Carbohydrates
Many aspects of the chemistry of carbohydrates are not specific to this class of compounds but are merely examples of the simple chemical reactions we have already met.
Therefore, against the usual practice, we have not attempted a full treatment of carbohydrate chemistry and biochemistry in this chapter. We want to avoid giving the impression that the reactions described here are something special to this group of compounds.
- Instead, we have deliberately used carbohydrates as examples of reactions in earlier chapters, and you will find suitable cross-references.
- Carbohydrates are among the most abundant constituents of plants, animals, and microorganisms. Polymeric carbohydrates function as important food reserves and as structural components in cell walls.
- Animals and most microorganisms are dependent upon the carbohydrates produced by plants for their very existence.
- Carbohydrates are the first products formed in photosynthesis and are the products from which plants synthesize their food reserves, as well as other chemical constituents.
- These materials then become the foodstuffs of other organisms. The main pathways of carbohydrate biosynthesis and degradation comprise an important component of intermediary metabolism that is essential for all organisms.
The name carbohydrate was introduced because many of the compounds had the general formula Cx(H2O) y, and thus appeared to be hydrates of carbon. The terminology is now commonly used in a much broader sense to denote polyhydroxy aldehydes and ketones, and their derivatives.
Sugars or saccharides are other terms used in a rather broad sense to cover carbohydrate materials. Though these words link directly to compounds with sweetening properties, the application of the terms extends considerably beyond this. A monosaccharide is a carbohydrate usually in the range C3 –C9 whereas an oligosaccharide covers small polymers comprised of 2–10 monosaccharide units. The term polysaccharide is used for larger polymers.
Monosaccharides
Six-carbon sugars (hexoses) and five-carbon sugars (pentoses) are the most frequently encountered monosaccharide carbohydrate units in nature. Primary examples of these two classes are the hexoses glucose and fructose, and the pentose ribose. Note the suffix -ose as a general indicator of carbohydrate nature.
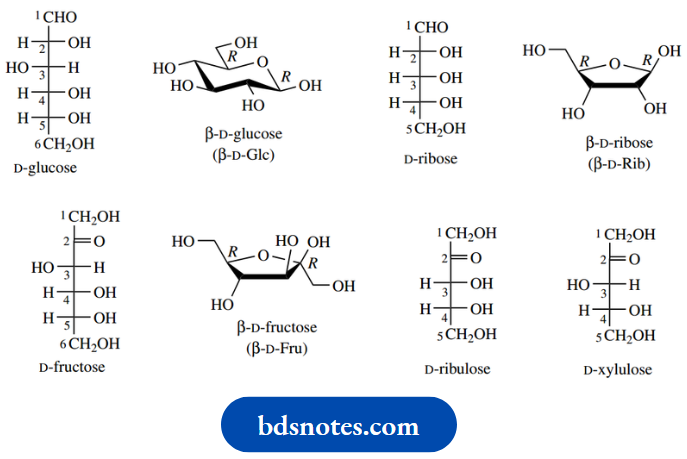
The structures above show some of the fundamental features of carbohydrates. Initially, we have drawn these compounds in the form of Fischer projections, a depiction developed for these compounds to indicate conveniently the stereochemistry at each chiral centre.
- The Fischer projection is drawn as a vertical carbon chain with the group of highest oxidation state, i.e. the carbonyl group, closest to the top, and numbering takes place from the topmost carbon.
- The carbonyl group in glucose and ribose is an aldehyde; such compounds are termed aldoses.
- Fructose, by contrast, has a ketone group and is therefore classified as a ketose. Glucose could also be termed an aldohexose and fructose a ketohexose, whereas ribose would be an aldopentose, names which indicate both the number of carbons and the nature of the carbonyl group.
- Another aspect of nomenclature is the use of the suffix -ulose to indicate a ketose. Fructose could thus be referred to as a hexulose, though we are more likely to see this suffix in the names of specific sugars,
Example:
Ribulose is a ketose isomer of the aldose ribose.
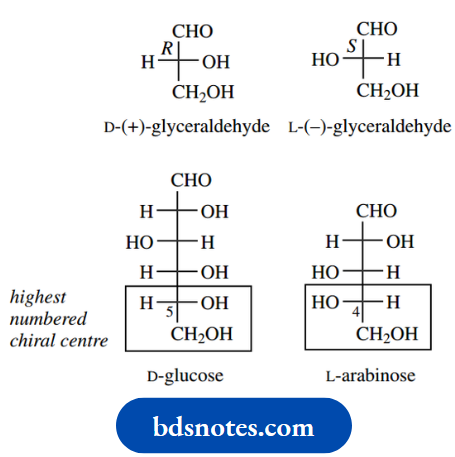
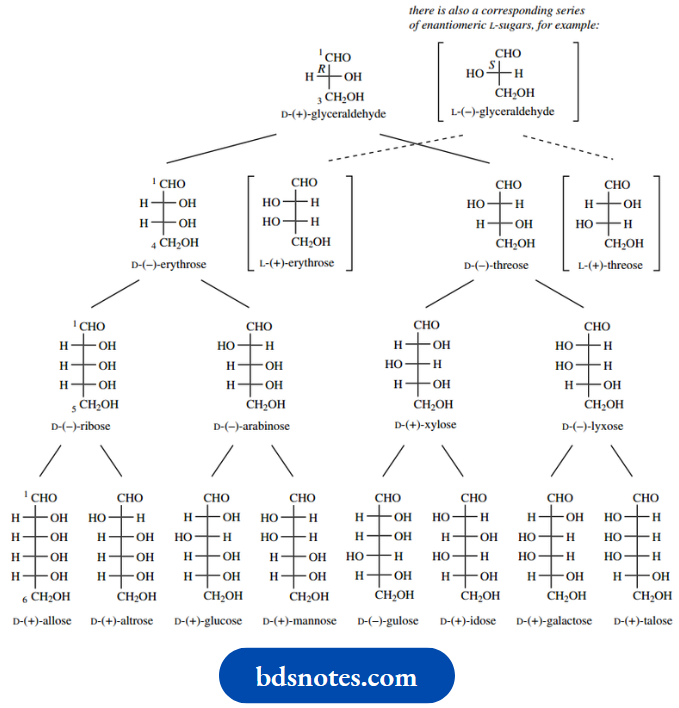
Each of these compounds has a prefix D- with the name. As we saw, this indicates that the configuration at the highest numbered chiral centre is the same as that in D-( R)-(+)-glyceraldehyde; the alternative stereochemistry would be related to L-( S)-(−)-glyceraldehyde and consequently be part of an L-sugar
Structures of the various D-aldoses in the range C3 –C6 are shown below. These compounds are multifunctional structures, having a carbonyl group and several hydroxyls, usually with two or more chiral centres.
