Drugs Acting On Central Nervous System Sedatives And Hypnotics Introduction
In general, sedative-hypnotics are drugs used to slow down mental and physical functions of the body. These are also referred to as CNS depressants.
- Sedatives are chemical agents that tend to produce a calming effect, relax muscles, and relieve feelings of tension, anxiety, and irritability without causing drowsiness or sleep. Example: Potassium bromide.
- At higher doses, most of these sedative drugs will also produce drowsiness and eventually produce sleep.
- Drugs that have such a sleep-inducing effect or produce sleep similar to that of natural sleep are called hypnotic drugs or hypnotics, commonly known as sleeping pills, and are a class of psycho-depressive drugs. Example: Thiopentone sodium.
- There is no sharp distinction between sedatives and hypnotics and the same drug may have both actions depending on the method of use and the dose employed. At other times, the drug may act as a sedative while at a higher dose, the same drug may act as hypnotic.
However, the combination of the terms sedative-hypnotic appropriately identifiers the pharmacological effects of these drugs.
- In reality, almost any drug that calms, soothes, and reduces anxiety is also capable of relieving insomnia and is frequently used in pre-anesthetic medication and as an adjunctive therapy in psychiatry.
- Although narcotics and sedative-hypnotics share many of the same actions, the latter drugs have no practical pain-relieving properties.
- Unlike narcotics, intoxicating doses of sedative-hypnotics almost always result in impaired judgment, slurred speech, and loss of motor function.
Sedatives And Hypnotics Classification Of Sedatives And Hypnotics
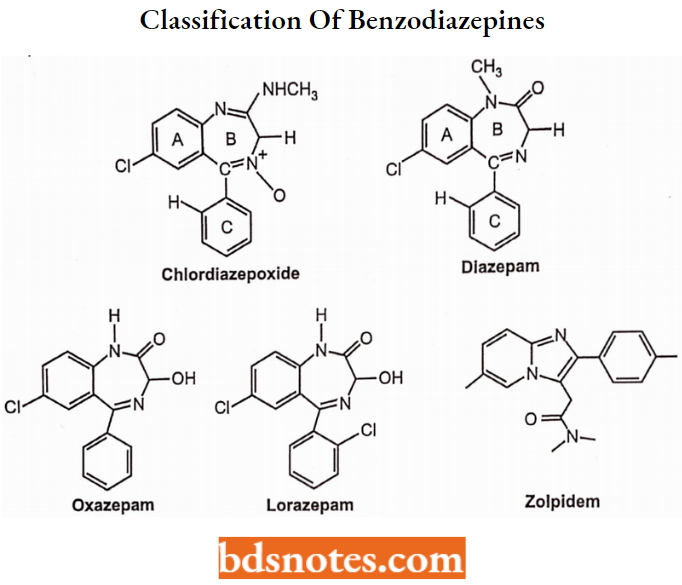
- Benzodizepine derivatives: Chlordiazepoxide, Diazepam, Oxazepam, Chlorazepate,
Alprazolam, Lorazepam, Zolpidem. - Barbiturates: Barbital, Phenobarbital, Mephobarbital, Amobarbital, Butabarbital, Pentobarbital, Secobarbital.
- Miscellaneous:
- Amides and imides: Glutethimide.
- Aldehydes and their derivatives: Paraldehyde, Triclofos sodium.
- Alcohols and their carbamate derivatives: Ethchlorvynol, meprobamate.
Sedatives And Hypnotics Benzodiazepines
Benzodiazepines have sedative, hypnotic, anti-anxiety, anticonvulsant, and muscle relaxant properties.
- Benzodiazepines have been used therapeutically since the early 1950s and were initially part of a class of therapeutic agents referred to as tranquilizers.
- The “new” benzodiazepine agents were safer than the older barbiturate agents with far less addiction potential.
- Presently, benzodiazepines are referred to as sedative-hypnotic agents and have some different therapeutic uses including the treatment of anxiety disorders, insomnia seizure disorders, alcohol withdrawal symptoms, and conscious sedation anesthesia.
However, the use of BZDs is often controversial or general as they are widely acknowledged to be addictive, and withdrawal symptoms can occur after 4-6 weeks of continuous use.
- This led to the recommendation that they should not be used as hypnotics or anxiolytics for longer than 4 weeks.
- They replaced barbiturates and meprobamate in the treatment of anxiety because they are safer (less side effects and dependence) and more effective. At low doses they as sedatives and at high doses they produce a hypnotic effect.
Classification of Benzodiazepines
Classification of benzodiazepines is classified according to the duration of action into:
- Short-acting (3-8 hours): Triazolam, Oxazepam.
- Intermediate (10-20 hours): Alprazolam, Lorazepam, Estazolam, Term are useful azepam.
- Long acting: (1-3 days): Diazepam, Chlordiazepoxide, Flurazepam, Quazepam, Clorazepate.
Mechanism of Action of Benzodiazepine
Mechanism of action: The great breakthrough in our understanding of the mechanism of action of BZDs came in the mid-1970s.
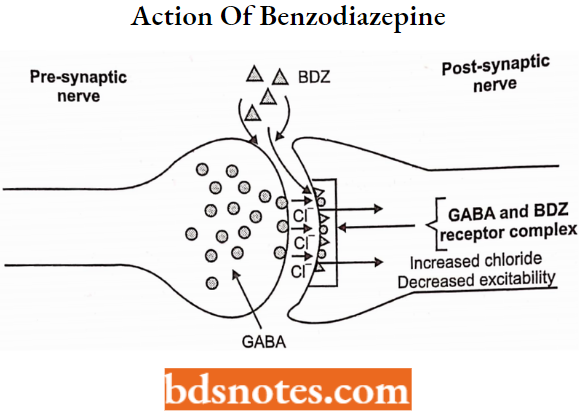
- When biologists at Hoffman-La Roche demonstrated that BZDs exert their psychotropic effects by Gamma-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the mammalian central nervous system (CNS).
- Eliciting its physiological effects through interaction with several distinct classes of cell-surface receptors: GABAA, GABAB, and GABAC receptors. The GABAA receptor is the most abundant and is a member of the superfamily of ligand-gated ion channels.
- The interaction of GABA with this receptor determines the opening of the intrinsic chloride ion selective channel, which is followed by an increase in chloride flux, with the result of hyperpolarization of the neuronal cell membrane and a concomitant decrease in neuronal transmission.
The GABAA receptor complex also has other distinct, high-affinity binding sites able to modulate the channel function, such as the benzodiazepine receptor (BzR), the picrotoxin site, the barbiturate site and sites that bind neurosteroids, and ethanol.
- All of these other sites are “allosteric” and function as modulatory sites that regulate GABA affinity.
- Among the allosteric receptor sites, the sites for the benzodiazepines (BZD) are of prime importance since these ligands have been employed widely as anxiolytic or anticonvulsant agents since the 1960s.
- Studies of molecular biology have suggested that the GABAA/BZR complex is an entameric protein polymer constituted principally from a, p, and y subunits.
- At hetero, a total of 16 subunits (6α, 4β, 3γ, 1δ, and 2ρ) have been isolated and identified from the CNS (15 of these have been found in the mammalian CNS).
- Recent studies of recombinant GABAA/BZR have shown that the presence of α, β and γ subunits is necessary to constitute a fully functional benzodiazepine receptor or GABA or chloride ions channel that mimics the pharmacological, biochemical, and electrophysiological properties of a native receptor.
Structural Activity of Benzodiazepine
Benzodiazepines are bicyclic heterocycles that contain a benzene nucleus being fused to a seven-membered hetero ring having two nitrogen atoms, five carbon atoms, and a maximum possible number of cumulative double bonds.
“Benzo” prefix benzene ring fused onto the diazepin ring. “R” labels denote common locations of side chains, which give different benzodiazepines with different properties.
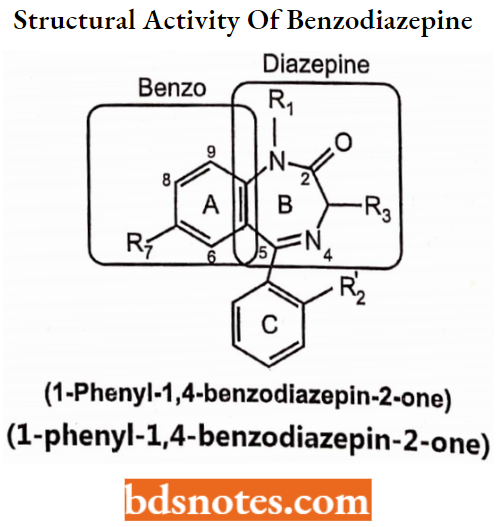
Rings A, B, and C are required for BDZ-receptor binding activity:
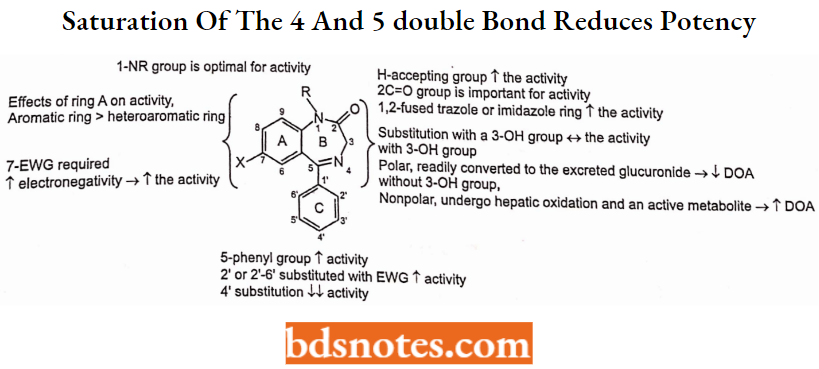
Ring A participates in “pi-pi stacking” interactions with a complimentary functionality on the receptor.
- An electron-withdrawing group or electronegative group at R7 (Usually Cl, Br, N02, or CN) is required for optimal receptor affinity and enhances the activity.
NO2 > Br > CF3 > Cl - Substitution on other positions (6,8 and 9) of this ring may decrease activity.
- Ring C contributes to BDZ-receptor binding through hydrophobic and steric interactions:
- Position R2 may be unsubstituted or contain halogen atoms. Halogenations (F, Cl) generally increase BDZ activity.
- Substitution on other positions of this ring may decrease activity (4 ).
Ring B is required for optimal BDZ-receptor binding,
- R1 can be H, CH3, or relatively small alkyl groups that show optimal activity
- Amide can be replaced with an amidine group as in chlordiazepoxide.
- Amide can be replaced with heterocycle such as imidazole or triazole resulting in pharmacologically active benzodiazepine derivative with the highest affinity.
- A proton-accepting group (carbonyl oxygen) at 2-position is necessary to interact with receptor histidine residue that acts as a proton donor and helps in ligand binding. Electron donating group must be in the same” plane as the electronegative group on ring A, favoring a coplanar spatial orientation of two moieties Substitution of O with S effect selective binding GABA BZR sub-populations but anxiolytic activity is maintained.
- Neither the amide C=0 nor N-alkyl groups (R1) directly contribute to binding. If replacement of the carbonyl function with two hydrogens in position-2 gives medazepam, less effective than diazepam.
- Substitution of 3-position methylene or imine nitrogen is sterically unfavorable. Derivatives having 3-hydroxy moiety have comparable potency to non-hydroxylated analogs but are excreted faster.
- The 4-5-imino group is not required for activity.
- Saturation of the 4,5-double bond reduces potency, as does a shift of the unsaturation into the 3,4-position.
N-l-Substituted-3-Unsubstituted Benzodiazepines (“Diazepams”)
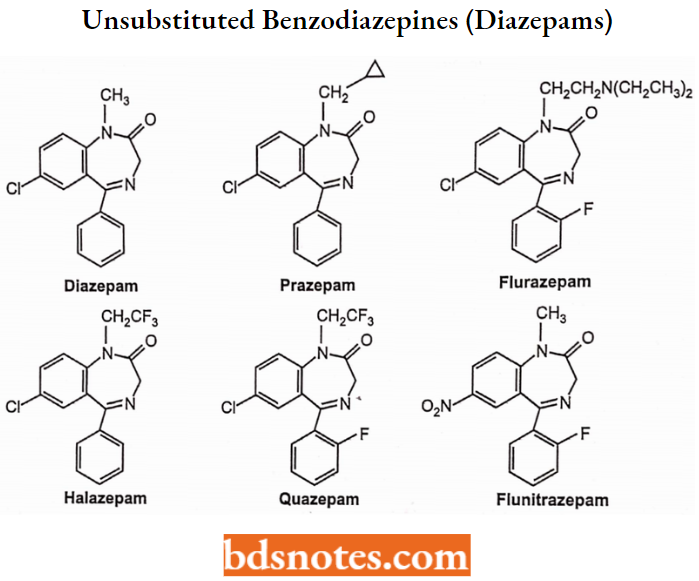
Aminidino-N-oxide Benzodiazepines (Chlordiazepoxide)
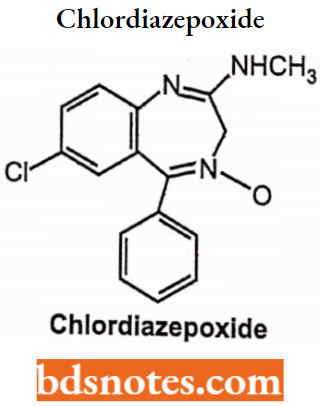
3-Carboxyl-N-l-Unsubstituted Benzodiazepines (Clorazepate)
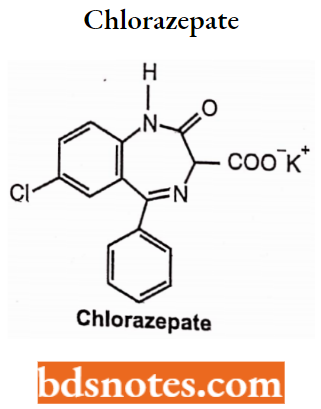
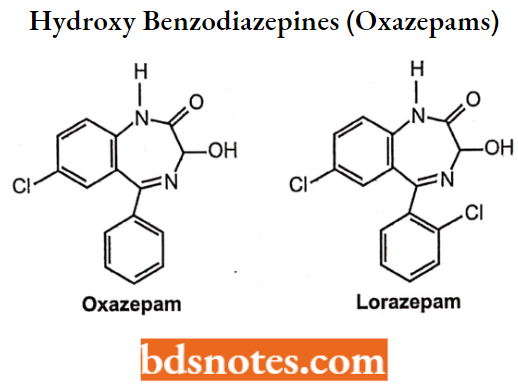
N-l-Substituted-3-Hydroxy Benzodiazepines (“N-Alkyl-Oxazepams”)
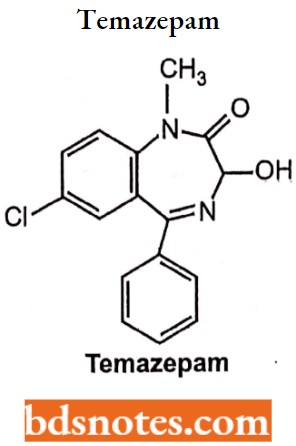
N-l-Unsubstituted-3-Hydroxy Benzodiazepines (“Oxazepams”)
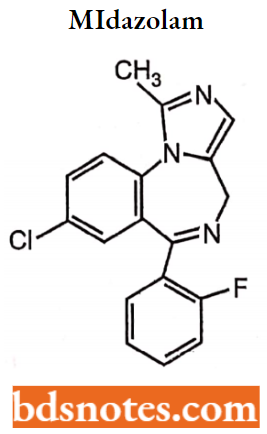
Imidazo-Benzodiazepines
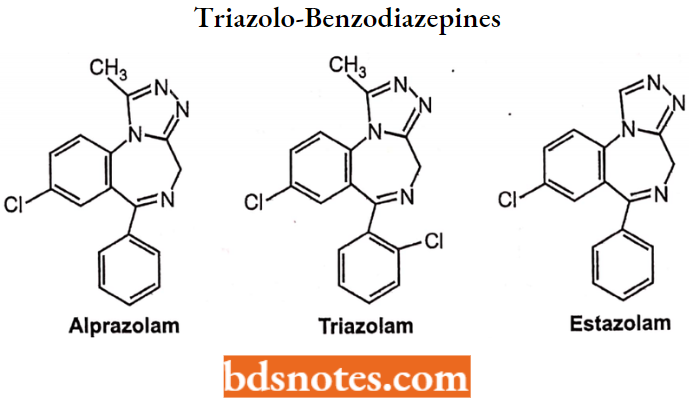
Triazolo-Benzodiazepines
Clinical Uses of Benzodiazepines
- Anxiety disorders: Generalized anxiety disorder, acute anxiety, panic disorder, phobias (social, simple), post-traumatic stress disorder, and obsessive-compulsive disorder.
- Anxiety associated with medical illness: Gastrointestinal, cardiovascular, and somatoform disorder.
- Insomnia.
- Convulsive disorders.
- Acute status epileptics: Neonatal seizures or febrile, tetanus, convulsions preeclampsia.
- Adjunct to other anticonvulsants.
- Amnestic (before surgery or procedure).
- Spastic disorders and other types of acute muscle spasms, multiple sclerosis, cerebral palsy, and paraplegia secondary to spinal trauma.
- Involuntary movement disorders, restless leg syndrome, akathisia associated with neuroleptic use, myoclonus, and chloroform disorders.
- Detoxification from alcohol and other substances.
- Agitation or anxiety associated with other psychiatric conditions, psychotic illness, acute mania, anxiety associated with depression, impulse control disorders, catatonia, or mutism.
- Diagnostic studies such as magnetic resonance imaging, computed tomography, and endoscopic cardio version chemotherapy.
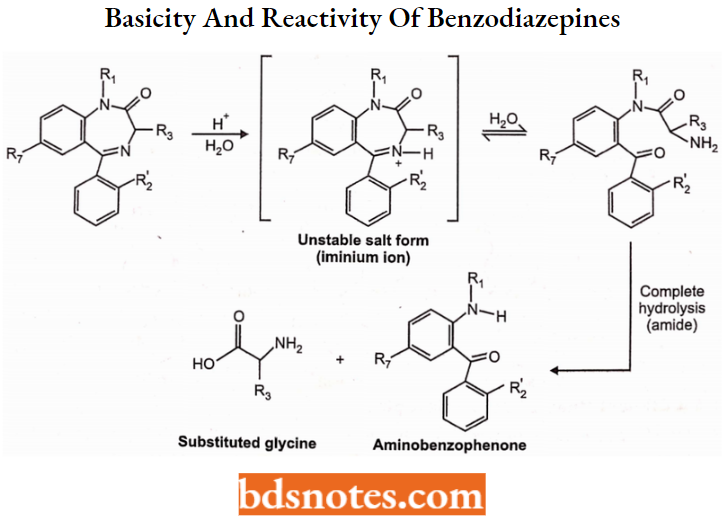
Basicity and Reactivity of Benzodiazepines
The BDZs are weak organic bases with the most basic nitrogen being the imine N4 (amide at positions 1,2 is non-basic). Thus BDZ salts can only be formed with strong acids.
- Unfortunately, such strong acid salts are unstable and readily undergo sequential hydrolysis, first at the imine bond and then at the amide to yield inactive products.
- The first hydrolysis reaction (imine hydrolysis) is reversible; however, the second (amide hydrolysis) eliminates GABA receptor activity.
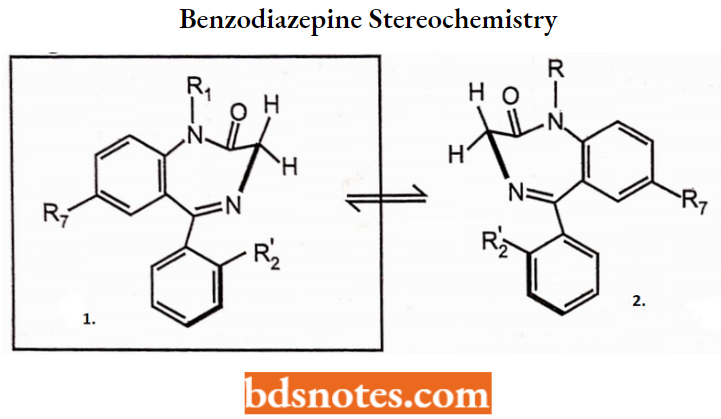
Benzodiazepine Stereochemistry
Most BDZs do not have a chiral center, but the 7-membered B ring in these compounds may adopt one of two energetically preferred boat conformations (1 and 2 below) which are enantiomeric relative to each other.
Some studies suggest conformation is preferred for BZD receptor binding:
Benzodiazepine Lipophilicity:
As a class benzodiazepines are relatively lipophilic compounds due to their high hydrocarbon content and presence of halogen atoms. Also, most (not all) benzodiazepines do not behave as acids or bases under physiologic conditions and thus are not ionized.
