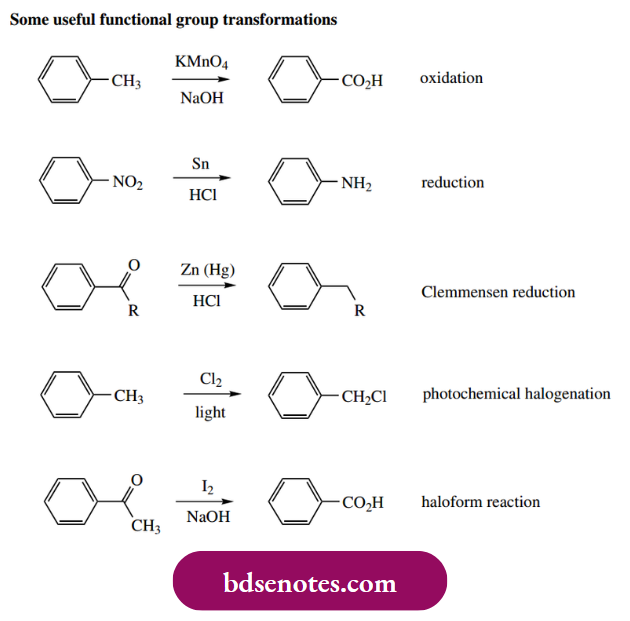
- The substituents that can be introduced by electrophilic substitution appear somewhat limited, but there exist standard chemical processes for converting these into other functional groups, thereby extending significantly the scope for this type of process. A few of these are shown below, though they will not be elaborated upon here.
Electrophilic Substitution On Polycyclic Aromatic Compounds
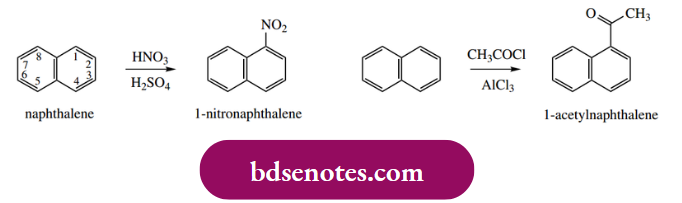
Fused-ring cyclic hydrocarbons such as naphthalene and anthracene display the enhanced stability and reactivity associated with simple aromatic compounds like benzene. We have briefly looked at the π electron systems and their aromatic status in Chapter 2. In this short section, we wish to demonstrate how the principles developed above for rationalizing the behaviour of benzene compounds can be extended to the more complex ring systems.
We observe that nitration of naphthalene using nitric acid–sulfuric acid gives predominantly 1- nitro naphthalene (sometimes α-nitro naphthalene), and that Friedel–Crafts acylation with acetyl chloride–AlCl3 gives mainly 11-acetyl naphthalene
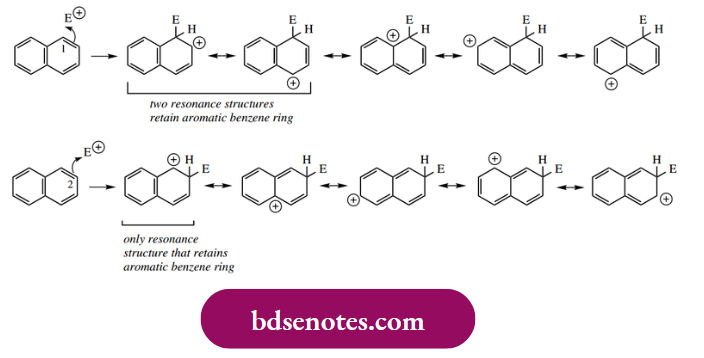
This behaviour can readily be explained. Let us simply consider the resonance structures for the intermediate cation following an attack of electrophile at position 1 ( α position) or at position 2 ( β position).
On drawing these out, we find that two of the five structures retain a benzene ring if an attack occurs at position 1. For attack at position 2, only one resonance structure has a benzene ring. We know that a benzene ring has special stability, so we can predict that the intermediate cation with more benzenoid resonance structures should be the more stable. This fits with the observation that electrophilic substitution occurs predominantly at position 1.
Using the same reasoning, it is not difficult to see why anthracene becomes substituted on the central ring. The intermediate cation then benefits from the stability of two benzene rings, which is

substantially more than for a single naphthalene ring. Anthracene undergoes aromatic substitution more readily than naphthalene, and can frequently lead to disubstitution, with both substituents on the central ring.
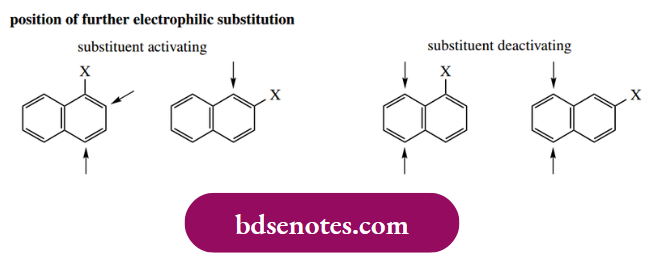
- We can also rationalize how a substituent on naphthalene will direct further substitution. If we have an activating group at position 1, the electrophilic attack will occur on the same ring and at positions 2 or 4.
- Consideration of resonance structures shows that the benzene ring can be retained whilst providing favourable structures in which an electron-releasing group minimizes the charge. Further, those groups that are electron-releasing through lone pair donation, e.g. NH2 or OH, are ideally placed to delocalize charge. This is shown in the case of para and ortho attacks.
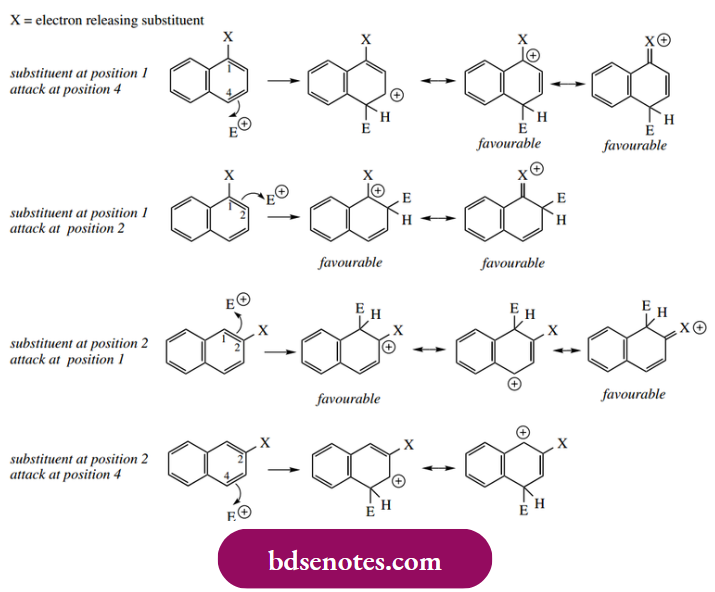
- Should the activating substituent be at position 2, further substitution will be almost exclusively at position 1; this follows from consideration of resonance structures, where the 2-substituent has minimal effect if an attack occurs at position 4. Of course, this would equate to a meta attack, which we know is unfavourable for an ortho and para director.
- Deactivating (electron-withdrawing) substituents do just that: they deactivate the ring to which they are attached and hinder any further attack. Hence, further electrophilic substitution occurs on the other ring whether the substituent is at C-1 or C-2.
- Further substitution occurs at positions 5 or 8, the positions most susceptible to attack. These trends are summarized below, though we recommend deducing the reactivity rather than committing it to memory.
Leave a Reply