You will notice that we are comparing the stereochemistry in the different possible diastereoisomers for compounds containing several chiral centres. There is a corresponding series of enantiomeric L-sugars; only a few of these are shown.
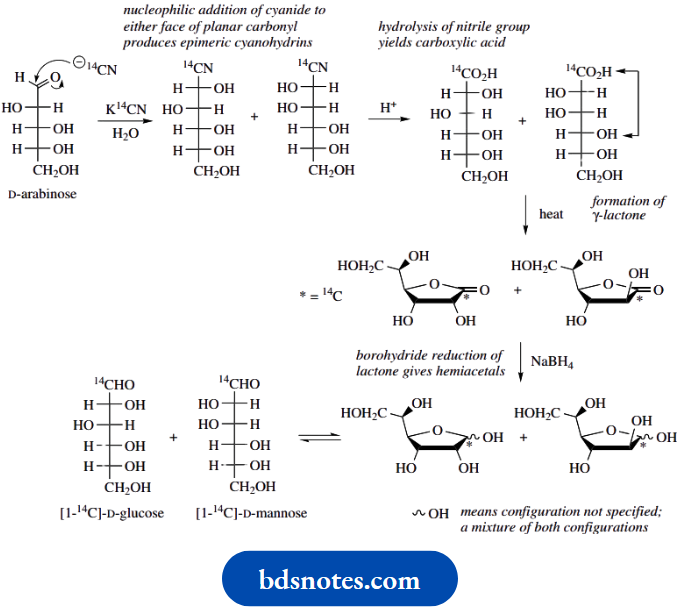
Synthesis of 14C -labelled glucose
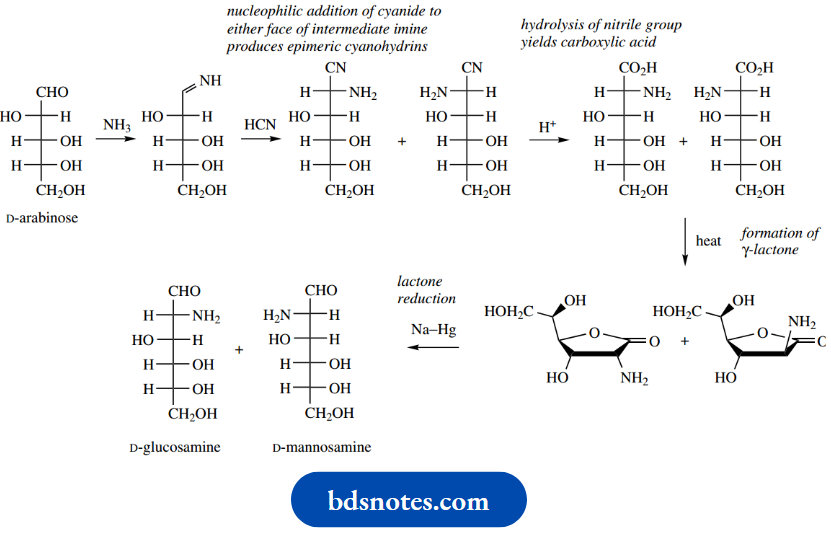
A sequence known as the Kiliani–Fischer synthesis was developed primarily for extending an aldose chain by one carbon and was one way in which configurational relationships between different sugars could be established.
A major application of this sequence nowadays is to employ it for the synthesis of 14C-labelled sugars, which in turn may be used to explore the role of sugars in metabolic reactions. The synthesis of 14C-labelled D-glucose starts with the pentose D-arabinose and 14C-labelled potassium cyanide, which react together to form a cyanohydrin.
Since cyanide can attack the planar carbonyl group from either side, the cyanohydrin product will be a mixture of two diastereoisomers that are epimeric at the new chiral centre. The two epimers are usually formed in unequal amounts because of a chiral influence from the rest of the arabinose structure during the attack of the nucleophile.
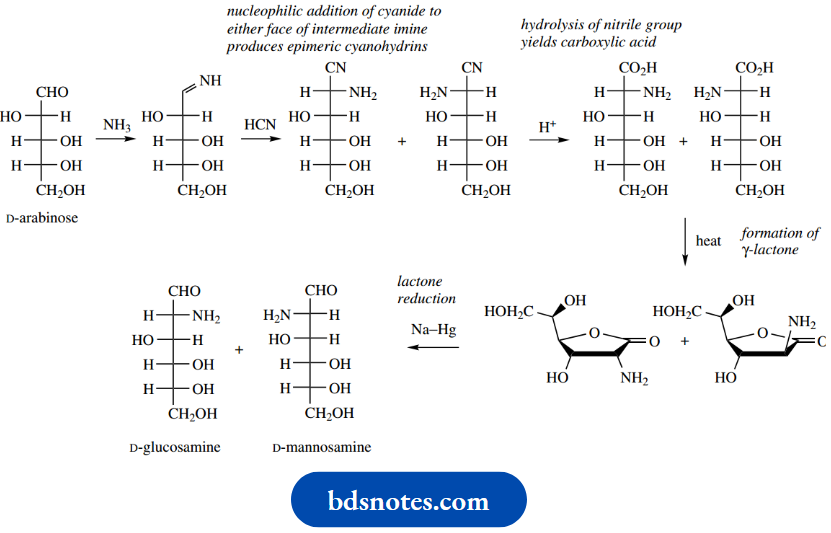
The nitrile groups in the product mixture are then hydrolysed to carboxylic acids ( Upon heating, the acids readily form cyclic esters (lactones) through the reaction of the hydroxyl group on C-4 with the carboxylic acid, the five-membered ring being most favoured (see
The pair of lactones is then reduced using sodium amalgam under acidic conditions to yield aldehydes, though it has been found that this reaction can also be achieved using aqueous sodium borohydride.
Sodium borohydride reacts readily with lactones, though it is not usually effective in reducing esters. It is also normally difficult to stop at an aldehyde intermediate (see Section 7.11), but the reduction of a lactone gives initially a hemiacetal; ring opening of the hemiacetal then leads to the aldehyde.
The product will be a mixture of the two epimeric sugars D-glucose and D-mannose, which will be labelled with 14C in the aldehyde function. Separation of the diastereoisomeric products may be achieved via fractional crystallization or by chromatography and may be carried out at either the cyanohydrin stage or the final product stage.