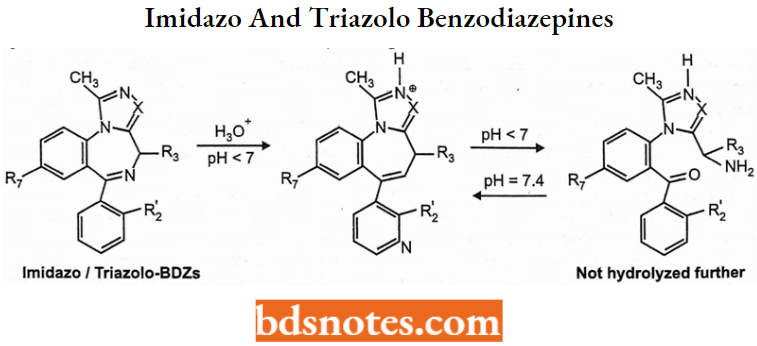
Basicity and Reactivity of “Imidazo- and Triazolo-Benzodiazepines”:
The tricyclic benzodiazepines have a more basic nitrogen atom (or more) in their additional ring structure (imidazole or triazole ring).
- These nitrogen atoms typically are not sufficiently basic to be protonated (ionized) at physiologic pH, but they are sufficiently basic to yield water-soluble salts when treated with strong acids.
- The salts formed from the heterocyclic benzodiazepines are more stable. When placed in aqueous media the heterocyclic salts may undergo imine hydrolysis similar to the traditional agents.
- However, no further hydrolysis (to inactive products) can occur since the heterocyclic compounds no longer have an acid labile amide group; in these compounds, the amide was replaced with the heterocyclic group.
Thus at acidic pH imine hydrolysis may occur as shown below but the reaction does not proceed, and at physiologic pH (post-injection) reformation of the benzodiazepine ring is favored as shown below:
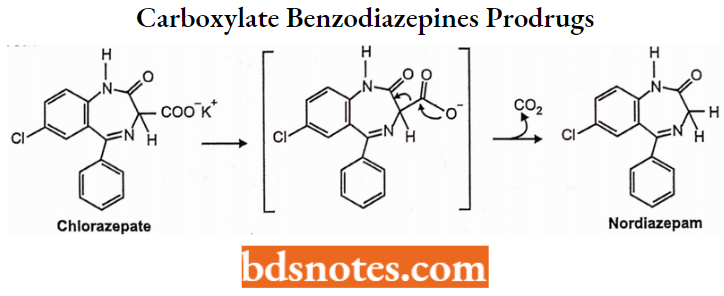
Clorazepate and 3-Carboxylate Benzodiazepines: Prodrugs:
- The 3-carboxylate benzodiazepines are unique in that they contain a 3-COO K+ functionality which allows for water solubility. These drugs, of which chlorazepate is the prototype, function as water-soluble prodrugs for the more traditional benzodiazepines.
- When administered (orally) they are readily protonated in the upper GI tract and spontaneously decarboxylate (loss of C02) as shown below to yield an active benzodiazepine that is absorbed from the gut. This is a chemical reaction and not an enzyme-catalyzed reaction.
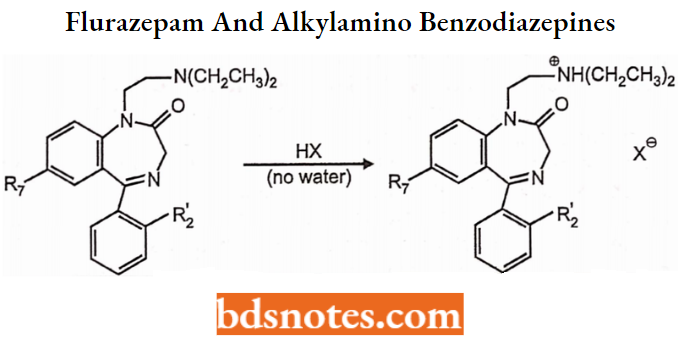
Flurazepam and 1-Aikylamino Benzodiazepines:
- Flurazepam differs from other traditional benzodiazepines in that it contains a 1-(diethylamino)ethyl side chain. The nitrogen atom in this side chain is a typically tertiary amine and is relatively basic (pKa about 9).
- Thus this nitrogen serves as a basic center for salt formation as shown below:
Sedatives And Hypnotics Benzodiazepine Pharmacokinetics
Oral administration: Most BDZs are relatively weak bases and relatively lipophilic, resulting in fairly rapid and complete absorption from the GI tract.
- Absorption from other sites (IM) may be more erratic due to the lipophilicity of these compounds. Generally, the rate of absorption from the GI tract is dependent on the lipophilicity of the BDZ.
- For example, diazepam is absorbed very rapidly, while oxazepam and clonazepam are absorbed more slowly.
Distribution: BDZs are highly protein bound (70-99%) but rapidly distribute to CNS. They cross the BBB by passive diffusion, thus the rate of CNS distribution correlates with lipophilicity (diazepam is “appropriately” lipophilic).
Duration of Action: The duration of BDZ action is dependent largely on the rate and nature of metabolism, which is dependent on the structure of the drug.
- Differences in duration are accounted for by structural differences involving primary N-l and C-3- substitution and metabolism as detailed in the Metabolism Section.
- For example, compare diazepam and flurazepam (both N-substituted, non-C3-OH benzodiazepines) to oxazepam and lorazepam (N-unsubstituted, C3-OH benzodiazepines).
- It is also important to note that BDZs do not induce the metabolism of other drugs (unlike barbiturates). BDZs and their metabolites may accumulate, especially upon repeated dosing.
This may result in a delay in the appearance of adverse reactions, and extension of the clinical effect beyond discontinuation of the drug. This creates concerns for hepatically impaired patients, the elderly, etc.
Metabolism of Benzodiazepines
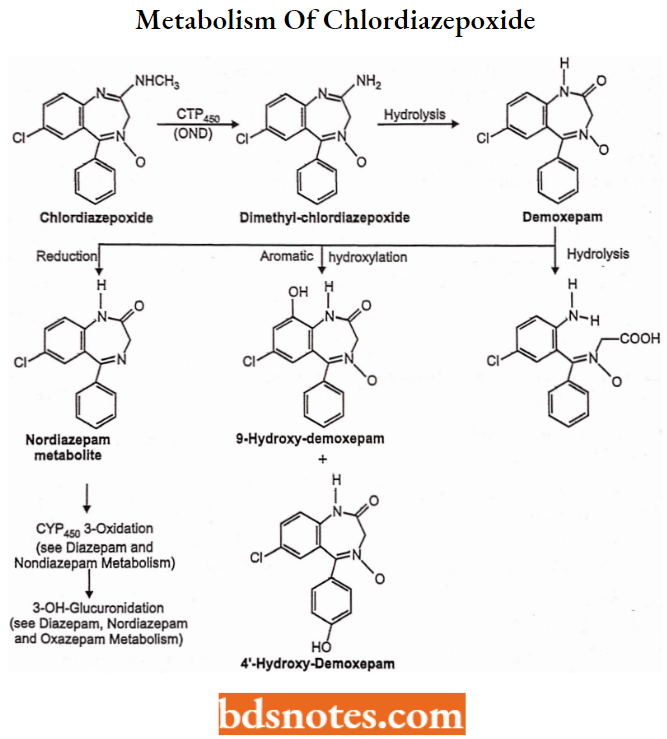
Metabolism of Chlordiazepoxide or Amidine BDZs: (Intermid Onset, Long Duration) Chlordiazepoxide is well absorbed after oral administration, but slowly and erratically absorbed from IM injection sites.
This drug is metabolized to active benzodiazepine metabolites in a series of reactions beginning with CYP-mediated OND-demethylation of the amidine group.
This “desmethyl” metabolite is slowly hydrolyzed to demoxepam which can undergo three different reactions:
- Hydrolysis to an inactive ring-opened form
- Aromatic hydroxylation to phenol metabolites that retain some activity and
- Reduction to an active “nordiazepam” metabolite. The nordiazepam metabolite may be further oxidized to an active “oxazepam” metabolite that can be conjugated as a glucuronide which is inactive and eliminated.
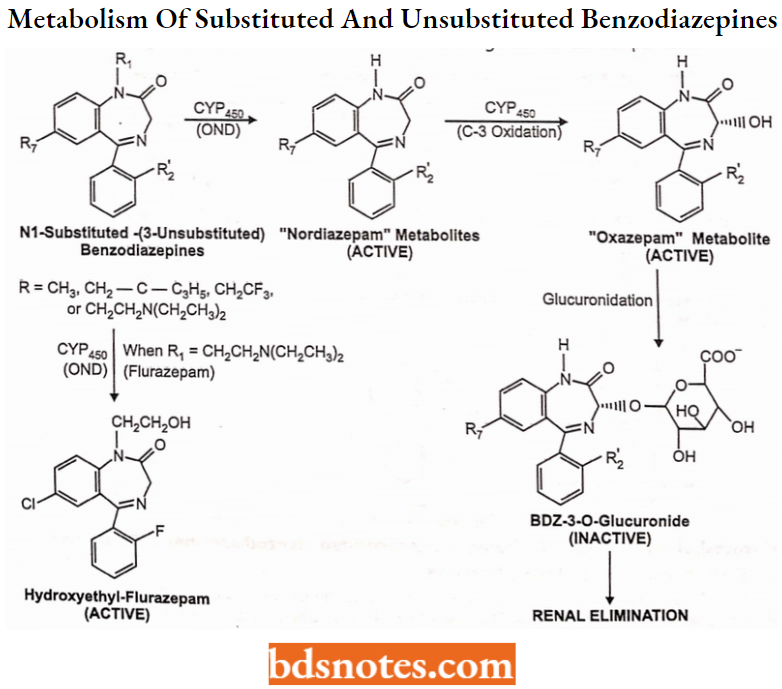
Metabolism of N-l-Substituted-3-Unsubstituted Benzodiazepines (“Diazepams”):
Slow to fast onset and Long Duration
The N-l-substituted-3-unsubstituted benzodiazepines or ‘”diazepams” are variably absorbed and distributed to the CNS based on their solubility and lipophilicity.
- All of these share the common property of long duration. This is a result of the formation of several active metabolites including the corresponding “nordiazepams” and “oxazepams” as shown.
- Each of these N-i-substituted-3-unsubstftuted benzodiazepines undergo sequential oxidative metabolism, first at N-l to yield active “nordiazepam” metabolites, then at the C-3 position to yield active “oxazepam” metabolites.
- Each of these metabolites can penetrate the BBS enter the CNS and bind to BZ receptors on the GABA complex and thus are active. Each of these metabolites is also lipophilic enough to be reabsorbed from the kidney or during biliary cycling.
- Only after these benzodiazepines are metabolized to glucuronide conjugates is BZ-activity lost and the drug eliminated.
Thus these compounds have long “effective” half-lives, it is important to note that the clearance of the N-lsubstituted-3-unsubstituted benzodiazepines, as well as the amidine and 3-carboxyl benzodiazepines discussed above, is dependent on oxidative metabolism (largely hepatic metabolism).
- Thus elderly patients and others with impaired hepatic function will clear these drugs more slowly than benzodiazepines that do not require oxidation for clearance (see the “oxazepams” below).
- In other words, these benzodiazepines will have substantially longer half-lives and may accumulate in patients with impaired or limited hepatic function.
- It is also important to note that ONLY benzodiazepines with “oxidizable” N-1 substituent undergo N-l oxidative dealkylation and that is why the carbon substituent bound to N-1 contains at least one (usually two) hydrogen atom.
- Benzodiazepine derivatives lacking hydrogen substituents on the carbon bound to N-1 could not undergo this metabolic process.
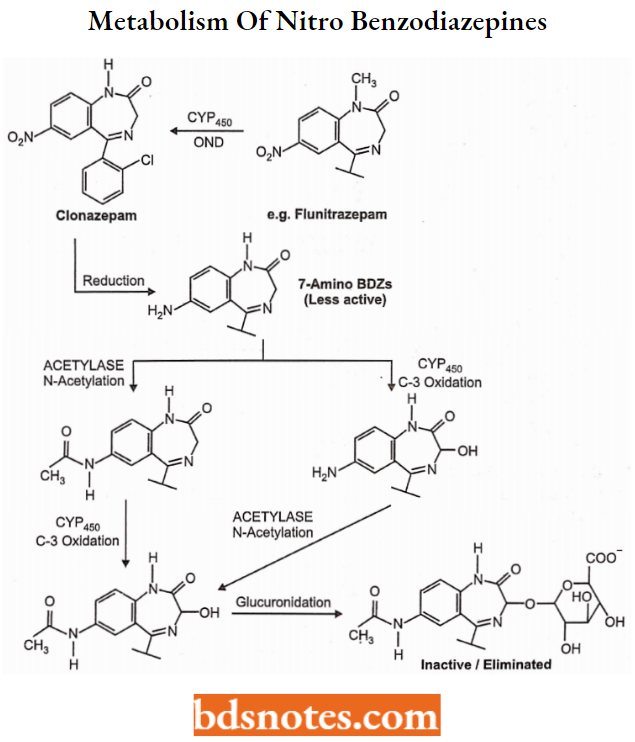
Metabolism of 7-Nitro-benzodiazepines: Slow to intermediate onset and Long Duration
As indicated in the metabolic scheme below, the 7-nitro-benzodiazepines can undergo the same metabolic reactions as other benzodiazepines of comparable functionality including OND at N-l (when N-l substituent is present), C-3-Oxidation and glucuronide conjugation of the C-OH metabolite.
- Again, the intermediate “nordiazepam” and “oxazepam” metabolites are active as BZ-receptor ligands, and these compounds thus have a relatively long duration of action.
- The 7-nitro benzodiazepines also can undergo a reduction of the 7-nitro group to the corresponding aniline (7-amino) as shown in the figure below.
The 7-amino metabolites are typically less active than the parent nitro compounds (see structure-activity requirement), and can be conjugated by acetylation (also less active). Thus complex, inactive metabolites eventually form from these compounds:
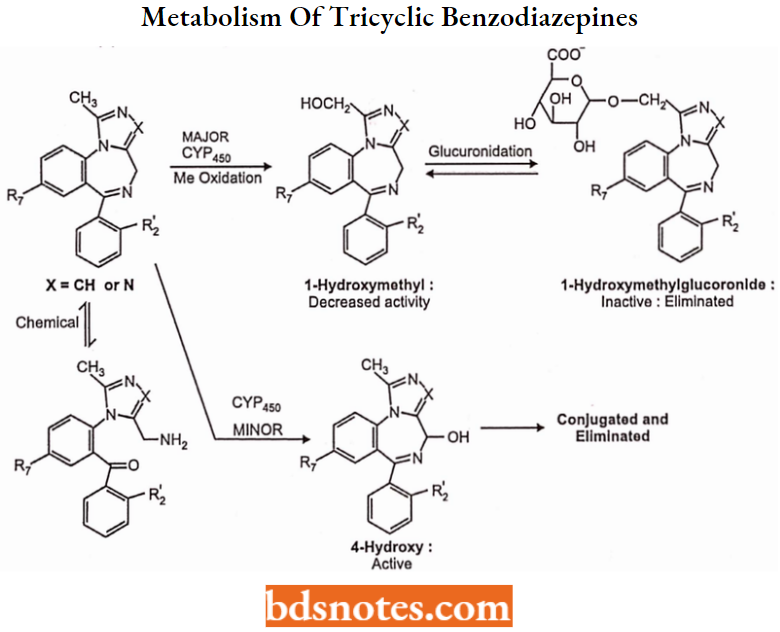
Metabolism of Tricyclic Benzodiazepines: Relatively rapid onset, Intermediate to short duration
These compounds are relatively lipophilic and thus are rapidly absorbed and diffused into the CNS. Metabolism of tricyclic benzodiazepines typically follows a different pathway than the “traditional” benzodiazepines.
- All of these compounds except estazolam have a “benzylic type” methyl group at the 1-position of the imidazole or triazole ring and this group is subject to rapid cytochrome-mediated oxidation as shown below.
- This 1-hydroxymethyl metabolite retains activity, but in most cases (except alprazolam) is rapidly conjugated as an inactive glucuronide and eliminated. Thus these compounds have intermediate to short duration of action.
- These compounds may also be oxidized at the available carbon in the benzodiazepine ring (as shown below), but this pathway appears to be relatively minor.
Also, some levels of the “ring-opened” benzophenone may exist in plasma (and appear in urine).
Benzodiazepine Derivatives
Chlordiazepoxide 7-chloro-2-(methylamino)-5-phenyl-3H-l,4-benzodiazepine 4-oxide hydrochloride.
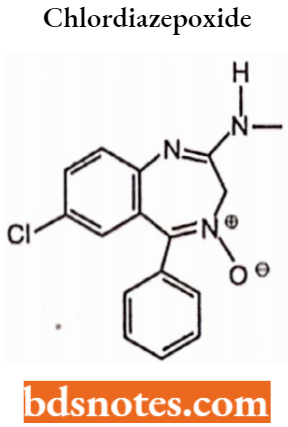
Chlordiazepoxide is a sedative and hypnotic drug belonging to the benzodiazepine class. The half-life of the drug has a medium to long half-life but its metabolite has a very long half-life.
Properties: The drug has amnesic, anticonvulsant, anxiolytic, hypnotic, sedative, and skeletal muscle relaxant properties.
MOA: Allosteric GABAA enhancer.
Uses: It is used to treat anxiety, insomnia, and withdrawal symptoms from alcohol and or drug abuse.
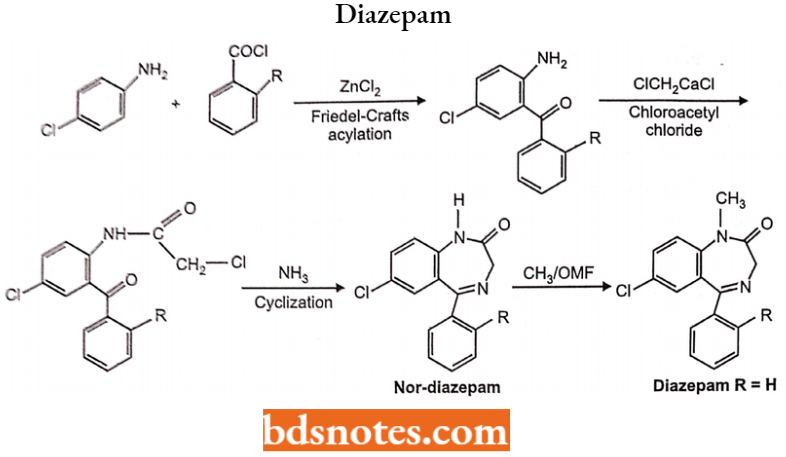
Diazepam 7-chloro-l,3-dihydro-l-methyl-5-phenyl-l,4-benzodiazepine-2-one
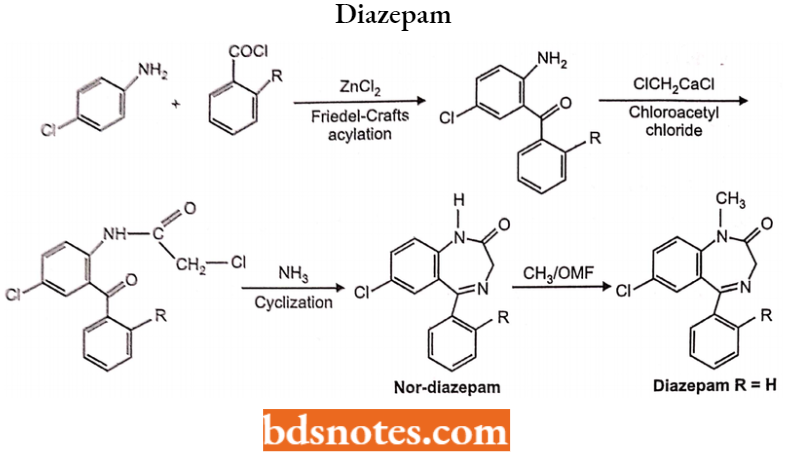
Long-acting benzodiazepine (>20 hrs), due to high blood protein binding of 98.5% reduces the rate of elimination, and its metabolic product is also active.
Properties: It has anxiolytic, anticonvulsant, hypnotic-sedative, skeletal muscle relaxant, and amnestic properties.
Uses: It is used for anxiety, panic attacks, insomnia, seizures, muscle spasms (such as in tetanus cases), restless legs syndrome, alcohol withdrawal, and opiate withdrawal syndrome.
- It is also used as a premedication for inducing sedation, anxiolysis, or amnesia before certain medical procedures (For Example., endoscopy).
- Diazepam is the drug of choice for treating benzodiazepine dependence with its long half-life allowing easier dose reduction.
- Not used for long-term epilepsy due to the development of tolerance and avoidance during pregnancy.
MOA: Allosteric GABAA enhancer.
Synthesis: Friedel-Crafts acylation of 4-chloro aniline with corresponding benzoyl chloride in the presence of Lewis acid affects benzophenone derivative.
Acetylation of an amino group with chloroacetyl chloride gives the chloroacetamide. Heating with ammonia undergoes a cyclization reaction to form non-diazepam; N-methylation methyl iodide affords diazepam.
Oxazepam:
Oxazepam is a short acting benzodiazepine.
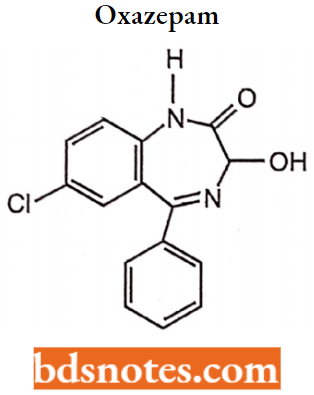
Properties: It is a metabolite of diazepam, prazepam, and temazepam and has moderate amnesic, anxiolytic, anticonvulsant, hypnotic, sedative, and skeletal muscle relaxant properties compared to other benzodiazepines.
Uses: It is used for the treatment of anxiety, and insomnia and in the control of symptoms of alcohol withdrawal syndrome.
MOA: Allosteric GABAA enhancer.
Chlorazepate:
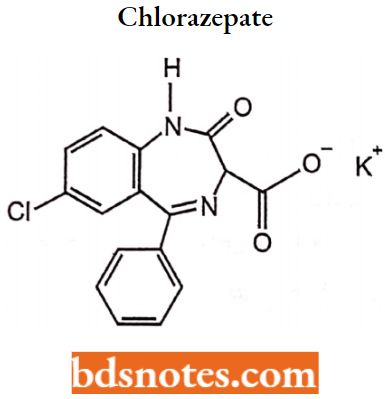
Chlorazepate is a benzodiazepine derivative. It is available as clorazepate dipotassium.
Uses: Chlorazepate is used for symptomatic relief of anxiety associated with neurosis, and psychoneurosis.
MOA: Allosteric GABAA enhancer.
Lorazepam Chemical, (E) – 7 – chloro – 5 – (2 – chlorophenyl) – 3 – hydroxy – 1h – benzo[1,4] diazepine – 2(3H) – one.
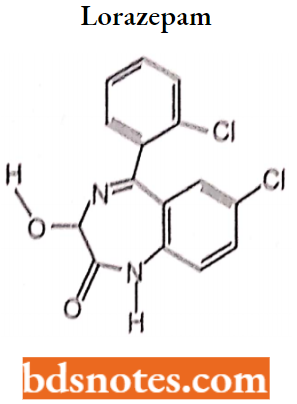
Uses: It is used to treat anxiety disorders, trouble sleeping, active seizures including status epileptics, alcohol withdrawal, and chemotherapy-induced nausea and vomiting. It can be given by 4 and the effect is shown between one and thirty minutes which lasts for up to a day.
Alprnzolnm 8-chloro-l-methyl-6-phenyl-4H-s-triazolo [4,3-aJ [1,4] benzodiazepine.
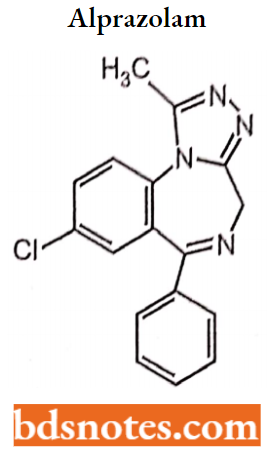
Alprazolam is a triazolo analogue of 1,4-benzodiazepine. It belongs to intermediate acting benzodiazepine.
Properties: It has potent anxiolytic, amnestic, hypnotic, anticonvulsant, skeletal muscle relaxant, and sedative properties.
Uses: Alprazolam is used for the treatment of anxiety disorders and panic attacks can cause fetal abnormalities and should not be used in pregnancy. It is excreted in breast milk and should not be used by women who are nursing.
MOA: Alloteric GABAA enhancer.
Other Non-Benzodiazepine Sedatives
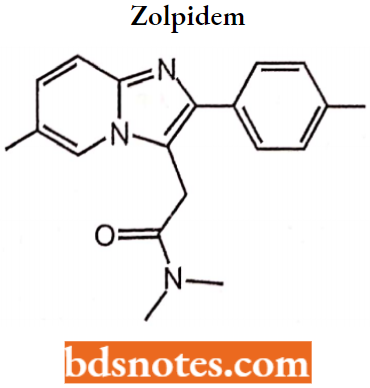
Zolpidem: Zolpidem is not a benzodiazepine in structure, but it acts on a subset of the benzodiazepine receptor family.
Lorazepam, oxazepam, and alprazolam have low hepatic metabolism and do not have active metabolites. Diazepam and chlorazepate are rapidly absorbed benzodiazepine agents and have the most rapid onset of action but also the greatest abuse or dependence potential.
- Chlordiazepoxide, clonazepam, clorazepate, and diazepam are considered long-acting benzodiazepine agents and are associated with accumulation which may result in sedation, cognitive impairment, and psychomotor retardation.
- Alprazolam and lorazepam are considered short-acting benzodiazepine agents and have been associated with increased anxiety, insomnia and rebound effects upon discontinuation.
- All benzodiazepine agents result in dependence or tolerance with long-term use.
- Benzodiazepine therapy should be discontinued by a slow taper to avoid withdrawal effects including: “sleep disturbance, irritability, increased tension and anxiety, panic attacks, hand tremor, sweating, difficulty in concentration, dry retching and nausea, some weight loss, palpitations, headache, muscular pain” and may also include more serious effects including seizures and psychotic reactions.
Transitioning patients from long-acting to short-acting agents may allow for more flexibility and aid in discontinuing therapy.
Sedatives And Hypnotics Barbiturate
Barbiturates are a group of drugs in the class known as sedative-hypnotics. Barbiturates were first introduced in 1903 and became increasingly popular in the 1960s and 1970s as treatments for anxiety, insomnia, or seizure disorders.
- The abuse of barbiturates increased similarly. Barbiturate use and abuse have declined dramatically since the 1970s, primarily due to the advent of safer benzodiazepines.
- Barbiturates all have a similar structure (barbituric acid), but differing side chains influence the drug’s potency, duration of effect, and rapidity of symptom onset.
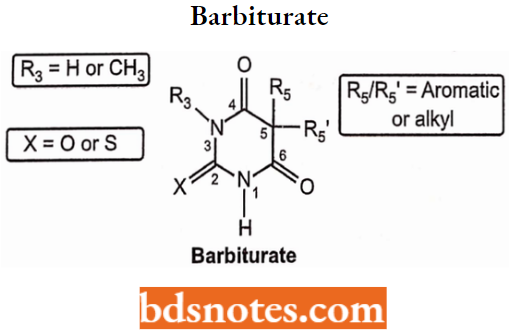
Barbiturate General Structure and Numbering
Barbiturates contain a “balance” of hydrophilic (2,4,6-pyrimidinetrione ring structure) and lipophilic (5,5-substituents) functionality. The overall hydrophilic (polar) or lipophilic (non-polar) character of the barbiturates is a function of:
- The hydrophilicity of the pyrimidinetrione ring which is a function of the number of N-substituents and the pKa of the acidic proton(s), and
- The overall size and structure of the two substituents at the 5-position.
Classification of Barbiturates
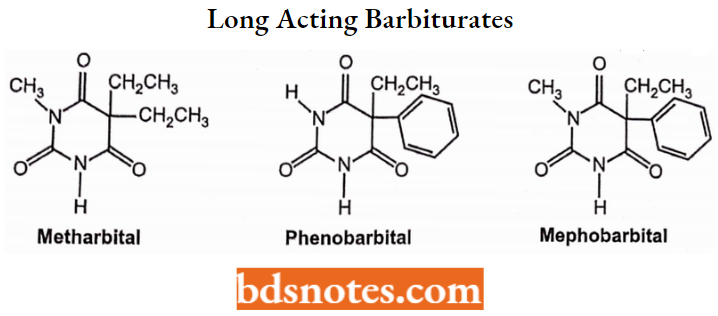
Long-Acting Barbiturates: Relatively slow onset (30-60 minutes) and relatively long duration (10-16 hours).
Structure: N-H or N-Methyl and C-5 side chains consisting of two ethyl groups, or an ethyl and phenyl group.
Properties: Relatively low lipophilicity and low plasma protein binding (<40%):
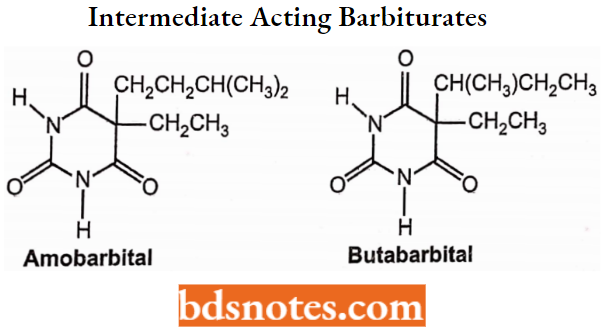
Intermediate-Acting Barbiturates:
Relatively slow onset (45-60 minutes) and intermediate duration (6-8 hours).
Structure: N-H and C-5 side substituents consisting of ethyl or allyl group and a 3 to 5-carbon atom unit.
Properties: Intermediate lipophilicity and intermediate plasma protein binding (50%).
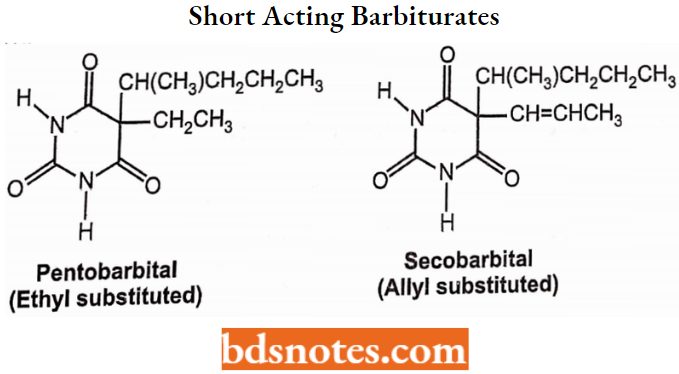
Short-Acting Barbiturates:
Relatively rapid onset (10-15 minutes) and relatively short duration (3-4 hours).
Structure: N-H and C-5 side chains consisting of ethyl or allyl and a 5-carbon unit.
Properties: High lipophilicity and high plasma protein binding (70%). Rapid distribution and redistribution.
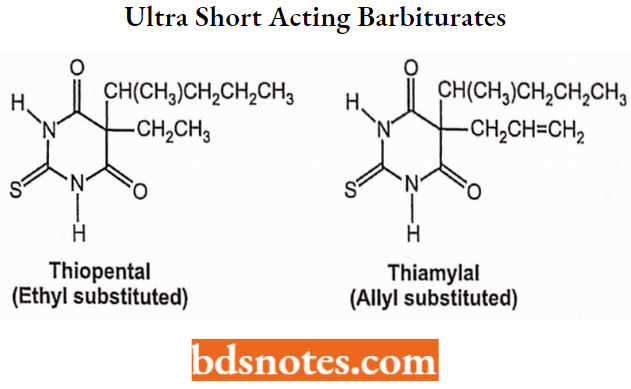
Ultra-Short-Acting Barbiturates:
Administered by injection (as salts): Immediate onset and very short duration.
Structure: N-H with a thiocarbonyl and C-5 side chains consisting of ethyl or allyl with a 5-carbon unit.
Properties: Very high lipophilicity and high plasma protein binding (>70%). Rapid distribution and redistribution.
Barbiturates and Mechanism of Action
Barbiturates potentiate the effect of GABA on the GABA-A receptor. The GABA-A receptor is a ligand-gated ion channel membrane receptor that allows for the flow of Cl– through the membrane in neurons.
GABA is the principal neurotransmitter for this receptor which upon binding causes the channel to open and create a negative change in the transmembrane potential. This makes it an inhibitory neurotransmitter.
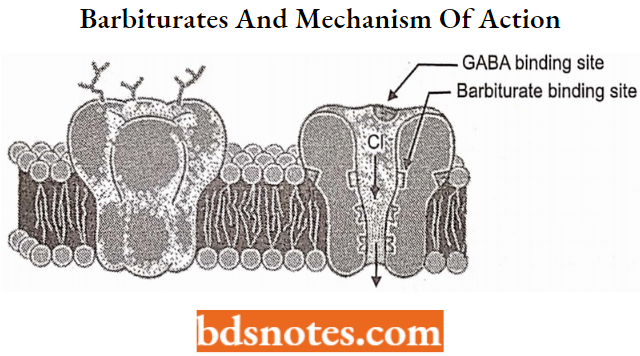
Barbiturates also block the AM PA receptor which is sensitive to glutamate, the excitatory neurotransmitter.
- Glutamate performs the opposite effect from GABA restricting ion flow and increasing the transmembrane action potential of the neuron.
- By blocking this action, barbiturates serve to increase the duration of the receptor response to GABA and extend the depressed condition of the cell.
Structure-Activity ReEalionship of Barbiturates (SAR)
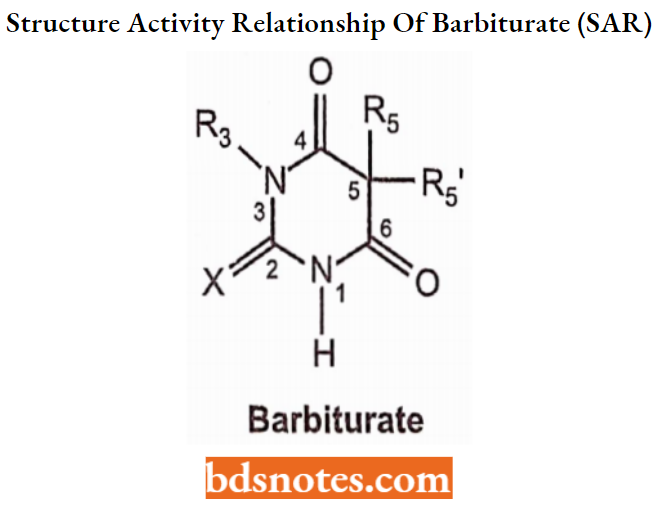
One of the ways of making potentially biologically active compounds is modification on C-5 of barbituric acid. The combination of barbituric acid moiety with other pharmacophoric groups gives the possibility to synthesize numerous derivatives with potential biological effects.
- Two active hydrogen atoms at position 5:5 have appropriate substituent (alkyl or aryl) groups to produce hypnotic activity.
- The total number of carbon atoms present In the two groups at carbon 5 must not be less than 4 and more than 10 for optimal therapeutic results.
- Only one of the substituent groups at position 05 may be a closed chain. Methylepentobarbital.
- The branched-chain isomer exhibits greater activity and shorter duration. The greater the branching, the more potent the drug For Example., pentobarbital > amobarbital.
- Double bonds in the alkyl substituent groups produce compounds more readily vulnerable to tissue oxidation; hence, they are short-acting, For Example. Pentobarbital sodium.
- Aromatic and alicyclic moieties exert greater potency than the corresponding aliphatic moiety having the same number of carbon atoms. Methylepentobarbital
- Short chains at C-5 resist oxidation and hence are long-acting. Long chains are readily oxidized and thus produce short-acting barbiturates. Pentobarbital Sodium Barbital.
- The inclusion of a halogen atom in the C-5 alkyl moiety enhances activity.
- The inclusion of polar groups (For Example., OH, CO, COOH, NH2, RNH, and SO3H) in the C-5 alkyl moiety reduces potency considerably.
- Methylation of one of the imide hydrogens enhances onset and reduces the duration of action.
- The replacement of an O-atom with an S-atom, at the C-2 position of the barbiturates significantly enhances the lipid solubility profile.
- The resulting modified versions of the barbiturates thus obtained exert a rapid onset of activity because they attain maximal thiobarbiturate -brain levels. Therefore, such drugs as ‘thiopental sodium’ find their profuse and abundant application as ‘intravenous anesthetics’. Thiopental sodium.
- The inclusion of more sulfur atoms (For Example., 2, 4-dithio; 2, 4, 6-trithio) decreases activity. Likewise, the introduction of imino group(s) into the barbituric acids abolishes activity (For Example., 2-imino; 4-imino; 2, 4-diimino and 2, 4, 6-triimino).
Clinical Uses of Barbiturates
Sedation: Although traditionally used as non-specific CNS depressants for daytime sedation, barbiturates have generally been replaced by benzodiazepines.
Hypnotic: Short-term treatment of insomnia, since barbiturates appear to lose their effectiveness in sleep induction and maintenance after 2 weeks. If insomnia persists, seek alternative therapy (including non-drug) for chronic insomnia.
Anticonvulsant (mephobarbital, phenobarbital): Treatment of partial and generalized tonic-clonic and cortical focal seizures.
Barbiturate Ionization, Acidity, and Salt Formation
Barbiturates containing at least one N-H hydrogen atom are acidic. Acidity results from the ability of the N to lose hydrogen and the stabilization of the resulting anionic charge of the conjugate base by resonance delocalization as shown below:
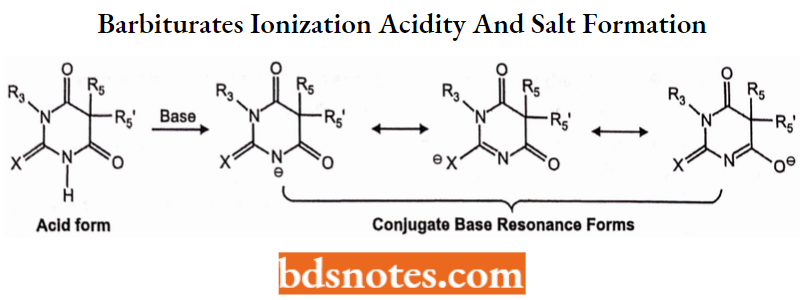
The relative acidity of different barbiturates is a function of the degree of N substitution and C-5-substitution as shown below (electron donors decrease acidity).
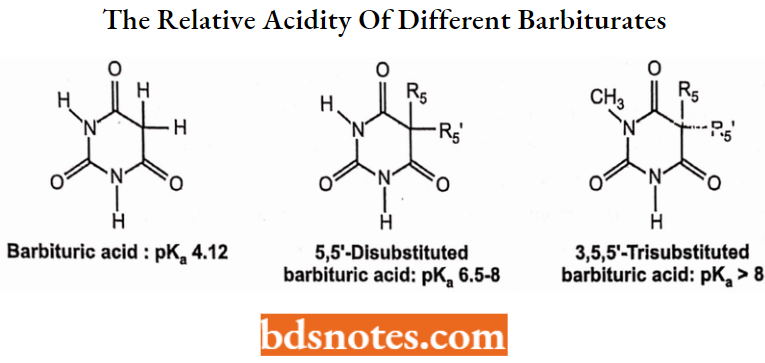
Barbituric acid (N- and C-5-unsubstituted) is highly acidic (but not active as a CNS depressant); See structures above,
- The addition of substituents at the 5-position decreases acidity (raises pKa) due to the electron-donating effects (+1) of the 5-alkyl groups: See structures above.
- Substitution at one ring nitrogen atom reduces acidity (raises pKa) due to the electron-donating effects (+1) of the N-alkyl group: See structures above.
- Substitution at BOTH ring nitrogen atoms eliminates both acidic protons (non-acidic).
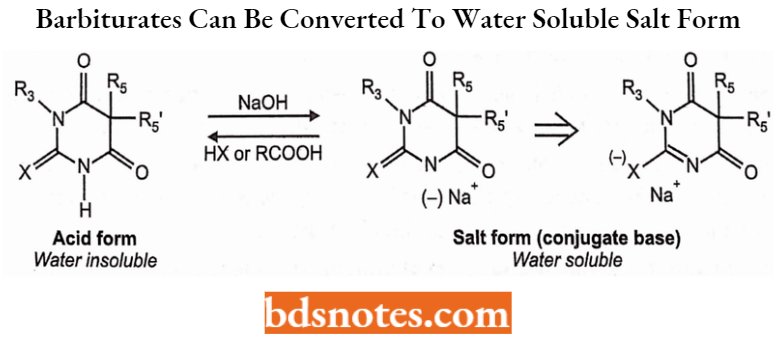
Due to the presence of one (or more) acidic protons, barbiturates can be converted to water-soluble salt forms by treatment with an appropriate base as shown below. Note that the charge resides primarily on the more electronegative oxygen atom;
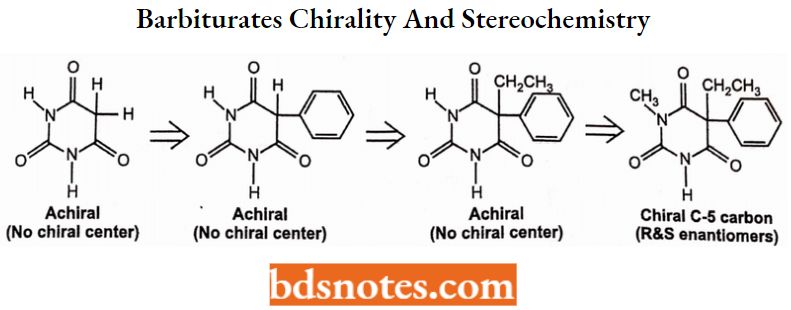
Barbiturate Chirality and Stereochemistry
The barbiturate ring system contains only one sp2 carbon atom and it is not chiral unless
- There are two different C-5 substituents and
- One ring of nitrogen is substituted as shown in the example below:
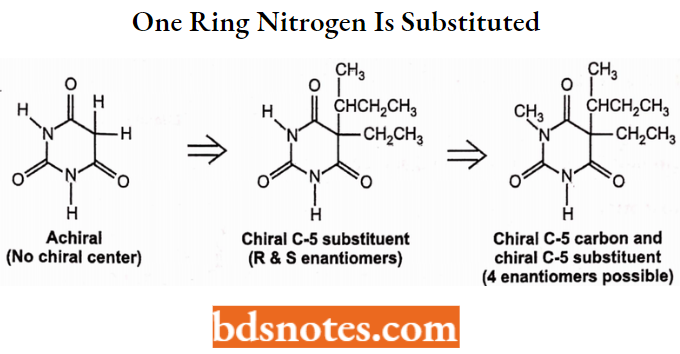
The C-5 substituents may contain chiral (unsymmetrically substituted sp2 carbon atom(s)) and in such cases the barbiturate is chiral. Some barbiturates have both a chiral C-5 atom and a chiral side chain as shown in one example below.