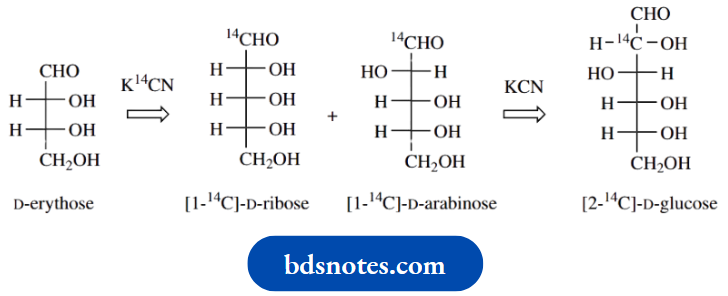
Note how the process may be modified to extend its versatility. Thus, using 14C-labelled potassium cyanide with D-erythrose yields a mixture of [1-14C]-D-ribose and [1-14C]-D-arabinose. The sequence could then be repeated on the latter product, using unlabelled KCN, to give [2-14C]-D-glucose.
Enolization and isomerization
In common with other aldehydes or ketones that have hydrogen on the α-carbon, enolization is possible, especially when sugars are treated with base. The additional presence of a hydroxyl on the α-carbon causes further isomerization. Thus, treatment of D-glucose with dilute aqueous sodium hydroxide at room temperature leads to an equilibrium mixture also containing D-mannose and D-fructose.
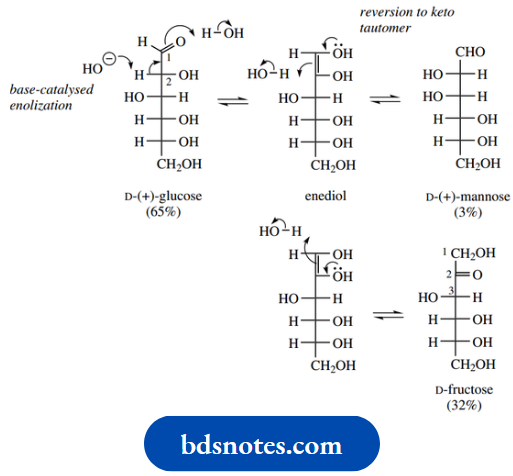
Removal of the α-hydrogen in D-glucose leads to enolization (we have omitted the enolate anion in the mechanism). Reversal of this process allows epimerization at C-2, since the enol function is planar, and a proton can be acquired from either face, giving D-mannose as well as D-glucose.
Alternatively, we can get isomerization to D-fructose. This is because the intermediate enol is an enediol; restoration of the carbonyl function can, therefore, provide either a C-1 carbonyl or a C-2 carbonyl. The equilibrium mixture using dilute aqueous sodium hydroxide at room temperature consists mainly of D-glucose and D-fructose, with smaller amounts of D-mannose. The same mixture would be obtained if either D-mannose or D-fructose were treated similarly.
Note that harsher conditions may lead to further changes, e.g. epimerization at C-3 in fructose, plus isomerization, or even reverse aldol reactions. In general, basic conditions must be employed with care if isomerizations are to be avoided. To preserve stereochemistry, it is usual to ensure that free carbonyl groups are converted to acetals or ketals (glycosides, see Section 12.4) before basic reagents are used. Isomerization of sugars via enediol intermediates features prominently in the glycolytic pathway of intermediary metabolism.
Cyclic hemiacetals and hemiketals
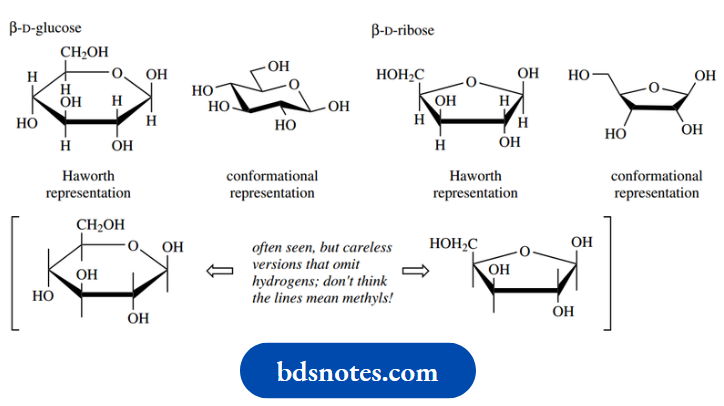
Monosaccharide structures may be depicted in openchain forms showing their carbonyl character, or in cyclic hemiacetal or hemiketal forms. Alongside the Fischer projections of glucose, ribose, and fructose shown earlier, we included an alternative representation of the compound in its cyclic form. The compounds exist predominantly in cyclic forms, which result from the nucleophilic attack of an appropriate hydroxyl onto the carbonyl.
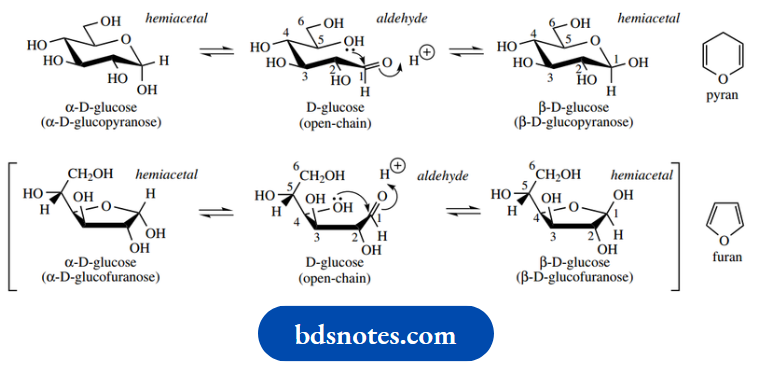
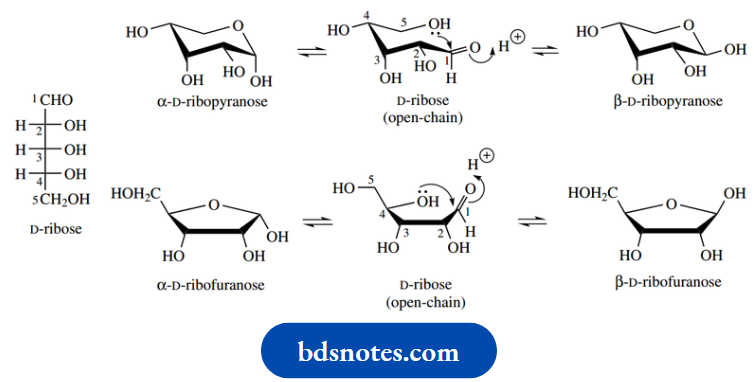
Both six-membered pyranose and five-membered furanose structures are encountered, a particular ring size usually being characteristic of any one sugar. Thus, although glucose has the potential to form both six-membered and five-membered rings, an aqueous solution consists almost completely of the six-membered hemiacetal form; five-membered rings are usually formed more rapidly, but six-membered rings are generally more stable and predominate at equilibrium. The names pyranose and furanose are derived from the oxygen heterocycles pyran and furan. Shown below is a reminder of how we can transform a Fischer projection of sugar into a cyclic form
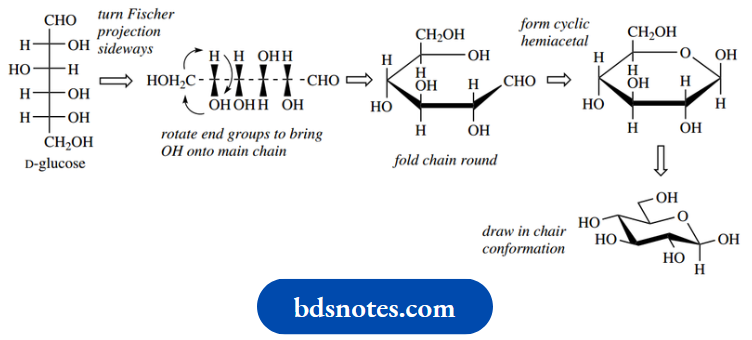
The pentose ribose is also able to form six-membered pyranose and five-membered furanose rings. In solution, ribose exists mainly (76%) in the pyranose form; interestingly, however, when we meet ribose in combination with other entities, e.g. nucleosides, it is almost always found in furanose form.