Metabolism of Barbiturates
Barbiturates are distributed throughout the body with the highest concentrations occurring in the brain, liver, and kidneys.
- In general, duration of action is dependent upon lipid solubility and extent of protein binding with the short-acting barbiturates showing the most lipid solubility and percentage of protein binding.
- The short and intermediate-acting barbiturates are nearly entirely metabolized by the liver and excreted in the urine, while 25-50% of a dose of a long-acting barbiturate is excreted as an unchanged drug.
The half-life is variable with short-acting barbiturates being detectable in urine for 24 hours and the long-acting drugs detectable for 2-3 weeks following ingestion.
1. Omega and Omega-1 Oxidation:
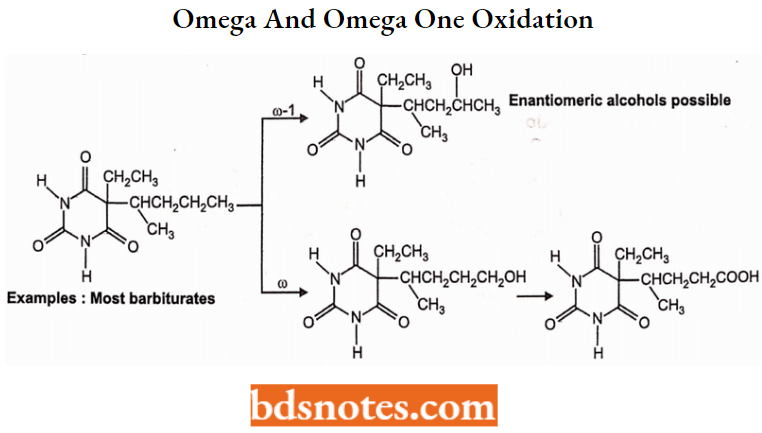
2. Alkene and Allylic Oxidation:
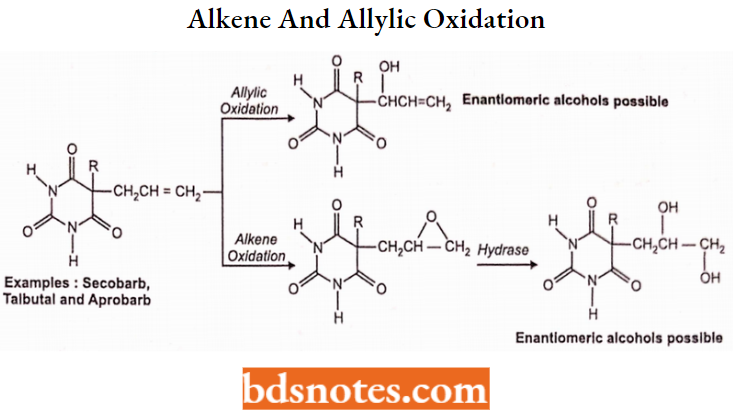
Examples: Secobarb, Talbutala nd aprobarb.
3. N-oxidation:
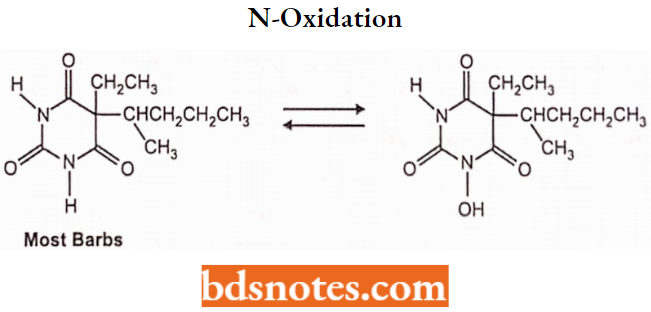
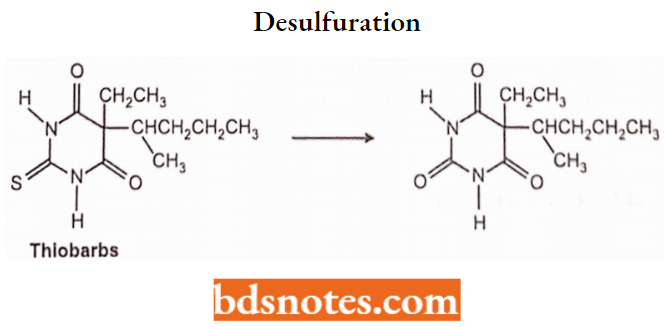
5. Oxidation N-dealkylation:
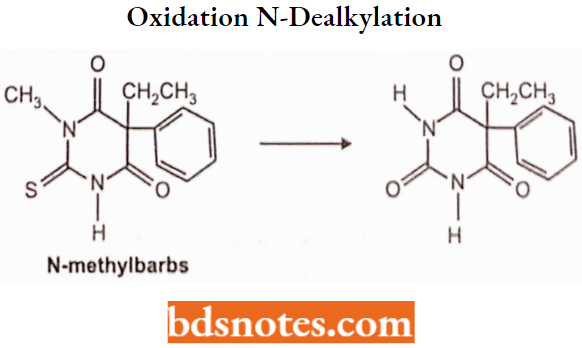
6. Hydrolysis:
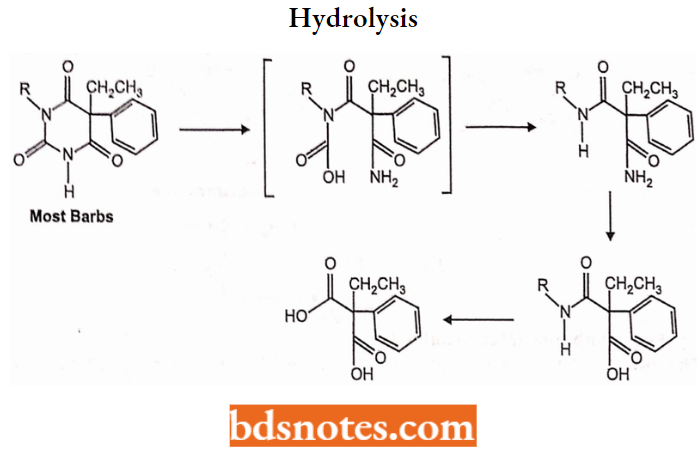
Barbiturate Derivatives
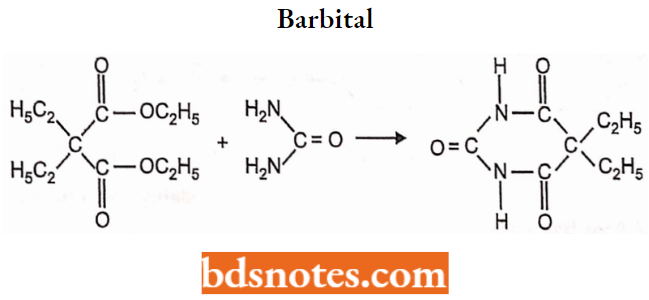
Barbital
Chemistry: Barbital is 5,5-diethylbarbituric acid.
Properties: Barbital is a white crystalline powder. It is slightly soluble in water but freely soluble in aqueous solutions of alkali hydroxides and carbonates. It is prepared by condensation of ethyl diethylmalonate with urea.
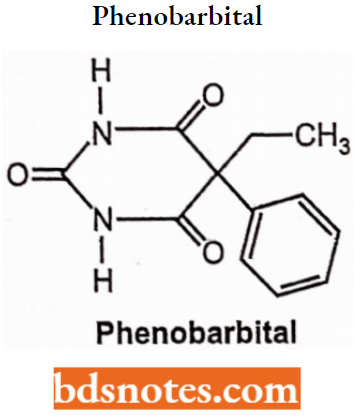
Phenobarbitone or Phenobarbital, 5-ethyl-5-phenylbarbituric acid. It occurs as sodium salt.
Properties: Phenobarbitone sodium is hygroscopic, bitter taste, water-soluble, odorless, white crystalline powder.
Synthesis: Phenobarbitone is synthesized by the following steps:
In the first step, a p-keto ester (Ethyl oxalophenylacetate) is prepared by Claisen condensation reaction of ethyl phenylacetate with ethyl oxalate in the presence of sodium.
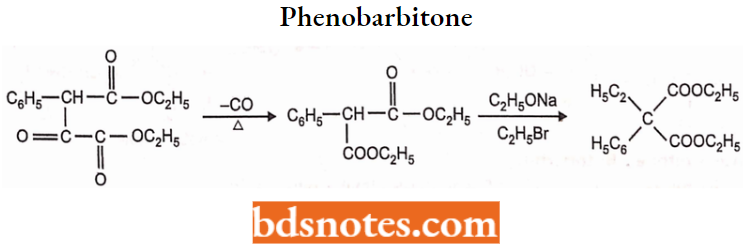
The ethyl oxalophenylacetate is then decomposed by distillation to form ethylphenylmalonate. The ethyl phenylmalonate is further ethylated to form ethyl ethylphenylmalonate with ethyl bromide and ethanol.
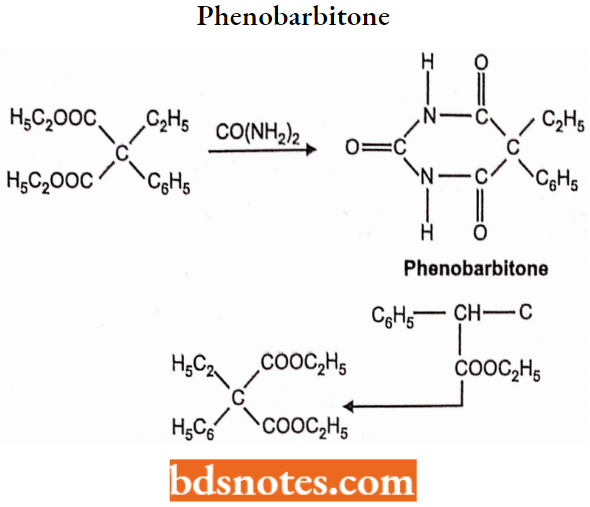
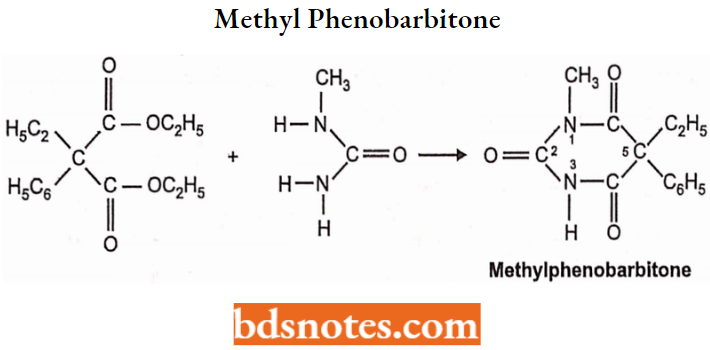
Methyl Phenobarbitone (Mephobarbital):
Chemistry: Methyl phenobarbitone is 5-ethyl-l-methyl-5-phenylbarbituric acid synthesis.
Mephobarbital is prepared by condensing ethylphenyl ethyl malonate with monomethyl urea.
Properties: Mephobarbital is a white crystalline, water-insoluble powder. It is soluble in aqueous solutions of alkali hydroxides and carbonates.
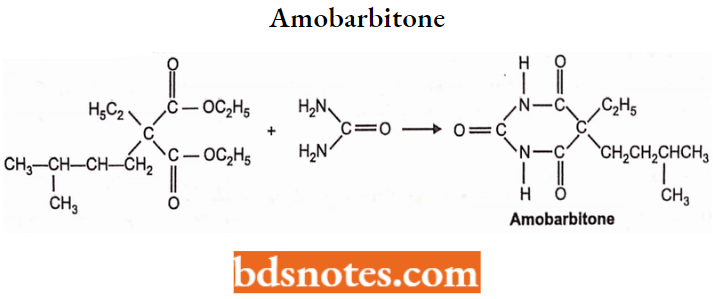
Amobarbitone or Amobarbital:
Chemistry: Amobarbitone is 5-ethyl-5-isopentylbarbituric acid.
Properties: It occurs as a white crystalline powder. It is slightly soluble in water but freely soluble in alkali hydroxide and carbonate solutions. It is prepared by condensation of ethyl isopentyl ethyl malonate with urea.
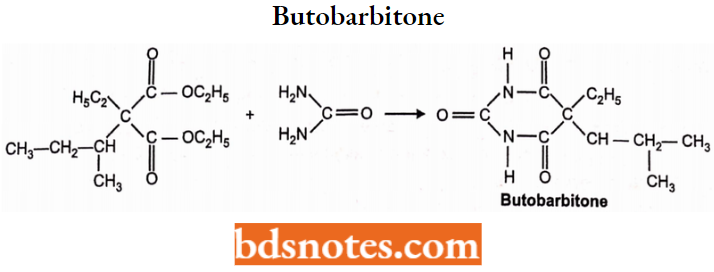
Butobarbitone or Butobarbital:
Chemistry: Butobarbitone is 5-isobutyl-5-ethylbarbituric acid.
Properties: It is a white crystalline powder, slightly soluble in water. It is prepared by condensation of ethyl isobutyl ethylmalonate with urea.
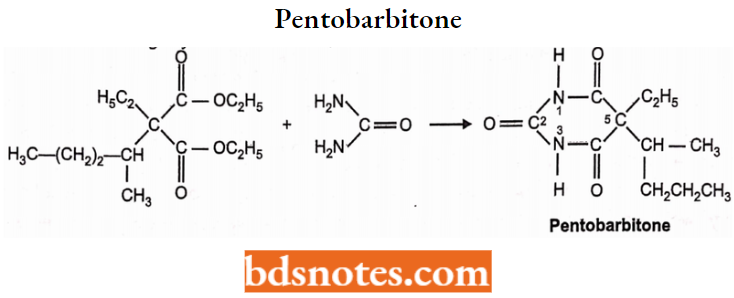
Pentobarbitone or Pentobarbital:
Chemistry: Pentobarbitone is 5-ethyl-5-(l-methyl butyl) barbituric acid. Pento-barbitone is available as pentobarbitone sodium salt. It is prepared by condensing ethyl 1-methyl butyl ethyl malonate with urea.
Properties: Pentobarbitone and its sodium salt is available as a white, crystalline powder. Pentobarbitone is slightly soluble in water, whereas its sodium is a white powder. salt is freely soluble in water.
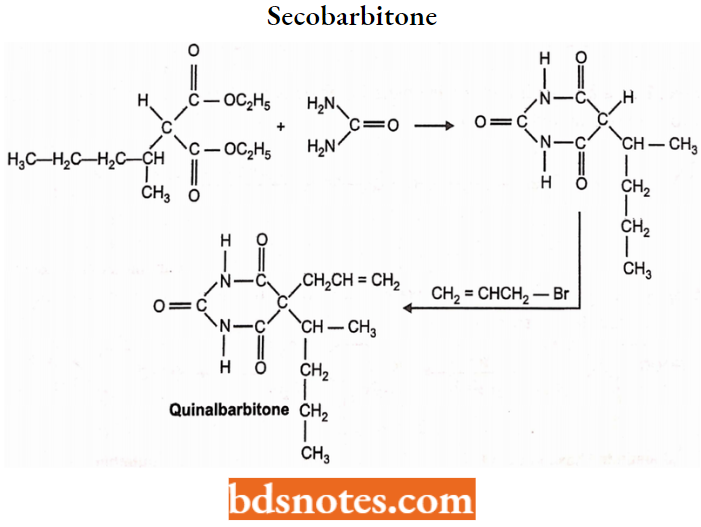
Secobarbitone or Secobarbital:
Chemistry: It is also known as Quinalbarbitone. Quinalbarbitone is (RS)-5-allyl-5-(1-methyl butyl) barbituric acid. It is prepared by condensing an equimolar mixture of urea with ethyl 1-methylbutylmalonate and alkyl bromide.
Properties: Quinalbarbitone occurs as its sodium salt. Quinalbarbitone sodium is a white
powder. It is freely soluble in water.
Sedatives And Hypnotics Non-Barbiturate
Numerous heterocyclic derivatives with low toxicity for hypnotic and sedative properties were synthesized. The following are some most important \ non-barbiturate sedative hypnotics among piperidines, quinazolinones, aldehydes, benzodiazepines, etc.
Amides Aand Lmides
Glutethimide 2-ethyl-2-phenyl-glutarimide.
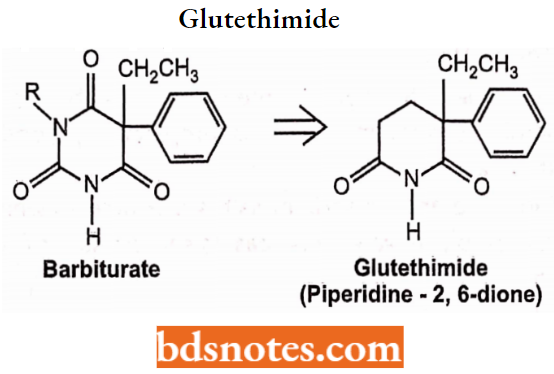
Structure, Chemistry, and Actions: A barbiturate analog lacking one of the “amide components” of the typical barbiturate ring system.
- Glutethimide is similar to barbiturates including the presence of an acidic imide group (pKa 9.2). It produces CNS depression similar to barbiturates.
- Glutethimide exhibits pronounced anticholinergic activity, which is manifested by mydriasis, inhibition of salivary secretions, and decreased intestinal motility.
It has generally been replaced by safer and more effective agents. Glutethimide is prepared by the following steps:
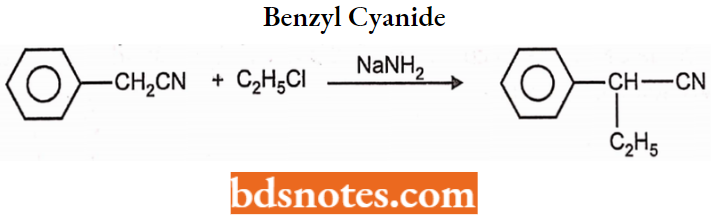
Step 1: Glutethimide is prepared by treating benzyl cyanide with ethyl chloride in the Barbiturate presence of sodamide to yield a-ethyl benzyl cyanide.
Step 2: The above-formed a-ethyl benzyl cyanide is condensed with a-bromopropionic ester to form substituted hexanoic acid.
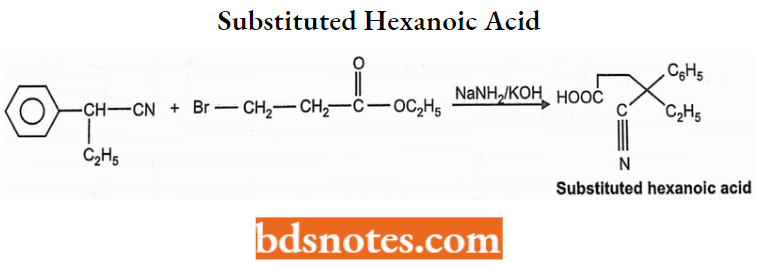
Step 3: Cyclization. Substituted hexanoic acid forms the amide on treatment with 80% H2S04 which spontaneously cyclizes to glutethimide.
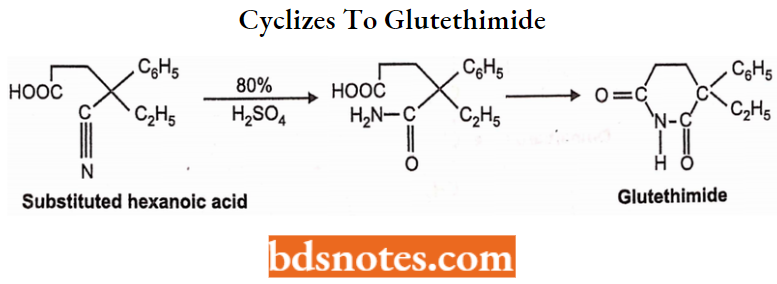
Properties: Gultethemide is a colorless or white-colored, water-insoluble powder. It should be stored in light-protected containers.