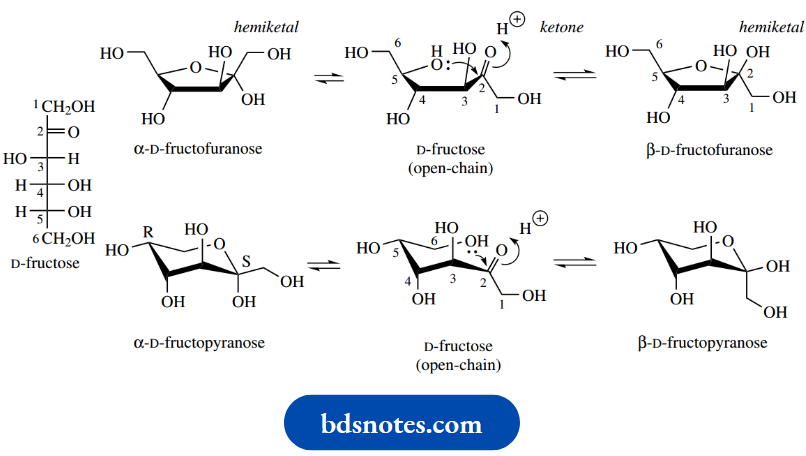
Fructose is a ketose and, therefore, forms hemiketal ring structures. Like ribose, it is usually found in combination as a five-membered furanose ring, though the simple sugar in solution exists primarily in the pyranose form.
The anomeric centre
Since the carbonyl group is planar and may be attacked from either side, two epimeric structures (anomers) are possible in each case, and in solution, the two forms are frequently in equilibrium because hemiacetal or hemiketal formation is reversible. The two anomers are designated α or β by comparison of the chiralities at the anomeric centre and the highest-numbered chiral centre. If these are the same (RS convention), the anomer is termed β, or α if they are different.
In practice, this translates to the anomeric hydroxyl being ‘up’ in the case of β-D-sugars and α-L-sugars. It is interesting to note that the descriptors α or β were originally assigned to the two forms of glucose based on the order in which they crystallized out from the solution. Without changing the nomenclature for these two compounds, α or β are now assigned on a much more rigid stereochemical basis.
By convention, the ring form of sugars is drawn with the ring oxygen to the rear and the anomeric carbon furthest right. Wedges and the bold bond help to emphasize how we are looking at the chair-like pyranose ring. However, to speed up the drawing of structures we tend to omit these, and then the lower bonds always represent the nearest part of the ring.
Since there are two anomeric forms, and these are often in equilibrium via the acyclic carbonyl compound, we can use a new type of bond to indicate that the configuration is not specified and could be of either stereochemistry. This is the wavy or wiggly bond, and to display our indecision further we usually site it halfway between the two possible positions.
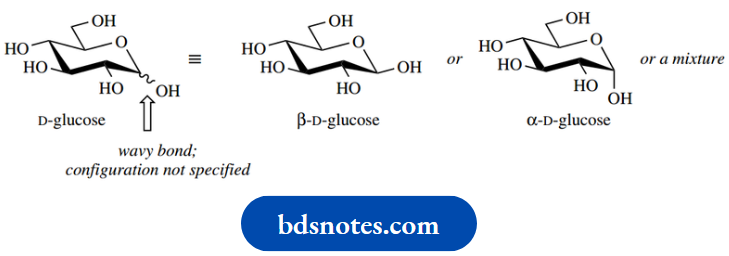
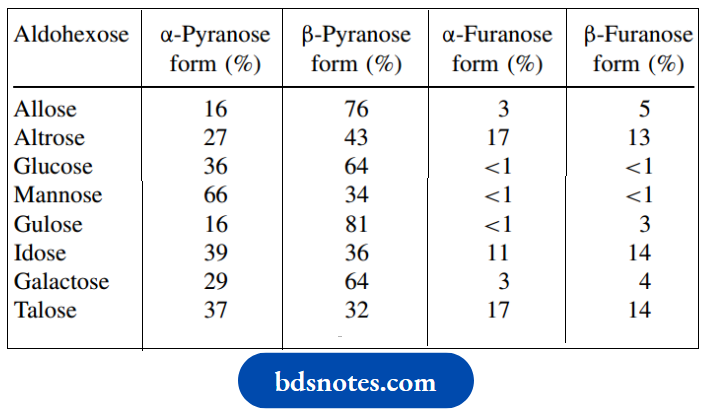
It follows that, when we dissolve a sugar such as glucose or ribose in water, we create a mixture of various equilibrating structures. The relative proportions of pyranose and furanose forms, and their respective anomers for the eight aldohexoses, are shown in In each case, the proportion of non-cyclic form is very small.
The most stable conformation of the cyclic sugar is mainly determined by a minimization of steric interactions, i.e. the maximum number of equatorial substituents (see Section 3.3.2). It follows that the preferred conformation for β-D-glucose will be that with all substituents equatorial; the alternative has all substituents axial. Carbohydrate chemists have introduced a neat way of referring to the two.