Uses: Gultethimide is used as a hypnotic in all types of insomnia. It induces sleep without depressing respiration.
Absorption or Distribution: It is erratically absorbed from the GI tract giving peak plasma concentration within 1-6 hours after administration. The average plasma half-life is 10 to 12 hours. About 50% of the drug is bound to plasma proteins; protein binding results in part from modest acidity (imide) Glutethimide stimulates hepatic microsomal enzymes.
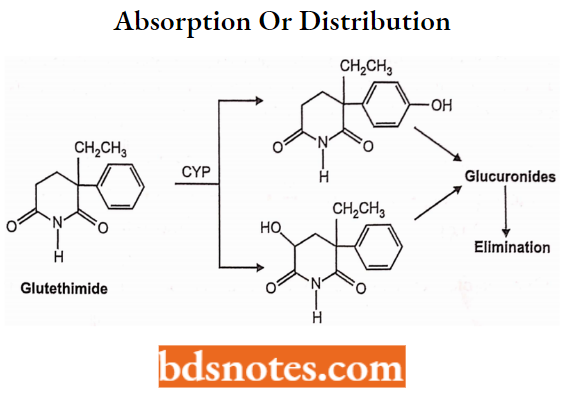
Metabolism or Execretion: Glutethimide is a racemate; both isomers are hydroxylated and both hydroxylated metabolites are reported to be active as sedatives. These metabolites are conjugated with glucuronic acid. The glucuronides pass into the enterohepatic circulation and are excreted in the urine (< 2% unchanged).
Alcohols and their Carbamate Derivatives
- The very simple alcohol ethanol has a long history of use as a sedative and hypnotic. Its modes of action were described under the anesthetic heading and are said to apply to other alcohols.
- It is widely used in self-medication as a sedative-hypnotic. Because this use has so many hazards, it is seldom a preferred agent medically.
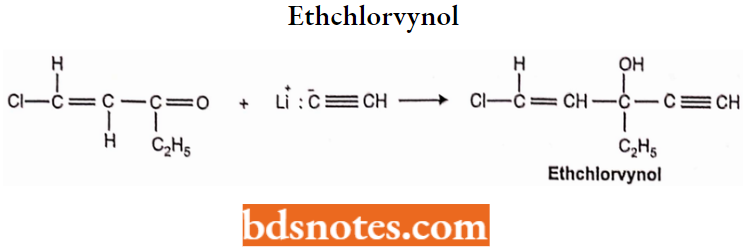
Ethchlorvynol: Ethchlorvynol is l-chloro-3-ethyl-l-penten-4-yn-3-ol. It is prepared from ethyl chlorovinyl ketone by following chemical reactions under strict anhydrous conditions.
- Ethchlorvynol is a mild sedative-hypnotic with a quick onset and short duration of action (t1/2 5.6 hours).
- Because of its highly lipophilic character, it is extensively metabolized to its secondary alcohol (90%) before its excretion. It reportedly induces microsomal hepatic enzymes.
Ethchlorvynol Properties: Ethchlorvynol is a yellow-colored liquid with a characteristic odor. It is light.
Ethchlorvynol Uses: Ethchlorvynol is a short-term hypnotic used to treat insomnia. It has a rapid onset, and a short duration of action-sensitive drug hence should be protected from light.
Meprobamate:
Meprobamate Chemistry: Meprobamate is 2-methyl-2-propyl trimethylene dicarbamate. Meprobamate is a propanediol derivative.
Meprobamate Properties: Meprobamate is an odorless, white-colored crystalline aggregate with a bitter taste. It is insoluble in water but soluble in alcohol and slightly soluble in ether.
Step 1: Meprobamate is prepared by condensing 2-methyl 2-n-propyl-l,3-propanediol with phosgene at 0°C to get chloroformate diester.
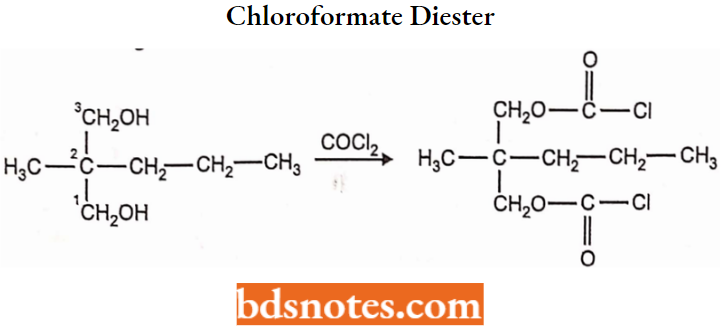
Step 2: The chloroformate diester is subjected to ammonolysis to form meprobamate.
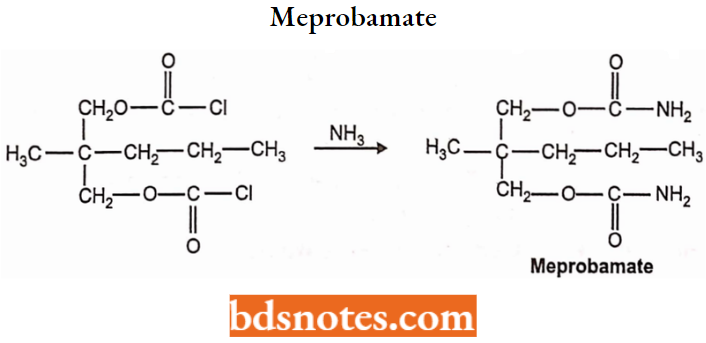
Meprobamate Uses: Meprobamate is used to induce sleep in anxiety and tensive patients. It also possesses anticonvulsant and muscle relaxant properties.
Aldehydes and their Derivatives
Paraldehyde:
Paraldehyde Chemistry: Paraldehyde is a cyclic trimer of acetaldehyde. Chemically paraldehyde is 2,4,6-trimethyl-l, 3, 5-trioxane. It is prepared by condensing 3 molecules of acetaldehyde in the presence of small quantities of a catalyst (S02 HCI or ZnCl2).
Paraldehyde Properties: Paraldehyde is available as a colorless or pale yellow color liquid. It has a strong characteristic odor and is soluble in water. Paraldehyde should be stored in airtight, light-protected containers.
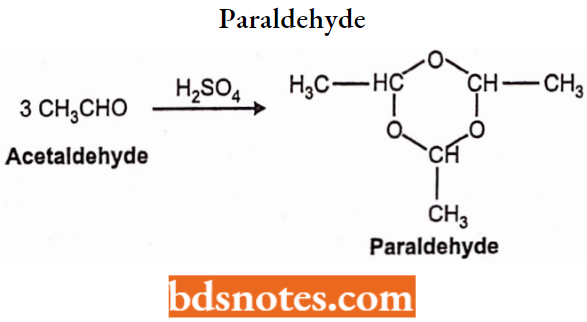
Paraldehyde Uses: Paraldehyde is one of the oldest hypnotics. It is used as a hypnotic and sedative.
Triclofos Sodium:
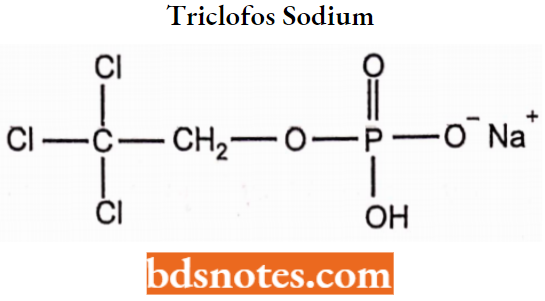
Triclofos is 2,2,2-trichloroethylhydrogen orthophosphate, which occurs as its sodium salt. Triclofos sodium is a hygroscopic, white-colored, water-soluble powder. Triclofos is used as a hypnotic and sedative.
Drugs Acting On Central Nervous System Antipsychotics
Antipsychotics Introduction
Psychoactive or psychotropic drugs are also known as tranquilizers. These drugs are used in the treatment of psychiatric disorders i.e. abnormalities of mental function.
- The psychoactive drugs render the patient calm and peaceful by reducing agitation and anxiety.
- Psychoactive drugs do not cure mental disorders but the available drugs do control most symptomatic manifestations and behavioral deviances, facilitate the patient’s tendency toward remission, and improve the capacity of the patient for social, occupational, and familial adjustment.
- The primary characteristic feature of these drugs is that they alter the mental state and behavior predictably. Tranquilizers fall into two main classes, major and minor.
- Major tranquilizers, which are also known as antipsychotic agents, or neuroleptics, are so called because they are used to treat major states of mental disturbance in schizophrenics and other psychotic patients.
Minor tranquilizers, which are also known as antianxiety agents or anxiolytics, are used to treat milder states of anxiety and tension in healthy individuals or people with less serious mental disorders.
Antipsychotics Phenothiazine
Phenothiazine Introduction
Phenothiazine derivatives are chemically characterized by a lipophilic fused tricyclic system (the phenothiazine nucleus) linked through the nitrogen atom of the central ring to a hydrophilic aminoalkyl substituent (the tertiary basic side chain).
- First synthesized in 1950, chlorpromazine was the first drug developed with specific antipsychotic action, and would serve as the prototype for the phenothiazine class of drugs Phenothiazines act exclusively on specific postsynaptic receptors and block the post-synaptic dopamine receptors.
- They work on the positive symptoms of psychosis such as hallucinations, delusions, disorganized speech, looseness of association, and bizarre behavior.
Phenothiazines are chemically constituted by a lipophilic, linearly fused tricyclic system having a hydrophilic basic aminoalkyl chain. The following is the general structure of antipsychotic drugs.
Phenothiazine Mechanism of Action:
Evidence supports the hypothesis that the etiology of psychotic disorders lies in neurochemical defects of dopaminergic and serotonergic pathways in the brain. This hypothesis is supported by the fact that the primary pharmacological action of antipsychotic agents is the antagonism of dopamine and or serotonin receptors in the CNS.
Phenothiazine Physicochemical Properties:
- The phenothiazine heterocycle has a high degree of lipophilicity on these antipsychotics, which is balanced (solubility) by the cationized (at physiologic pH) amine function
- H2O solubility of the antipsychotics phenothiazines is increased for oral dosage formulation by treatment with an acid
- The phenothiazines possess two potentially basic functional groups:
- The N10-amine which is very weakly basic (pKb>10) because of the electron-withdrawing effects of the 2 benzene rings attached to it is not appreciably cationized at physiological pH.
- The side chain tertiary amine function confers strong organic basicity on the antipsychotic phenothiazines.
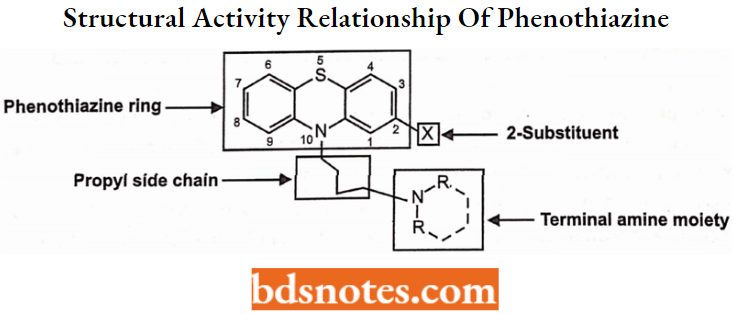
Structural Activity Relationship of Phenothiazine:
Phenothiazines are the derivatives of phenothiazine tricyclic heterocyclic moiety. The central ring possesses nitrogen and sulfur heteroatoms.
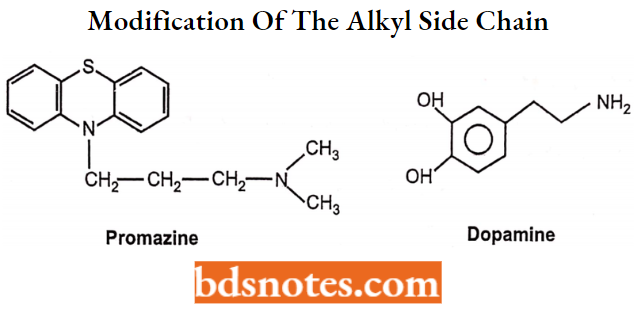
Modification of the Alkyl Side Chain:
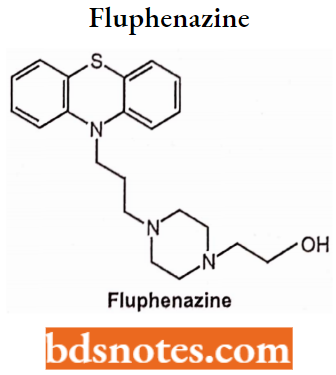
The maximum antipsychotic property or potency is observed when nitrogen of phenothiazine being present on the side chain nitrogen is connected by 3-carbon between two ‘N’ atoms because it resembles that of the dopamine (DA) structure.
An increase or decrease in the length of the alkyl side chain from 3 carbons, decreases the activity. Shortening of the chain to 2-carbons results in a change in receptor affinity from DA to CNS histamine receptors. Molecules with a 2-carbon side chain are the antagonists.
The introduction of a methyl group at a-carbon decreases antipsychotic activity and produces imipramine-like activity. Substitution at the (3-position of the side chain with a small group like methyl will decrease the antipsychotic potency but increase the anti-histaminic activity.
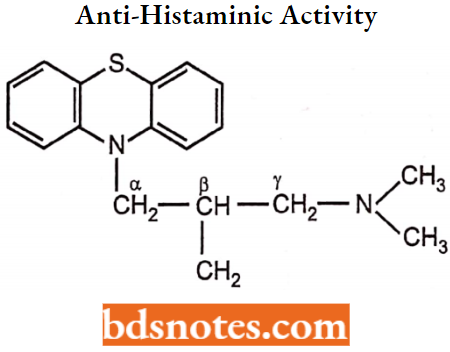
But substitution at the α-position of the side chain with a larger group decreases both anti-psychotic and anti-histaminic activity. Substitution at y-position increases anticholinergic activity and decreases dopaminergic antagonism.
Amino Group Modification:
3° nitrogen shows maximum potency whereas 2″ and V show reduced or abolished activity, i.e. 3° > 2° >1°.
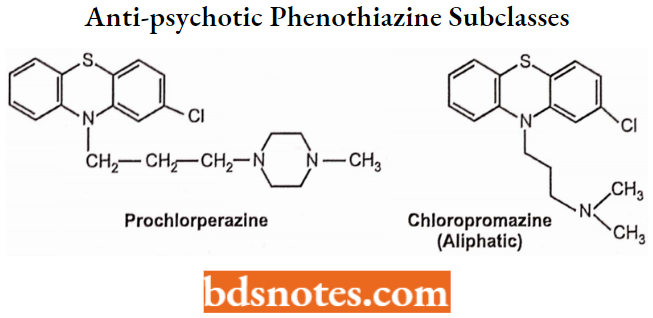
N-alkylation with more than one carbon atom decreases activity. Activity is increased when the dimethyl amino group is replaced by pyrrolidinyl, morpholinyl, or thiomorpholinyl groups, i.e., Structural modification of the side chain amine function yields three anti-psychotic phenothiazine subclasses.
Introduction of -OH, CH3, CH3CH3OH at C-4 of piperazine results in increased activity.
The quaternary amino group contains molecules as inactive because they are positively charged and cannot pass or cross the blood-brain barrier. The pharmacologic/therapeutic profiles of these three classes of antipsychotics differ as follows:
Antipsychotic potency: Piperazines > Piperidines > Aliphatics.
EPS frequency: Piperazines > Piperidines > Aliphatics.
Sedation: Aliphatics = Piperidines > Piperazines.
Hypotension: Aliphatics > Piperidines > Piperazines.
Phenothiazine Ring Modification:
The presence of an electron-withdrawing group at the C-2 position on the phenyl ring increases antipsychotic activity. The order of potency is in position 2 > 3 > 4 > 1.
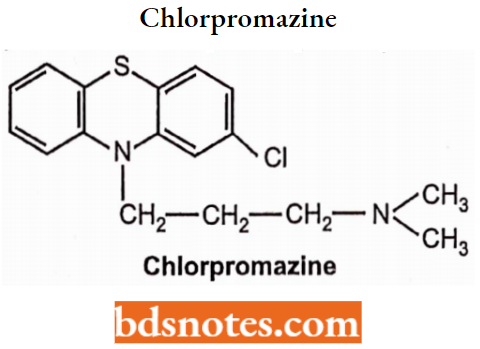
The potency of various groups increases in the following order,
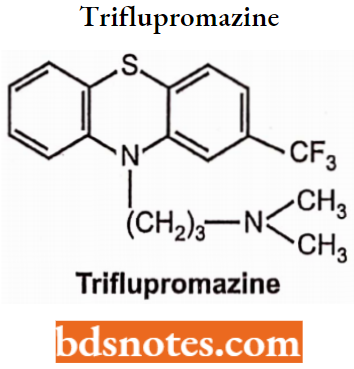
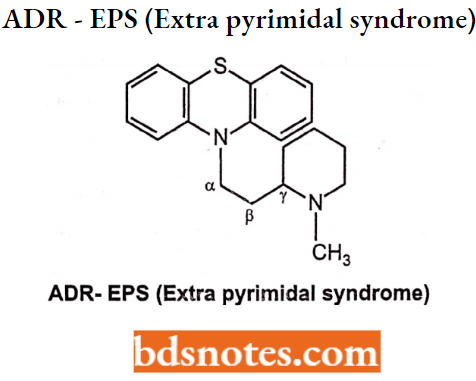
Disubstitution or trisubstitution of the C-2 substituted drugs results in harmful potency. CF3 is more potent than Cl but EPS (Extrapyramidal symptoms) appears. Hence chlorpromazine is much used than triflupromazine.
- Substitution at the C-l position decreases antipsychotic activity because it interferes with the bending of the side chain, (i.e., alkyl). Substitution at the C-4 position may also interfere with S-binding to the receptor.
- Multiple disubstitution on the ring system decreases potency (disubstitution on the phenothiazine ring decreases neuroleptic potency).
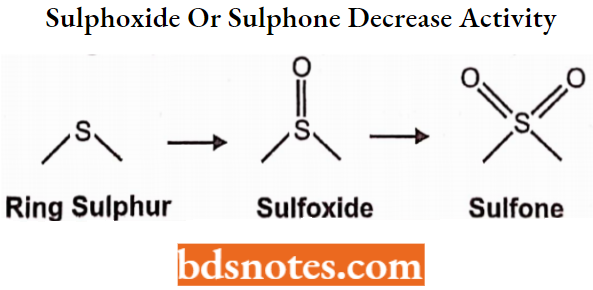
- Conversion of ring sulfur into sulphoxide or sulphone decreases activity
Antipsychotics Phenothiazine Derivative
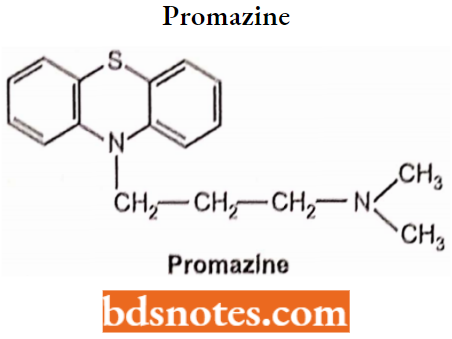
Promazine Hydrochloride
Promazine Hydrochloride Chemistry: Promazine hydrochloride is a phenothiazine derivative. Chemically it is 10-[3-(dimethylamino)-propyl] phenothiazine. Promazine is prepared by condensing 3-chloro-N, N dimethylpropylamine with phenothiazine in the presence of sodium hydride.
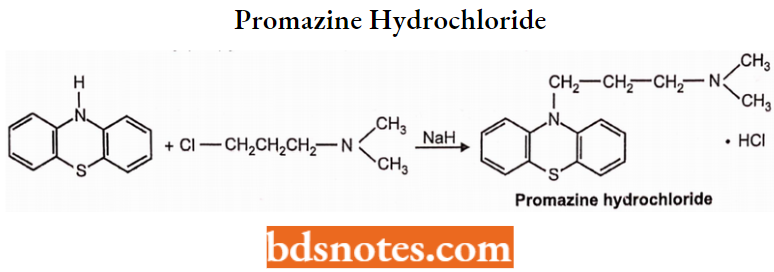
Promazine Hydrochloride Properties: Promazine is available as a hydrochloride salt. Promazine HCI is a white or slightly yellow crystalline powder and is freely soluble in water and chloroform. It should be protected from air.
Promazine Hydrochloride Uses: Promazine has antipsychotic properties and is also used to control nausea and vomiting.
Chlorpromazine Hydrochloride
Chemistry: Chlorpromazine hydrochloride is a phenothiazine derivative and has a chemical formula of 2-chloro-10-[3-(-dimethylamino) propyl] phenothiazine monohydro-hloride. Chlorpromazine is synthesized by cyclization of 3-chlorodiphenylamine with sulfur in the presence of a small amount of iodine as a catalyst.