Equilibrium proportions of pyranose and furanose forms ofaldohexoses in water:
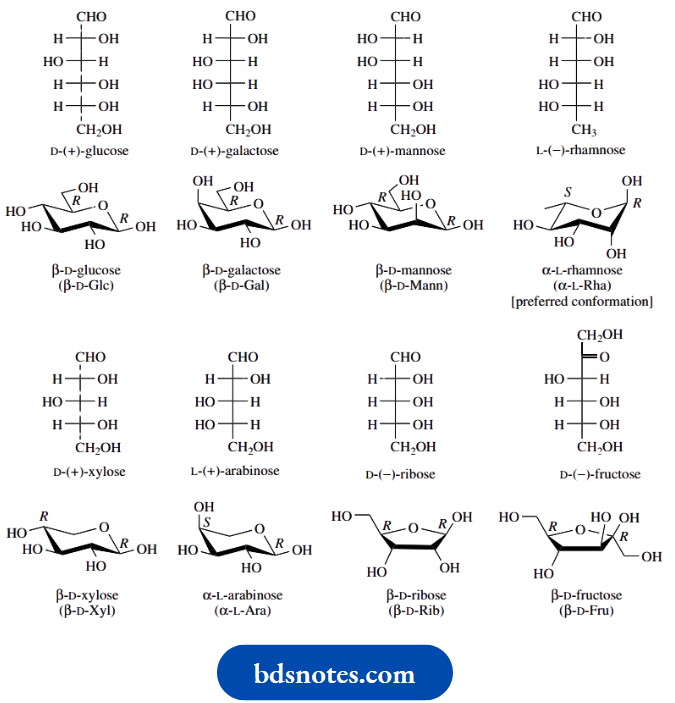
Ring size and a numeric form of common sugars
Sugars exist predominantly in cyclic hemiacetal or hemiketal forms, and whilst both six-membered pyranose and five-membered furanose structures are encountered, a particular ring size is usually characteristic for any one sugar, especially when it is found in combination with other entities in natural structures. The most commonly encountered monosaccharides and their usual anomers are shown here. By convention, the ring form is drawn with the ring oxygen to the rear and the anomeric carbon furthest right. Also shown are the accepted abbreviations for these sugars.
The two anomers are designated α or β by comparison of the chiralities at the anomeric centre and the highest-numbered chiral centre. If these are the same (RS convention), the anomer is termed β, or α if they are different.
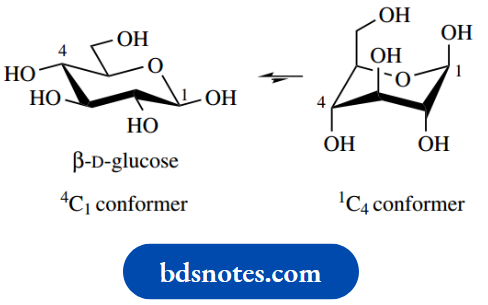
Note that the D and L prefixes are assigned based on the chirality (as depicted in Fischer projections) at the highest numbered chiral centre and its relationship to D-( R)-(+)-glyceraldehyde or L-( S)-(−)-glyceraldehyde The stereochemistries of the various substituents may be deduced by considering the implications of the Fischer projection conformers, in that the left-hand conformer of glucose is termed 4C1, and the right-hand one 1C4.
The ‘C’ indicates chair conformation, the superscript numeral is the carbon atom that is above the plane of the ring, and the subscript numeral is which carbon atom is below the plane of the ring. For this description, we consider the pyranose ring as originally planar, distorted to a chair by pushing carbons 1 and 4 out of the plane.
At first glance, the preferred conformation for L-hexoses,
Example: α-L-rhamnose, appears different from that of the D-hexoses.
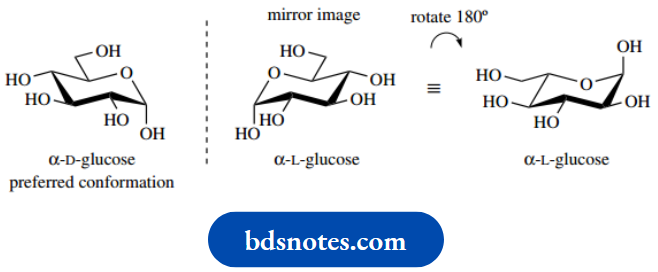
This is readily rationalized by considering the preferred conformation of α-L-glucose – the α-anomer is chosen simply because we can easily follow the anomeric substituent. Since α-L-glucose is the enantiomer of α-D-glucose, we can draw the mirror image representation, and then rotate this so that the heterocyclic oxygen comes to the required position.
Note that you may also encounter another version of the cyclic form referred to as the Haworth representation.
- This shows the ring as a planar system and is commonly used in biochemistry books. However, we know that five-membered and six-membered rings are certainly not planar.
- The Haworth representation nicely reflects the up–down relationships of the various substituent groups, but is quite uninformative about the shape of the molecule, and whether the substituents are equatorial or axial.
In really bad cases, authors omit the hydrogen atoms, giving an ambiguous structure – do lines mean methyl or hydrogen? Haworth representations may be easier to draw, but you are strongly encouraged to use the more informative conformational structures.
One of the consequences of forming a cyclic hemiacetal or hemiketal is that the nucleophilic hydroxyl adds to the carbonyl group and forms a new hydroxyl. This new group is susceptible to many normal chemical reactions of hydroxyls,
Example:
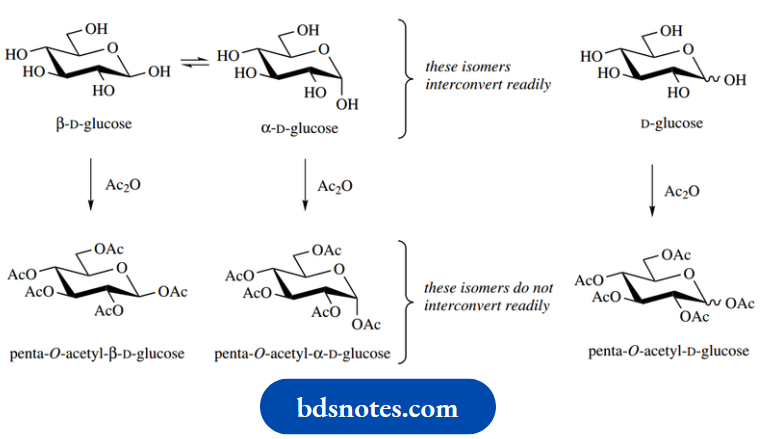
Esterification and this type of reaction effectively freezes the carbohydrate into one anomeric form, since the ring-opening and equilibration can now no longer take place.
Consider esterification of glucose with acetic anhydride β-D-Glucose will be acetylated to give the β-acetate, whereas α-D-glucose will specifically give the α-acetate. These two forms do not equilibrate merely by dissolving in a solvent, although they can be interconverted by some other means,
Example: Nucleophilic substitution reactions with acetate.
If we wish to consider esterification of the α– β mixture, we could use the unspecified wavy bond representation shown on the right.
Alditols
Reduction of the aldehyde or ketone group in sugar is readily achieved using a variety of reducing agents. Reduction occurs on the small amount of open-chain form present at equilibrium. As the openchain form is removed, the equilibrium is disturbed until total reduction is achieved. The products are polyhydroxy compounds termed alditols.
Reduction of aldoses is the more satisfactory reaction, in that a single product is formed. On the other hand, the reduction of ketoses generates a new chiral centre, and two epimeric alditols will result. Thus, treatment of D-glucose with sodium borohydride gives D-glucitol, also known as D-sorbitol.
It should be noted that LAH is not a satisfactory reducing agent for this reaction because of the several hydroxyl groups present
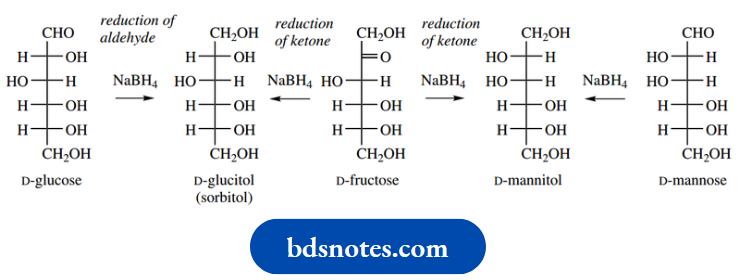
On the other hand, borohydride reduction of the ketose D-fructose will give a mixture of D-glucitol and its epimer, D-mannitol. A better approach to D-mannitol would be a reduction of the aldose D-mannose. D-glucitol (sorbitol) is found naturally in the ripe berries of the mountain ash (Sorbus aucuparia) but is prepared semi-synthetically from glucose.
It is half as sweet as sucrose, is not absorbed orally, and is not readily metabolized in the body. It finds particular use as a sweetener for diabetic products. D-Mannitol also occurs naturally in manna, the exudate of the manna ash Fraxinus ornus. This material has similar characteristics to sorbitol but is used principally as a diuretic. It is injected intravenously, is eliminated rapidly into the urine, and removes fluid by an osmotic effect.
Glycosides
The cyclic hemiacetal and hemiketal forms of monosaccharides are capable of reacting with an alcohol to form acetals and ketals. The acetal or ketal product is termed a glycoside, and the non-carbohydrate portion is referred to as an aglycone. In the nomenclature of glycosides we replace the suffix -ose in the sugar with -oside.
Simple glycosides may be synthesized by treating an alcoholic solution of the monosaccharide with an acidic catalyst, but the reaction mixture usually then contains a mixture of products.
This is an accepted problem with many carbohydrate reactions; it is often difficult to carry out selective transformations because of their multifunctional nature.
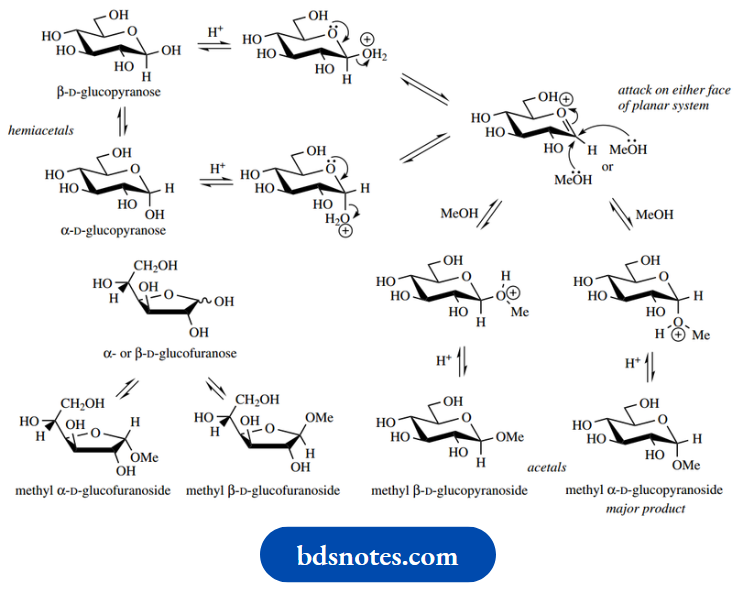
The reaction of glucose with methanol and gaseous HCl yields four acetal products, the α- and β-pyranosides and α- and β-furanosides, which may be separated. The pyranosides are the predominant components, and the major product is the α-pyranoside.
This is perhaps unexpected, in that the β-pyranoside has all its substituents equatorial, whereas the α-anomer has its anomeric substituent axial. This so-called anomeric effect arises from a favourable electronic stabilization in the axial anomer that is not possible in the equatorial anomer. It involves overlap from the ring oxygen lone pair, and to achieve this the lone pair and the substituent must be antiperiplanar.
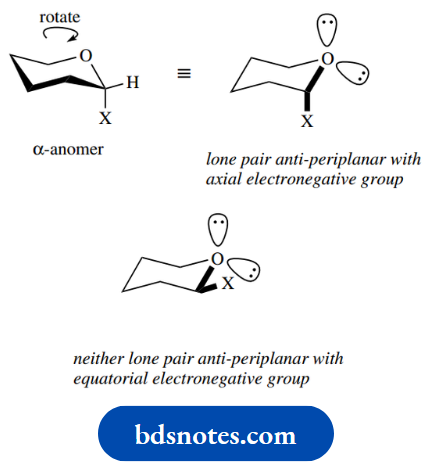
The anomeric effect is rather complex and will not be considered in any detail. It occurs when we have a heterocyclic ring (O, N, or S), with an electronegative substituent (halogen, OH, OR, OCOR, etc.) adjacent to the heteroatom, and favours the isomer in which the substituent is axial.