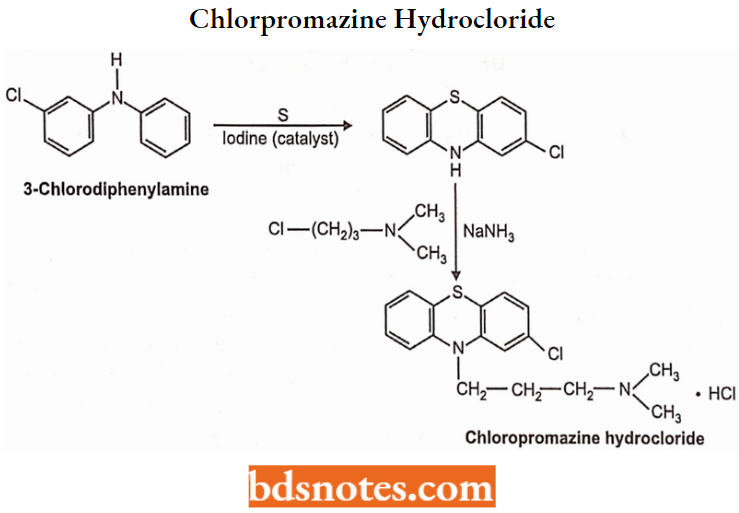
Chlorpromazine Hydrochloride Properties: Chlorpromazine hydrochloride is an odorless, white crystalline powder. It is freely soluble in water, alcohol, chloroform and insoluble in ether and benzene. It decomposes on exposure to air and light, hence it should be stored in airtight containers and protected from light.
Chlorpromazine Hydrochloride Mechanism of Action: Chlorpromazine blocks dopamine at D2 receptor sites in the mesolimbic medullary chemoreceptor trigger zone areas of the brain. It causes inhibitory post-synaptic effects by reducing the flow of dopamine as the dopaminergic ion channels are closed.
Chlorpromazine Hydrochloride Uses: Chlorpromazine is used in the management of psychotic conditions. It also controls excitement, aggression, and agitation. It has antiemetic, antipruritic, anti-histaminic and sedative properties.
Chlorpromazine Hydrochloride Side Effects:
Extrapyramidal symptoms, hypertension, orthostatic hypotension, blurred vision, dry mouth, anorexia, nausea, vomiting, constipation, diarrhea, weight gain, impotence, amenorrhea, photosensitivity.
Trifluopromazine
Trifluopromazine Chemistry: Trifluopromazine is a fluorinated phenothiazine derivative. Chemically, triflupromazine is 10-[3-(dimethylamino)propyl]-2-(trifluoromethyl) phenothiazine. Trifluopromazine is synthesized by condensing 2-(trifluoromethyl) phenothiazine with (3-chloropropyl) dimethylamine in dry benzene in the presence of sodamide.
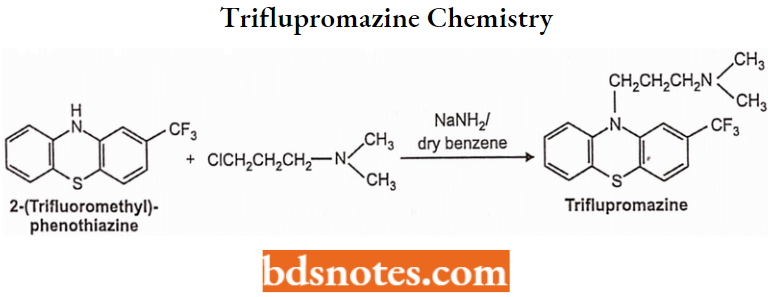
Trifluopromazine Properties: Promazine is available as hydrochloride salt. Promazine HCI is a white or slightly yellow crystalline powder and is freely soluble in water and chloroform. It should be protected from air.
Trifluopromazine Uses: Promazine has antipsychotic properties. It is also used to control nausea and vomiting.
Thioridazine Hydrochloride:
Thioridazine hydrochloride is a piperidine-typical antipsychotic drug belonging to the phenothiazine derivative. Chemically, 2-ethyl-10-(2-(l-methylpiperidin-2-yl)ethyl)-10 Hphenothiazine. R enantiomer has a higher affinity for the D2 receptor.
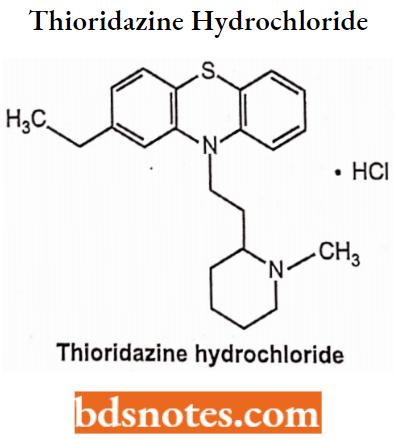
Thioridazine Hydrochloride: Uses: It is used in the treatment of schizophrenia and psychosis.
Thioridazine can cause the life-threatening side effects of irregular heartbeat that may sudden death.
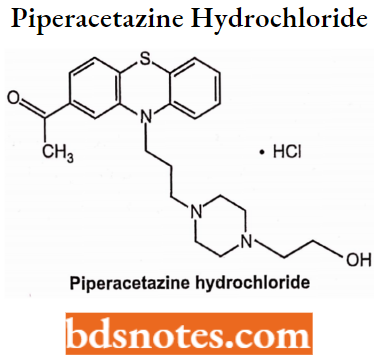
Piperacetazine Hydrochloride:
Piperacetazine hydrochloride is a piperazine antipsychotic prodrug belonging to the phenothiazine derivative used for the treatment of schizophrenia. In veterinary practice, the greatest value of piperacetazine is in anti-psychotic and behavior changes. Chemically, l-(10-(3-(4-(2-hydroxyethyl)piperazin-l-yl)propyl)-10H-phenothiazin-2-yl)ethanone.
Prochlorperazine:
Prochlorperazine Chemistry: Prochlorperazine is a phenothiazine derivative associated with piperazine. Chemically, prochlorperazine is 3-chloro-10-[3-(4-methyl-l-piperazinyl) phenothiazine. It occurs as maleate and mesylate salts. Prochlorperazine is prepared by refluxing l-(3-chloro propyl)-4-methylpiperazine with 2-chlorophenothiazine in the presence of sodamide in toluene.
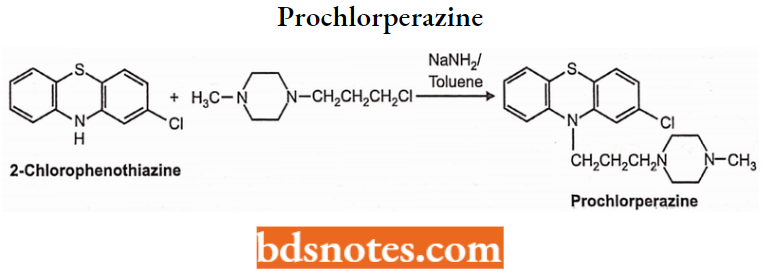
Prochlorperazine Properties: Prochlorperazine is a pale yellow-colored, viscous liquid and is very slightly soluble in water but freely soluble in alcohol.
Prochlorperazine: Uses: Prochlorperazine is an antipsychotic and tranquilizing agent. It is used to treat various psychiatric disorders such as schizophrenia, mania, involution psychoses, senile and tonic psychoses. It also has antiemetic properties.
Trifluperazine Hydrochloride:
Trifluperazine Hydrochloride Chemistry: Trifluperazine hydrochloride is a fluorinated phenothiazine derivative. Chemically, 2-(trifluoromethyl)-10-(3-(4-methylpiperazin-l-yl)propyl)-10H-phenothiazine.
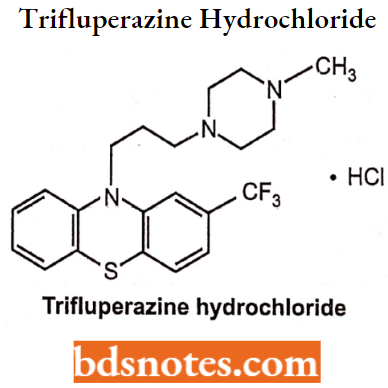
Trifluperazine Hydrochloride Uses: Triflupromazine is used to treat psychotic disorders, schizophrenia, anxiety, and other conditions.
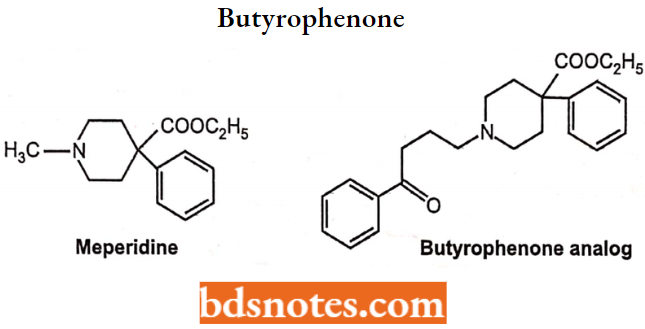
Fluorobutyrophenone:
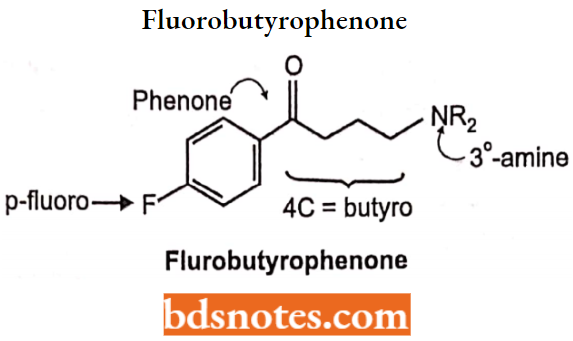
The fluorobutyrophenones belong to a much-studied class of compounds, with many compounds possessing high antipsychotic activity.
They were obtained by structure variation of the analgesic drug meperidine by substitution of the N-methyl by butyrophenone moiety to produce the butyrophenone analog which has similar activity as chlorpromazine.
Structure-activity Relationships of Fluorobutyrophenones:
The 4-aryl piperidine moiety is superimposable on the 2-phenylethylamine moiety of dopamine and, accordingly, could promote affinity for D2 receptors. The long N-alkyl substituent could help promote affinity and produce antagonistic activity.
The structural requirements for antipsychotic activity in the group are well worked out. General features are expressed in the following structure.
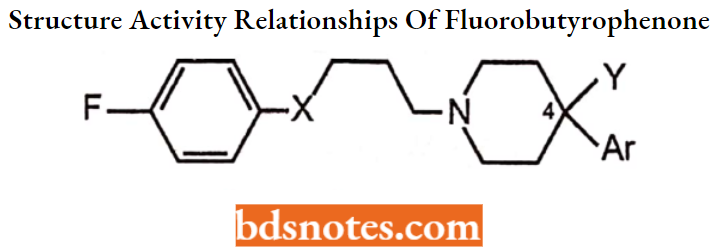
Para-F or similar electronegative substituent (For Example. CF3) provides maximal potency is seen when with an aromatic ring.
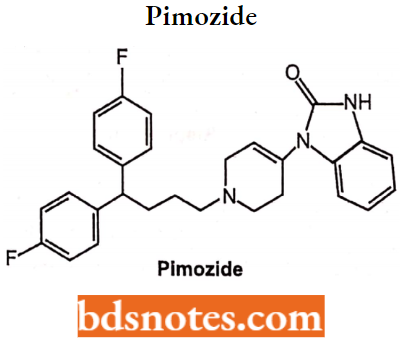
- X = C=0 (ketone) for optimal activity. The Carbonyl group of butyrophenones is necessary for antipsychotic activity. Replacement of the carbonyl group by functional groups such as X = C(H)OH or C(H)aryl structural feature yields therapeutically useful antipsychotics (For Example. Pimozide).
Altering length or branching of the three-carbon chain linking the keto and amino
group decreases antipsychotic activity i.e. Propylene bridge is required for
antipsychotic properties. Shortening lengthening or branching of the propylene
bridge decreases antipsychotic activity.
- Terminal basic amine function may vary in structure but is usually incorporated in a 6-membered heterocyclic ring.
- AR is an aromatic ring attached directly to or separated by one atom from position 4 of the six-membered ring.
- The Y group is variable and can enhance activity (Y = OH in haloperidol).
Fluorobutyrophenone Derivatives:
Haloperidol:
Haloperidol Chemistry: Haloperidol is a butyrophenone derivative with antipsychotic properties that has also been found effective in lowering levels of hyperactivity, agitation, and mania. It has a chemical formula of 4-[4-(p-chlorophenyl)-4-hydroxypiperidino]-4′-fluorobutyrophenone. Haloperidol is synthesized by condensing 4-(4-chlorophenyl-4-piperidinol with 4-chloro-4′-fluorobutyrophenone.
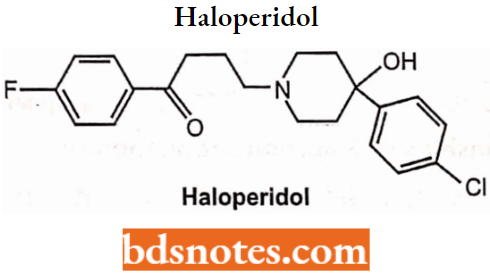
The mechanism inhibits the transport mechanism of cerebral monoamines, particularly by blocking the impulse transmission in dopaminergic neurons. Peak plasma levels of haloperidol reach within 2 to 6 hours of oral administration.
Haloperidol Uses: The compound is a potent antipsychotic useful in schizophrenia and in psychoses associated with brain damage. It is often chosen as the agent to terminate mania.
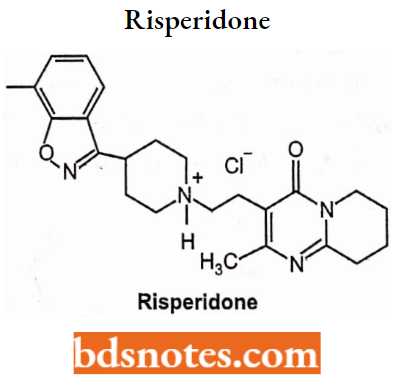
Risperidone:
Risperidone: Chemistry: Risperidone is a benzisoxazole derivative with a high affinity for central serotonergic 5-HT2, dopaminergic D2, and adrenergic α1-receptors in-vivo. HCl salt is used for oral formulation, schizophrenia, and reduced EPS.
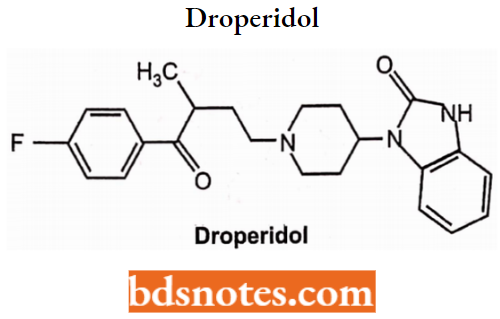
Droperidol:
Droperidol is a benzimidazolinone derivative of fluorobutyrophenones. Chemically, it is l-{l-[3-(p-Fluorobenzoyl)propyl]-l,2,5,6-tetrahydro-4-pyridyl}-2-benzimidazolinone. It is used alone as a pre-anesthetic neuroleptic or as an antiemetic.
Its most frequent use is in combination with the narcotic agent fentanyl pre-anesthetically. It is considered a short-acting sedating butyrophenone and is sometimes used in psychiatric emergencies as a sedative-neuroleptic.
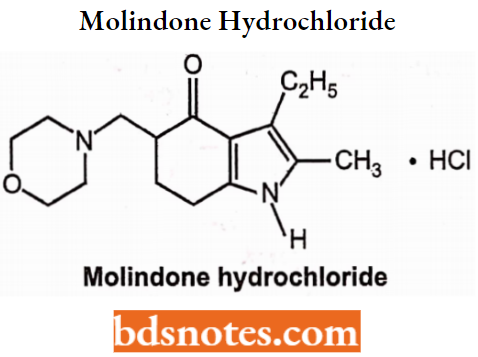
Beta Amino Ketones:
Molindone hydrochloride:
Molindone hydrochloride is a dihydroindolone compound that is not structurally related to the phenothiazines, the butyrophenones, or the thioxanthenes. Chemically, is 3-ethyl-6, 7-dihydro-2-methyl-5-(morpholinomethyl) indol-4 (5H)-one hydrochloride. It is a white to off-white crystalline powder, freely soluble in water and alcohol.
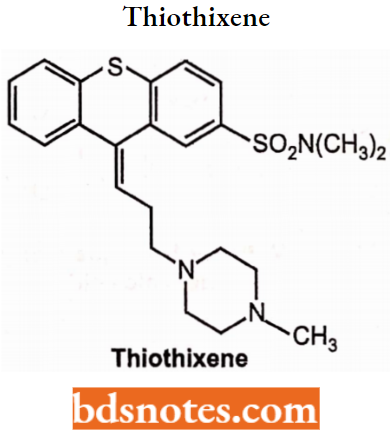
Thiothixene: The thioxanthenes differ from the phenothiazines by the replacement of nitrogen in the central ring with a carbon-linked side chain fixed in space in a rigid structural configuration.
- An N, N-dimethyl sulfonamide functional group is bonded to the thioxanthene nucleus. Chemically, it is N, N-dimethyl-9-[3-(4-methyl-l-piperazinyl)-propylidene] thioxanthene-2-sulfonamide, Z isomer (cis isomer) is more active than E isomer (trans).
- They act as dopamine 2 antagonists for Schizophrenia, other psychotic disorders, and bipolar disorder. Psychotic symptoms can also improve within 1 week, but it may take several weeks for full effect on behavior.
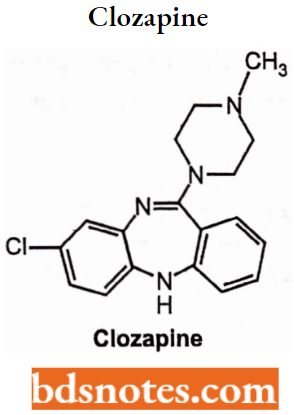
Clozapine: Clozapine are dibenzazepines or dibenzodiazepine derivatives and first a typical antipsychotic agent. They show weak D2 blocking action but act as strong D4 receptors and 5HT2 receptors.
Loxitane: Loxapine, a dibenzoxazepine compound, represents a subclass of tricyclic antipsychotic agents, chemically distinct from the thioxanthenes, butyrophenones, and phenothiazines.
- Chemically, it is 2-Chloro-11 (4-methyl-1-piperazinyl)dibenz[b,f][l,4] oxazepine. It is present in capsules as the succinate salt and in the concentrate and parenteral primarily as the hydrochloride salt.
- It is used for the treatment of schizophrenia. The major pharmacological mode of action involves dopamine D2 receptor antagonism, and to a lesser extent, blocking activity at D1 receptors as well.
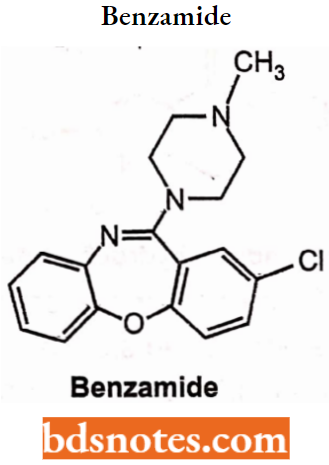
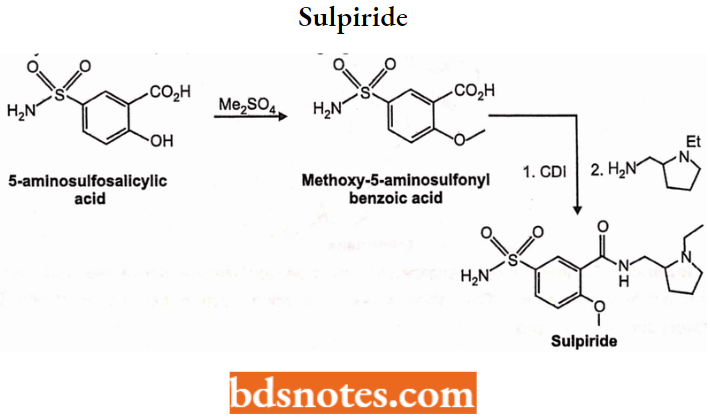
Sulpiride: Sulpiride is a typical antipsychotic benzamide derivative. It is used in the management of the symptoms of schizophrenia. Chemically, sulpiride is a selective antagonist at dopamine D2 and D3 receptors thereby reducing positive symptoms of psychosis.
Sulpiride can be synthesized from 5-aminosulfosalicylic acid. Methylating this with dimethylsulfate gives 2-methoxy-5-aminosulfonylbenzoic acid, which is transformed into an amide using 2-aminomethyl-l-ethylpyrrolidine as the amine component and carbonyl diimidazole (CDI) as a condensing agent.
Drugs Acting On Central Nervous System Anticonvulsants Introduction
Epilepsy is a common but serious brain disorder. It is universal, with no age, sex, geographical, social class, or racial boundaries. Epilepsy imposes a large economic burden on the healthcare systems of countries.