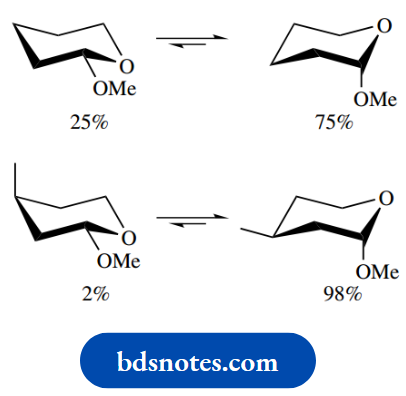
Thus, with the first of the simple acetals shown below, where we need to consider only conformational isomerism, some 75% of the axial conformer is present at equilibrium. Without the ring oxygen, we would see an equatorial isomer predominating. In the second example, the additional stability conferred by the equatorial methyl group increases even further the proportion of the conformer with the axial methoxyl.
We have noted that an aqueous solution of glucose exists as an equilibrium mixture containing some 64% of the β-anomer
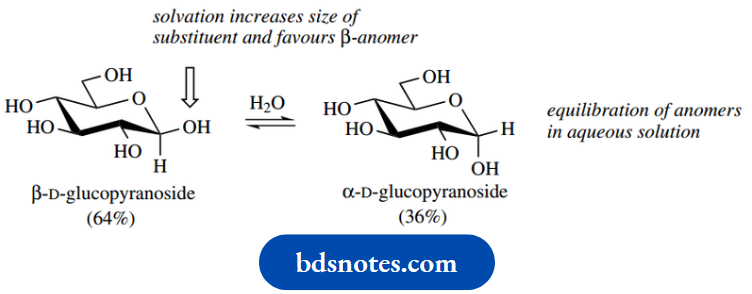
Based simply on steric effects, this proportion appears somewhat low, whereas because of the anomeric effect just described the proportion now seems rather high. Anomeric effects are observed to be solvent-dependent, and hydroxy compounds experience considerable solvation with water through hydrogen bonding.
This significantly increases the steric size of the substituent and reinforces the steric effects By considering the reversibility of the acetalforming reactions, treatment of either of the two methyl pyranosides with acidic methanol will produce the same equilibrium mixture. A related equilibration occurs with the anomers of glucose, as seen earlier.
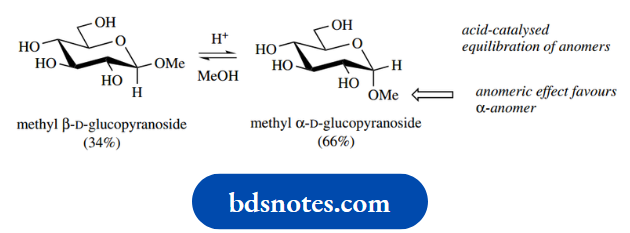
It should also be noted that hydrolysis of glycosides (acetals or ketals) will occur under acid-catalysed conditions if we have an excess of water present. This is a reversal of the process for glycoside formation, with the equilibrium favouring the aglycone plus sugar rather than the glycoside. The sugar product will again be the equilibrium mixture of anomers.
Acid-catalysed hydrolysis of glycosides
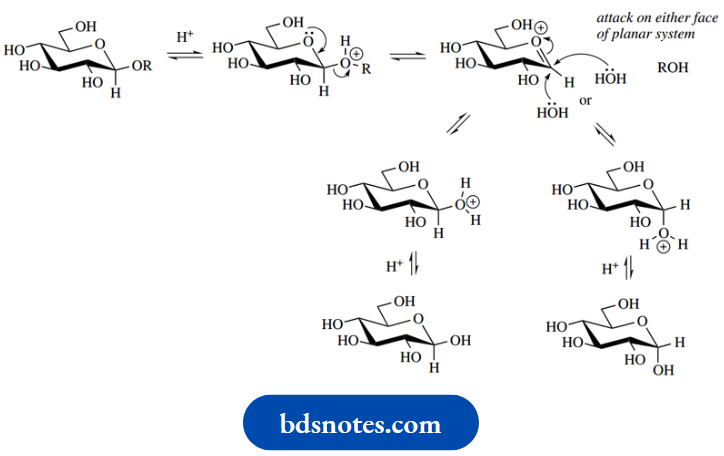
Hydrolysis of glycosides can also be achieved by the use of specific enzymes, e.g. β-glucosidase for β-glucosides and β-galactosidase for β-galactosides. These enzymes mimic the acid-catalysed processes, are commercially available, and may be used just like a chemical reagent.
Some examples of natural O-,S-,C- ,and N – glycosides
Many different types of glycoside structures are found in nature, especially in plants. Since the presence of a sugar unit in the structure provides polarity, glycosylation is likely a means by which an organism makes an aglycone water-soluble and transportable. Most of the natural glycosides are compounds in which the aglycone is an alcohol or a phenol, and such derivatives are termed O-glycosides. O-glycosides are thus acetals or ketals.
Less commonly, one encounters compounds in which a thiol (RSH) has been bonded to the sugar unit resulting in a thioacetal. These compounds are termed S-glycosides. Some examples of O- and S-glycosides are shown below.
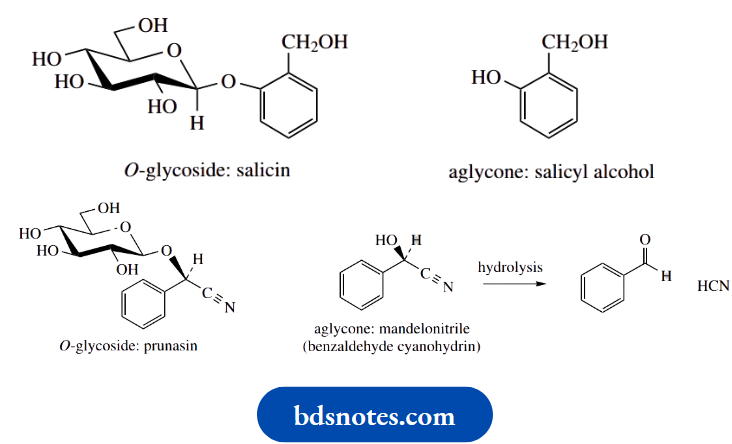
Salicin is an O-glycoside of a phenol, namely salicyl alcohol. Salicin is a natural antipyretic and analgesic found in willow bark and is the template from which aspirin (acetylsalicylic acid, was developed.
Prunasin from cherry laurel is an example of a cyanogenic glycoside, the hydrolysis of which leads to the release of toxic HCN ). It is the O-glucoside of the alcohol mandelonitrile, the trivial name for the cyanohydrin of benzaldehyde. It is the further hydrolysis of mandelonitrile that liberates HCN.