- There is also a hidden burden associated with stigma and discrimination against the patient and even his or her family in the community, workplace, school, and home.
- Many patients with epilepsy suffer severe emotional distress, behavioral disorders, and extreme social isolation.
What Is Epilepsy?
Epilepsy can be defined as “the occurrence of transient paroxysms of excessive or uncontrolled discharges of neurons, which may be due to several different causes leading to epileptic seizures”.
- The actual presentation or manifestation differs among individuals, depending upon the location of the origin of the epileptic discharges in the brain and their spread.
- A person should only be diagnosed as having “epilepsy” if there are recurrent manifestations; the first episode of a seizure is called a “single seizure” and not epilepsy.
- An epileptic seizure is an event in which an individual is not aware of the surroundings, either completely or partially.
- Various motor movements, such as shaking of limbs; sensory phenomena, such as electric shock-like sensation over a specific area; behavioral experiences, such as fear or confusion or autonomic disturbances such as excessive secretion of saliva or bladder or bowel incontinence could occur in association with this altered sensor.
Usually, it is very brief, lasting from a few seconds to minutes. Only in very rare cases will it be continuous, resulting in “status epilepticus”, i.e. a seizure lasting more than 30 minutes or recurrent seizures without the individual regaining consciousness between attacks.
Anticonvulsants Classification Of Epileptic Seizures
Epilepsy can broadly be divided into two categories: idiopathic where there is no known cause and secondary seizures where there is a known cause. Seizures can be either generalized or partial (or focal).
In generalized seizures, both halves of the brain are simultaneously affected. In partial seizures, the abnormal electrical discharge starts from a focus on one side of the brain.
Later, this may spread to the other side. This spread is called secondary generalization.
Generalized Seizures: In generalized seizures, patients suddenly stop what they are doing, the eyes and head turn to one side and the body becomes stiff.
- This is usually followed by several jerks of the hands and legs, groaning and frothing from the mouth. During the episode, the tongue may be bitten or severe injury can result from a fall or an accident Sometimes the patient may pass urine or stools.
- The body relaxes after a few minutes and the patient sleeps for a variable period. The patient is completely unaware of the seizure. Such seizures can also occur in sleep.
- Generalized seizures consist of many different seizure types, of which the primary generalized tonic-clonic seizure (GTCS) is the most common.
Tonic-clonic seizure: In a generalized tonic-clonic seizure the patient loses consciousness, falls, sometimes with a scream, and develops a generalized stiffness (the tonic phase).
- Breathing stops, as all the muscles of the trunk are in spasm, and the patient becomes cyanotic, the head is retracted, the arms flexed and the legs extended.
- After a while, this tonic phase is followed by the clonic phase, when the muscles alternately contract and relax, resulting in clonic movements. During this jerking the patient might bite his tongue, pass urine, or sometimes stool.
- The clonic phase may last several minutes. When all the jerking stops and the patient regains consciousness, he may feel very tired and have a headache and confusion.
He has no memory of what happened and may find himself on the floor in a strange position. Often he falls into a deep sleep.
- The frequency of the seizure may vary from one a day to one a month or once a year, or even once every few years. Either the tonic phase or the clonic phase can predominate in the seizure.
- Generalized tonic-clonic seizures can also occur due to secondary generalization in partial epilepsies.
Clonic seizures: These seizures are generalized seizures, where the tonic component is not present, only repetitive clonic jerks (clonic jerks are repetitive rhythmic flexing and stretching of limbs). When the frequency of jerks diminishes the amplitude of the jerks does not diminish.
Tonic seizures: Tonic seizures are sudden sustained muscle contractions, fixing the limbs in some strained position. There is an immediate loss of consciousness. Often there is deviation of eyes and head towards one side, sometimes rotation of the whole body.
Absence seizures: These are short periods of loss of consciousness lasting only a few seconds (not more than half a minute). They are of sudden onset, there are usually no, or only minimal motor manifestations.
There is a blank stare, a brief upward rotation of the eyes, and an interruption of ongoing activity. The child is unresponsive when spoken to. It is suddenly over, and the child continues what he was doing before the seizure. The child has no memory of these seizures.
Myoclonic seizures: These seizures consist of sudden, brief, shock-like muscle contractions, either occurring in one limb, or more widespread and bilateral. They may be single jerks, or jerks repeated over longer periods.
They are often seen in combination with other seizure types occurring in special epileptic syndromes.
Partial Seizures: Partial seizures are divided into two groups, simple partial seizures where consciousness is maintained, and complex partial seizures where there is an impairment of consciousness.
Simple partial seizures: In simple partial seizures, some patients may experience either motor or sensory phenomena. Such seizures arise from a specific area of the brain, with the patient being fully or partly aware of the event.
- In motor seizures, the focus is on the primary motor cortex. There are twitchings, starting in a distal part of the extremity, or the face. The twitching may remain localized, or spread up the whole extremity and even become completely generalized to involve the whole body.
- Sensory seizures have their focus in the post-central gyrus (primary sensory cortex). There might be feelings of tingling, pins, and needles, cold or heat, or numbness of a limb.
- Sometimes there may be strange feelings with visual signs, or hearing or smelling sensations. Autonomic seizures are associated with foci in the temporal lobe.
There may be a sensation rising from the epigastrium to the throat, palpitations, sweating, or flushing. The psychic symptoms may consist of changes in mood, memory, or thought (thinking).
There may be distorted perceptions (time, space, or person) or problems with language. Structured hallucinations could occur (music, scenes). These simple partial seizures are usually only recognized as epileptic seizures when they develop into generalized seizures.
Complex partial seizures: Here the patient has impaired consciousness, but NOT complete loss of consciousness. He is slightly aware of what is going on, but he cannot respond to anything, nor can he change his behavior during an attack.
- The seizure usually starts with an aura which can be of many types such as, a strange feeling in the stomach rising to the throat and head, or a sensation of light.
- Smell, sound, or taste or with changes in perception, For Example., of time (time seems to pass too slowly or too fast), of light or sound or space.
Sometimes the seizure occurs with hallucinations or with psychomotor symptoms such as automatisms For Example., pulling at the clothes, chewing, lip smacking, or repeated aimless movements.
There are different ways to treat epilepsy by different mechanisms:
- By inhibiting Na+ channels (reduction of electrical excitability of cell membranes).
- Example: Phenytoin, Carbamazepine, Valproate, Lamotrigine Phenytoin).
- By inhibiting GABA transaminase enzyme (Enhancement of GABA-ergic action).
- Example: Phenobarbital, Benzodiazepines, Vigabatrin, Gabapentin).
- By inhibition of calcium channel function (T-type calcium channels).
- Example: Ethosuximide, Gabapentin.
However, it is believed that the anticonvulsants suppress seizures by depressing the cerebral (motor) cortex of the brain, thereby raising the threshold of the central nervous system (CNS) to convulsive stimuli. Therefore, the person is less likely to undergo seizures.
Anticonvulsants Classification Of Anticomvulsamts
The anticonvulsants are classified as:
- Barbiturates: Phenobarbitone, Methabarbital.
- Hydantoins: Phenytoin, Mephenytoin, Ethotoin
- Oxazolidine diones: Trimethadione, Paramethadione
- Succinimides: Phensuximide, Methsuximide, Ethosuximide
- Urea and monoacylureas: Phenacemide, Carbamazepine
- Benzodiazepines: Clonazepam.
- Miscellaneous: Primidone, Valproic acid, Gabapentin, Felbamate.
Barbiturates:
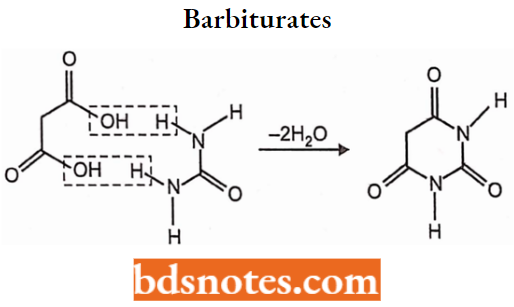
It has a pyrimidine derivative it is usually depicted as the cyclical ureide of malonic acid in either the keto or enol form. Barbituric acid is hypnotically inert but the introduction of organic substituents at position 5 endows the resultant drugs with the ability to depress consciousness.
- Diethylbarbituric acid or barbitone is the first sedative barbiturate. Barbitone was popular as a hypnotic for many years but it was too long-lasting for general use. Phenobarbitone was the next drug to be developed.
- Long-acting barbiturates such as phenobarbital (Luminal) and mephobarbital (Mebaral) are prescribed for two main reasons. When taken at bedtime, they help treat insomnia.
- When taken during the day, they have sedative effects that can aid in the treatment of tension and anxiety. These same effects have been found helpful in the treatment of convulsive conditions like epilepsy.
Phenobarbital has also been used in the treatment of delirium tremens during alcohol detoxification, although benzodiazepines have a more favorable safety profile and are more often used.
Barbiturates Mechanism of Action of Barbiturates:
Barbiturates act on GABA receptors in the central nervous system and increase the GABAergic inhibition of the central nervous system.
Barbiturates Structural Activity Relationship: Structural feature and structural activity relationship of antiepileptic drugs, the molecules should have at least one aryl or lipophilic unit.
- One or two hydrogen acceptor-donor atoms and an electron donor atom in a unique spatial arrangement to be recommended for antiepileptic activity, for example, mephobarbital, ethotoin, gabapentin, and zonisamide, etc. are characterized as their structural elements.
- These agents exert their action by different mechanisms. They include an enhancement of the GABA-ergic neurotransmission and effects on neuronal voltage-gated Na+ and or Ca2+ channels.
Barbiturates Structure: N-H or N-Methyl and C-5 side chains consisting of two ethyl groups, or an ethyl and phenyl group.
Barbiturates General Properties: Relatively low lipophilicity and low plasma protein binding.
Phenobarbital (Phenobarbitone):
Phenobarbital is the oldest currently available AED. Although it has long been considered one of the safest of the AED, the use of other medications with lesser sedative effects has been urged. The barbiturates are considered as the drugs of choice for the treatment of seizures only in infants.
The four barbituric acid derivatives that are clinically useful as AEDs are phenobarbitone (PBT), mephobarbital, metharbital, and primidone.
- The metharbital is methylated barbital and the mephobarbital is methylated PBT; both are demethylated.
- Chemically phenobarbital is 5-ethyl-5-phenyl-1H, 3H, 5H-pyrimidine-2(4,6-trione and the first antiepileptic drug ever developed.
Antiepileptic action is seen in most barbiturates, however, phenobarbital shows antiepileptic action in lower concentrations with an acceptable degree of sedation.
- Phenobarbital is orally administered in the treatment of grandal epilepsy.
- It is less effective in the treatment of petit mal and psychomotor epilepsies. The injectable form of the drug is used to treat other types of convulsions.
Barbiturates with phenytoin show better antiepileptics (which are better tolerable, especially about sedative effects).
Mechanism of Action of Barbiturates:
Phenobarbital (PBT) suppresses high-frequency recurring firing in neurons in culture by an action on Na+ ion conductance. Barbiturates block some Ca2+ ion currents (L-type and N-type).
- The PBT binds to an allosteric regulatory site on the GABA-BZD receptor, and it improves the GABA receptor-mediated current by extending the openings of the Cl” channels.
- The PBT also blocks excitatory responses stimulated by glutamate, mainly those mediated by activation of the AMPA receptor.
- Both the enrichment of GABA-mediated inhibition and the decline of glutamate-mediated excitation are seen with therapeutically applicable concentrations of PBT.
The PBT is valuable in the therapy of partial seizures and generalized tonic-clonic seizures, even though the drug is often tried for all seizure types, particularly when attacks are complicated to manage.
Barbiturates Adverse Effects:
The most common adverse effects associated with phenobarbital are sedation (although a degree of tolerance develops), dizziness, drowsiness, ataxia (lack of muscula and nystagmus (a rapid involuntary movement of the eyeball).
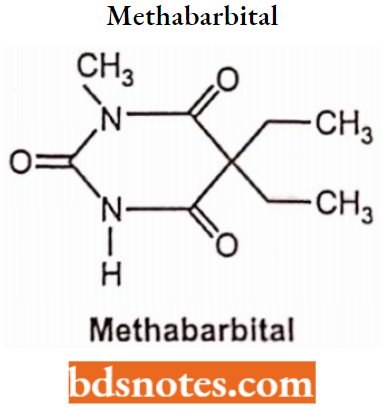
Methabarbital:
Phenobarbital’s success led to the development of other barbiturates as subsequent AEDs, including the N-methyl barbituric acid derivative mephobarbital (N-methylpheno-barbital), introduced in 1932.
- Like phenobarbital, both mephobarbital is water-insoluble weak acids with pKa values of 7.8 and 8.5, respectively.
- The introduction of the N-methyl group into the phenobarbital molecule breaks the symmetric axis possessed by barbituric acid or phenobarbital.
- Consequently, unlike phenobarbital, mephobarbital is a chiral compound containing one asymmetric carbon atom at position 5 of the molecule. It has been used clinically as racemic mixtures (equal parts of (R)- and (S)-enantiomers).
- Mephobarbital metabolism is stereoselective with (R)-methylphenobarbital being metabolized by cytochrome P450 (CYP)2C19-mediated aromatic hydroxylation (a genetic polymorphism-susceptible metabolic pathway coregulated by mephenytoin hydroxylation).
Whereas (S)-methylphenobarbital undergoes CYP2D6-mediated demethylation to form phenobarbital. In contrast to phenobarbital, mephobarbital is currently not widely used.
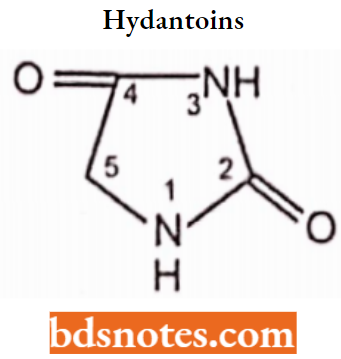
Hydantoins:
Hydantoins are cyclic monoacylureas. They possess an imidazoline-2,4-dione heterocyclic system and are structurally related to barbiturates, differing in lacking the 6-oxo moiety.
- Hydantoins are less acidic than barbiturates. Thus aqueous solutions of sodium salts provide strongly alkaline solutions.
- Clinically useful hydantoin possesses an aryl substituent at the 5-position and derivatives possessing lower alkyl substituents have antiabsence activity.
Structural Activity Relationship:
- A phenyl or other aromatic substitutions at C5 is essential for the activity.
- Alkyl substitutions at position C5 may contribute to sedation, a property that is absent in phenytoin.
- Among other hypnotics 1,3-disubstituted hydantoins exhibit activity against chemically induced convulsion. While it remains ineffective against electric shock-induced convulsion.
- A clinically useful hydantoin possesses an aryl substituent at the 5-position.
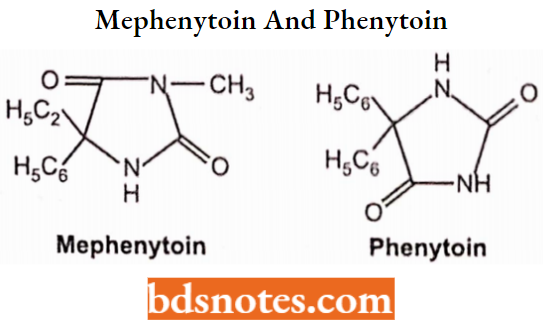
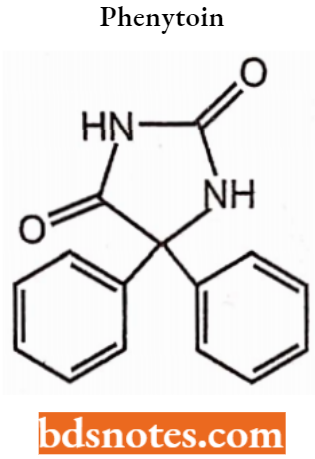
Phenytoin:
The derivatives of hydantoin (imidazolidine-2,4-dione) are well-known and clinically widely used in the therapy of epilepsy and cardiac arrhythmias. Phenytoin (5,5-diphenylhydantoin).
- One of the oldest anticonvulsants. is very effective in controlling a variety of seizure disorders, chemically 5,5-diphenylimidazolidine-2,4-dione. It is used to control certain types of seizures and to treat and prevent seizures.
- It works by decreasing abnormal electrical activity in the brain. These effects are due to a selective block of high-frequency neuronal activity.
- The drug targets the neuronal voltage-sensitive sodium channels (NVSC) to reproduce the normal ion potential and is known to block the release of neurotransmitters, such as serotonin and norepinephrine.
At an appropriate level, it inhibits monoamine oxidase activity and tends to alter the membrane potential. Phenytoin is used alone or in combination with phenobarbital in the treatment of grand mal and psychomotor epilepsy.
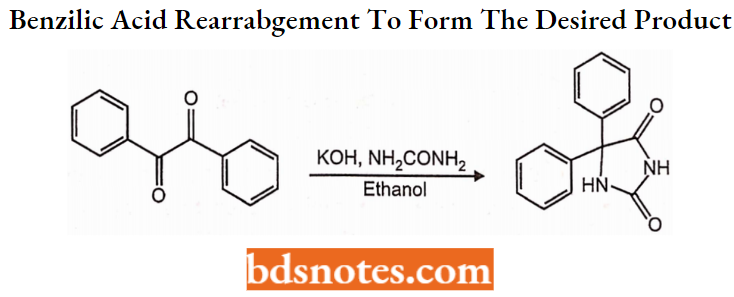
Phenytoin Synthesis:
5,5-diphenylimidazolidinedione is synthesized in two different ways. The first involves a base-catalyzed addition of urea to benzil followed by a benzilic acid rearrangement (1,2 phenyl migration) to form the desired product.
Phenytoin Adverse Effects:
Adverse effects associated with phenytoin include ataxia, nystagmus, and slurred speech.
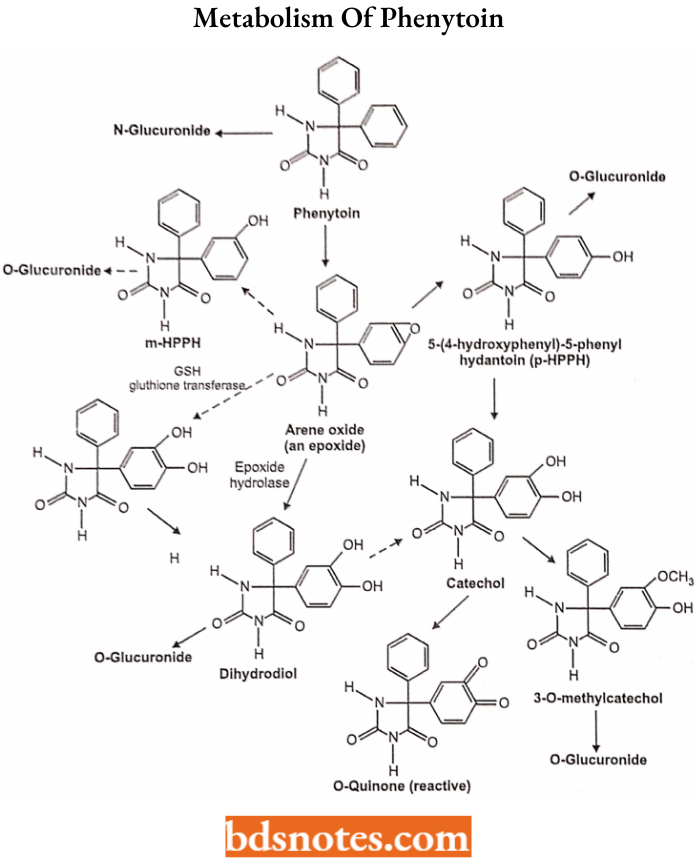
Phenytoin Metabolism of Phenytoin:
The principal metabolic pathway of phenytoin in humans is aromatic hydroxylation.
Mephenytoin: Mephenytoin is a compound belonging to the diphenylhydantoin (hydantoin derivative) compound containing a thiazolidinedione moiety substituted by a phenyl group.
Mephenytoin is known to target sodium channel protein type 5 subunit alpha. Cytochrome P450 2C19, Cytochrome P450 2C8, Cytochrome P450 2C9, Cytochrome P450 2B6, Cytochrome P450 1A2, and Cytochrome P450 2D6 are known to metabolize mephenytoin.. Chemically it is ethyl-3-methyl-5-phenylimidazolidine-2,4-dione.
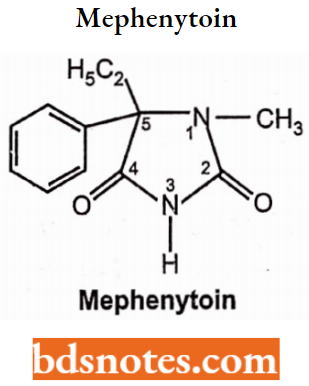
They have a marked anticonvulsive potency and are effective in grand mal seizure, focal epilepsy, and also in the case of status epilepticus without showing sedative-hypnotic properties.
Mephenytoin Mechanism of Action: The mechanism of action of mephenytoin is to block voltage-dependent neuronal sodium channels, and therefore limit repetitive firing of action potentials i.e. reduce the maximal activity of brain stem centers responsible for the tonic phase of tonic-clonic (grand mal) seizures.
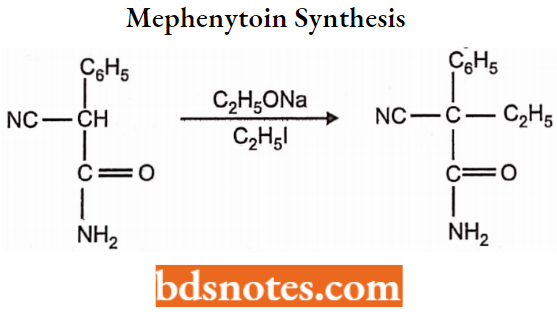
Mephenytoin Synthesis: Mephenytoin is prepared by the following steps:
In the first step, cyanoethylphenyl acetamide is prepared from cyanophenyl acetamide by treatment with sodium ethoxide and ethyliodide.
The product obtained by oxidation with an alkaline hypobromite solution converts the cyano group of cyanoethyl phenyl acetamide to the amide group, which isomerizes and cyclizes spontaneously.
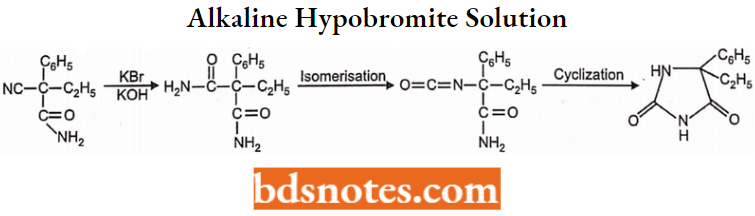
The above-cyclized product (5-ethyl 5-phenyl hydantoin) yields mephenytoin by methylation of the nitrogen atom (which is present between two carbonyl groups) with methyl sulfate or methyl iodide.
Oxazolidinediones:
Structural Activity Relationship: Replacement of the -NH group at position-1 of the hydantoin system with an oxygen atom yields the oxazolidinediones-2,4-dione system.
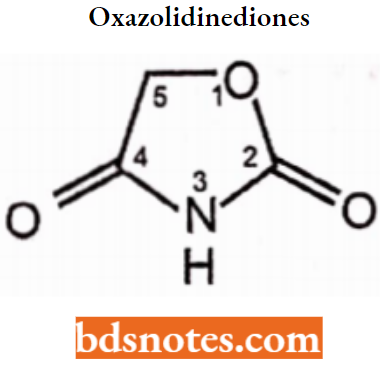
- The nature of the substitution on C-5 is essential for antiepileptic activity. Lower alkyl substitution towards anti-petitmal activity while acyl substituents towards antigrandmal activity.
- The N-alkyl substituent does not alter or afford the activity because clinically used agents undergo metabolism to produce N-dealkylation compounds.
Trimethadione: Trimethadione, the first oxazolidinedione, was introduced in 1945 and drug of choice for absence seizures until the introduction of succinimides in the 1950s.
- The use of the oxazolidinediones (trimethadione, paramethadione, and dimethadione) is now very limited. They contain an oxazolidine ring and have a similar structure to other antiepileptic agents introduced before 1960.
- These drugs are active against PTZ-induced seizures. Trimethadione is an oxazolidinedione derivative. Chemically it is 3,5,5-trimethyloxazolidine-2,4-dione.
Trimethadione raises the threshold for seizure discharges following repetitive thalamic stimulation. Its active metabolite methadone has the same effect on thalamic Ca2+ currents as ESM (reducing the T-type Ca2+ current).
- Thus, suppression of absence seizures is likely to depend on inhibiting the pacemaker action of thalamic neurons.
- The most common adverse effect is sedation and the unusual adverse effect is hemeralopia, a glare effect in which visual adaptation is impaired.
- Accumulation of methadone causes a very mild metabolic acidosis and should not be used during pregnancy.
Trimethadione Synthesis: Ethyl-a-hydroxy-α-methyl propionate is condensed with urea to yield 5, 5-dimethyloxazolidine-2, and 4-dione, which on treatment with a strong base followed by methylation with methyl sulfate gives trimethadione.
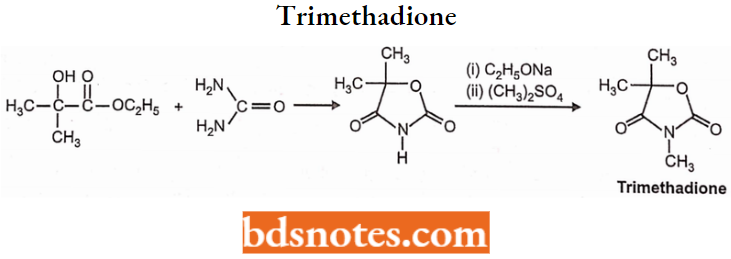
Paramethadione: Paramethadione is 5-ethyl-3, 5-dimethyl oxazolidine-2, 4-dione. Structurally it is very closely related to trimethadione. Paramethadione is used in the treatment of absence seizures.
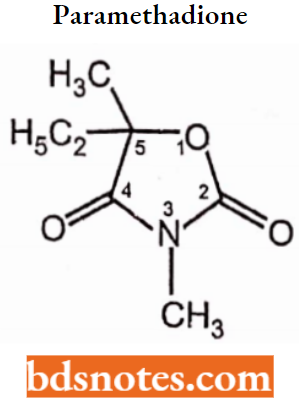
Succinimides: Oxazolidinediones are toxic hence to replace them with less toxic drugs succinimides were introduced in 1951 as antiepileptics.
- Succinimide is a cyclic imide of succinic acid or 2,5-pyrrolidinedione and its derivatives have been known for their antiepileptic activity, particularly against petitmal type of epilepsy.
- The mechanism of action of succinimides has been postulated that succinimides enhance inhibitory processes in the brain, by inhibiting T-type calcium channels and inhibiting the three-cycle per second thalamic ‘spike and wave’ discharge in absence seizures. Succinimide anticonvulsants are mostly used to treat absence seizures.
Ethosuximide: Ethosuximide is a succinimide anticonvulsant chemically designated as alpha-ethylalpha-methyl-succinimide i.e. (RS)-3-ethyl-3-methyl-pyrrolidine-2,5-dione), effective in the treatment of absence (petit mal) seizures.
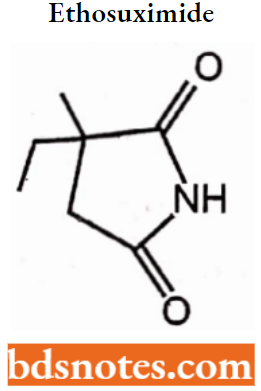
Ethosuximide Synthesis: Methylethylketone and cyanoacetic ester, undergo condensation. Then hydrogen cyanide is added. After acidic hydrolysis and decarboxylation of the synthesized dinitrile, 2-methyl-2-ethylsuccinic acid is formed.
Reacting this product with ammonia gives the diammonium salt, and heterocyclization into ethosuximide takes place during subsequent heating.
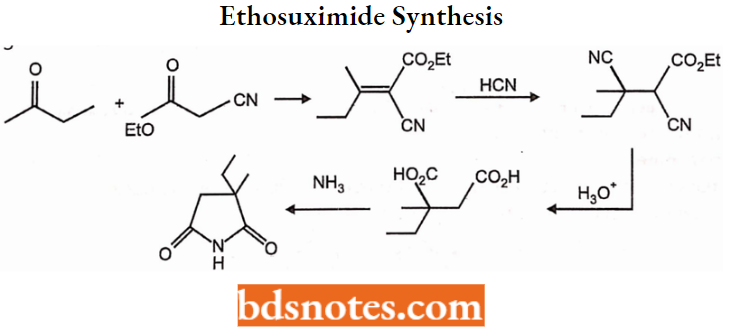
Ethosuximide Mechanism of Action:
- Its ability to decrease low-threshold calcium currents in thalamic neurons by T-type Ca2+ channel inhibition thereby the frequency of epileptiform attacks is reduced.
- Apparently by depression of the motor cortex and elevation of the threshold of the central nervous system to convulsive stimuli. It also inhibits Na+/K+ ATPase, depresses cerebral metabolic rate, and inhibits GABA aminotransferase.
Ethosuximide Adverse Reactions: Drowsiness, headache, fatigue, nausea, vomiting, idiosyncratic reactions and hematopoietic disturbances.
Phensuximide: Phensuximide is a 2,5-pyrrolidine-dione derivative. Chemically it is N-methyl-2- phenylsuccinimide. Phensuximide is a succinimide antiepileptic agent used to treat convulsions but it is reported to be less effective.
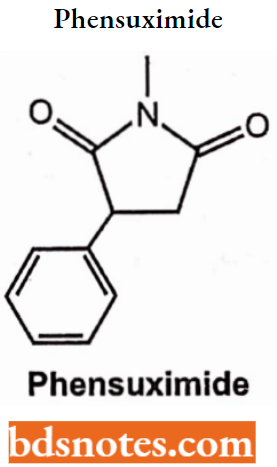
Methsuximide: Methosuximide is a succinimide antiepileptic drug. It is N, 2-dimethyl-2- phenyl succinimide. Phensuximide and methsuximide are phenyl succinimides that were developed before ethosuximide and used mainly as anti-absence drugs.
- Methsuximide has been used for partial seizures, but it is more toxic, Phensuximide is less effective than ethosuximide. Unlike ESM, these two compounds have some activity against seizures.
- The desmethyl (N-desmethylmethsuccimide) metabolite of methsuximide exerts a major anti-seizure effect.
Methsuximide Synthesis:
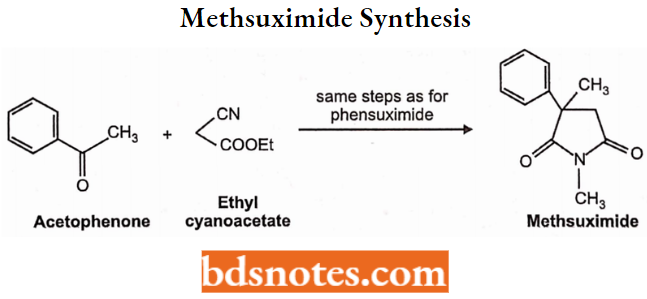
Urea and Monoacylureas:
Carbamazepine: Carbamazepine is an azepine derivative possessing dibenzazepine nucleus. Carbamazepine is 5H-dibenzazepine-5-carboxamide.
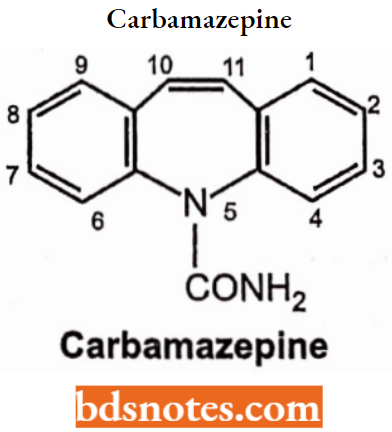
Carbamazepine (CBZ) is closely related to imipramine and other tricyclic antidepressants, it is a tricyclic compound useful in the management of bipolar depression.
- It was initially used for the therapy of trigeminal neuralgia but has been established as a useful antiepileptic agent as well. The ureide moiety (-N-CO-NH2) present in the heterocyclic ring exists in CBZ.
- The mechanism of action of CBZ was shown to be like that of Phenytoin. Like Phenytoin, CBZ exhibited activity against MES seizures. The CBZ blocks Na+ channels at therapeutic concentration and inhibits high-frequency recurring firing in neurons.
- It also operates presynaptically to reduce synaptic transmissions. These effects possibly account for the anti-epileptic action of CBZ.
- It interacts with adenosine receptors and also inhibits the uptake and release of norepinephrine from brain synaptosomes but does not control GABA uptake in the brain.
- The indication suggested that the postsynaptic action of GABA can be potentiated by CBZ.
It is the drug of choice for partial seizures and may be used for the treatment of generalized tonic-clonic seizures. It is also valuable in some patients with mania (bipolar disorder).
Carbamazepine Synthesis: It is prepared by the following steps:
10,11-Dihydro-5H-dibenzazepine by acetylation followed by bromination with N-bromosuccinimide gives N-acetyl-ll-bromo dibenzazepine.
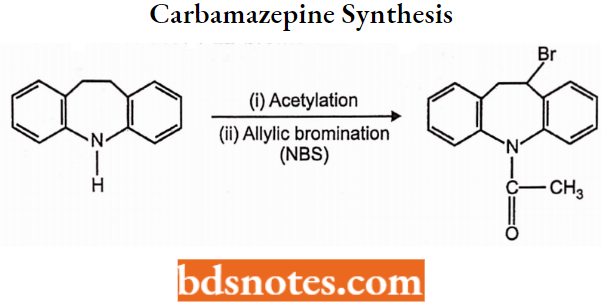
Dehydrohalogenation of N- acetyl-11-bromo dibenzazepine followed by saponification with potassium hydroxide in ethanol leads to dibenzazepine.
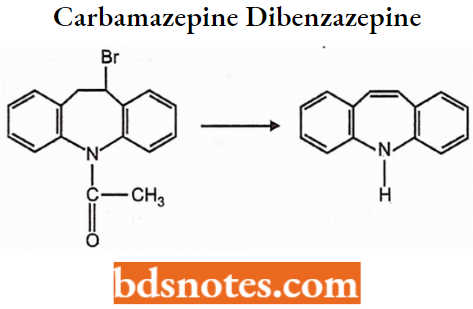
Treatment of dibenzazepine with phosgene followed by heating with ammonia produces carbamazepine.
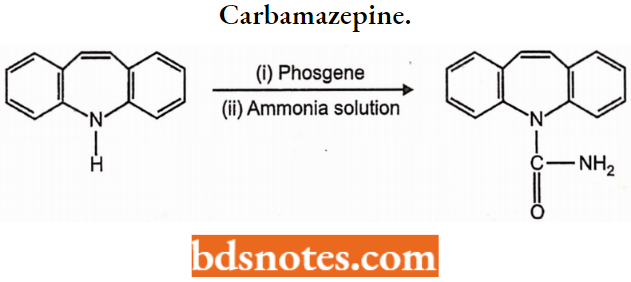
Carbamazepine Uses: Carbamazepine is an antiepileptic drug used to control grand mal and focal seizures. It is also used in the treatment of trigeminal neuralgia and the treatment of manic depression.
Clonazepam: Clonazepam is 5-(2-chlorophenyl)-3-dihydro-7-nitro-2H-1,4-benzodialzepin-2-one. Clonazepam is used in the treatment of grand mal epilepsy.
It is the alternate drug for the treatment of petit mal in patients who fail to respond to ethosuximide therapy. It suppresses various types of status epilepticus seizures because of its cardio-respiratory depressant effect.
Clonazepam Synthesis: p-Nitroaniline is treated with acetic anhydride to form an amide, thus protecting the amino group. Then followed by Friedel-Crafts acylation gives a diaryl ketone and removal of the amine protecting by hydrolysis of the amide in an aqueous base gives the free amine.
- Then treatment of the amine with chloroacetyl chloride gives an α-chloroamide. Nucleophilic displacement of the primary chloride by ammonia gives a primary amine.
- The reaction of the primary amine with the nearby ketone gives an imine and closes the seven-membered ring of clonazepam.
Clonazepam Adverse Effect: The primary side effect associated with clonazepam is central nervous system depression.
Valproic Acid:
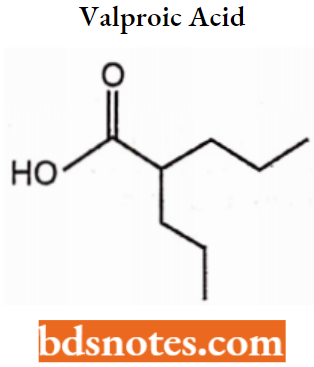
Valproic Acid Mechanism of Action:
Block of voltage-gated sodium channels in neuronal membranes prevents Na+ influx, which results in decreased axonal conductance by increasing the refractory period of the neuron and also inhibits GABA transaminase.
Valproic Acid Use: Complex partial seizures as monotherapy and/or adjuvant, Simple and complex absence seizures, Myoclonic seizures, Migraine prophylaxis, Bipolar mania.
Valproic Acid Synthesis:
Valproic acid may be synthesized from 4-heptanol by successive conversions to 4-bromoheptane with HBr, to 4-cyanoheptane with HCN and to 2-propyl pentanoic (valproic) acid by alkaline hydrolysis of 4-cyanoheptane.
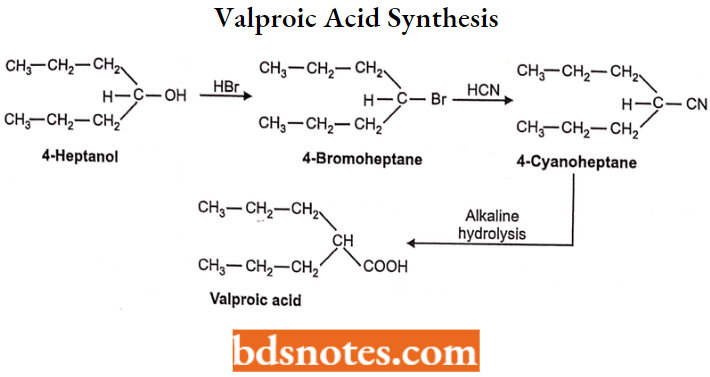
Valproic Acid Adverse Effects: Weight gain, pancreatitis, tremor, thrombocytopenia, headache, azoospermia, hirsutism, and hair color change.
Gabapentin: Gabapentin containing primary amino and carboxyl groups, is better represented as internal salt resulting from proton transfer from the acidic carboxyl group to the basic amino group.

Gabapentin is a derivative of GABA and is effective against partial seizures. It is found to be more effective as an antiepileptic drug and appears not to act on GABA receptors.
- It may change GABA metabolism, its non-synaptic release, or its reuptake by GABA transporters. An enhancement in brain GABA concentration is seen. Gabapentin is carried into the brain by the L-amino acid transporter.
- The drug is also connected to the subunit of voltage-sensitive Ca2+ channels. Gabapentin is active as an adjunct against partial and generalized tonic-clonic seizures.
- It is also effective in neuropathic pain and for post-therapeutic neuralgia in adults. The most frequent adverse effects are somnolence, ataxia, dizziness, headache, and tremor.
Primidone: Primidone is an antiepileptic agent related to barbiturates. Primidone is a diketone derived from hexahydropyrimidine. Chemically it is 5-ethyl-2, 3-dihydro-5-phenyl-4, 6- (1H, 5H)-pyrimidine dione.
- Primidone (2-deoxy PBT) was metabolized into phenobarbital and phenylethylmalonamide (PEMA). All these three compounds are active against convulsions.
- Primidone is useful against partial seizures and generalized tonic-clonic seizures and may be more effective than phenobarbital.
- It was considered to be the drug of choice for partial seizures, but the partial seizures in adults strongly suggest that CBZ and PHT are superior to primidone.
Primidone Synthesis: It is prepared from diethyl ethyl phenylmalonate by following steps: Diet yl ethyl phenylmalonate is converted into its amide derivative by the action of ammonia.
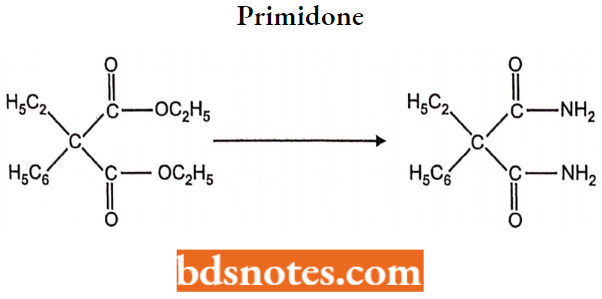
Felbamate: The mechanism of action suggested that it is an NMDA receptor blockade via the glycine binding site.
- Felbamate has a half-life of 20 hrs and is metabolized by hydroxylation and conjugation; a considerable amount of the drug is excreted unaffected in urine.
- When added to therapy with other antiepileptic drugs, felbamate enhanced plasma PHT and VLPA levels but reduced levels of CBZ. It is used in partial seizures and is also active against the seizures that happen in Lennox-Gastaut syndrome.
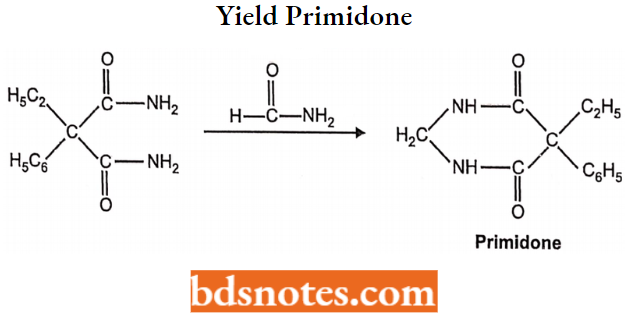
Leave a Reply