S-glycosides in nature are quite rare, but there is an important group called glucosinolates. These compounds are responsible for the pungent properties of mustard, horseradish and members of the cabbage family. One example is sinigrin, found in black mustard seeds. When seeds are crushed, enzymic hydrolysis liberates the aglycone, which subsequently rearranges to the pungent principle allylisothiocyanate.
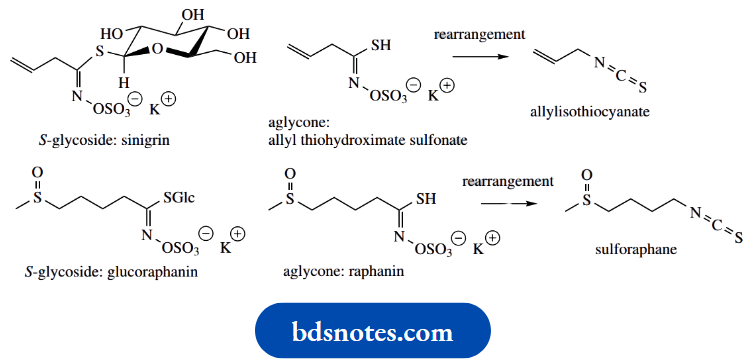
A related glucosinolate glucoraphanin is found in broccoli and is associated with the beneficial medicinal properties of this vegetable. This is hydrolysed to the isothiocyanate sulforaphane, which is believed to induce carcinogen-detoxifying enzyme systems.
Other natural glycosides are not acetals or ketals, but analogues in which the nucleophilic species has been an amine ( N-glycosides), or even some carbanionic species so that the sugar becomes attached to carbon ( C-glycosides). It should be noted that the presence of a C–C bond between the sugar and the aglycone means that C-glycosides are not cleaved by simple hydrolysis, but require an oxidative process.
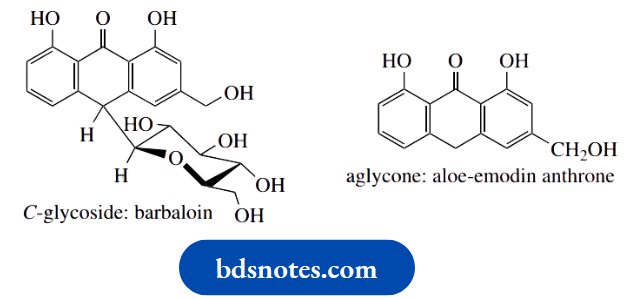
C-glycosides are typified by barbaloin, a component of the natural purgative drug cascara, but, as a group, the N-glycosides are perhaps the most important to biochemistry. N-glycosidic linkages are found in the nucleosides, components of DNA and RNA. In addition, nucleosides are essential parts of the structures of crucial biochemicals such as ATP, coenzyme A, NAD+, etc. The amine in these types of compounds is part of a purine or pyrimidine base.
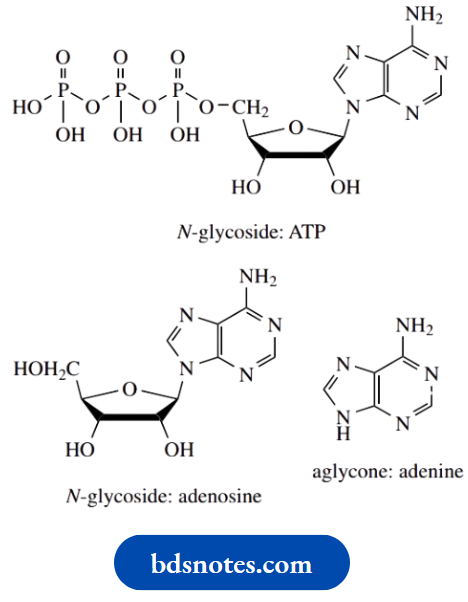
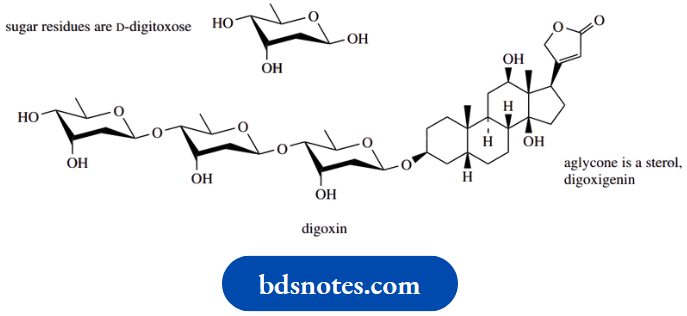
Perhaps the most significant group of glycoside derivatives are polysaccharides. In these structures, the aglyconeis itself another sugar, so that the polymer chain is composed of a series of sugar units joined by acetal or ketal linkages. Short carbohydrate polymers may also be found in some of the more complex O-glycosides,
Example: The heart drug digoxin from Digitalis lanata
Making a methyl glucopyranoside is relatively straightforward in that we can use the alcohol methanol as solvent, and, since it is thus present in large excess, this helps to disturb the equilibrium. The process is much less attractive for a more complex alcohol that is probably not available in excess and is unlikely to function as a suitable solvent.
Trying to join together two or more sugars would also be fraught with problems since each sugar contains several hydroxyl groups capable of acting as the nucleophile. These problems have been overcome by exploiting nucleophilic substitution for glycoside synthesis rather than the hemiacetal to acetal conversion we have been looking at, combined with the use of protecting groups to avoid unwanted couplings. A valuable reagent for adding a glucose unit onto a suitable nucleophile is acetobromoglucose.
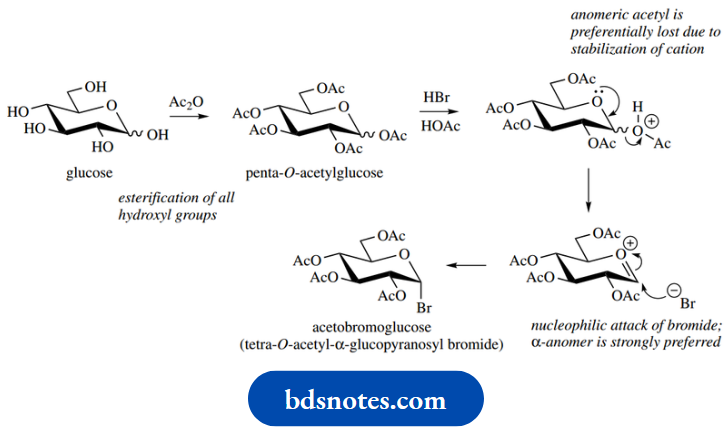
Glucose is first esterified to penta- O-acetylglucose using acetic anhydride. Note that the hemiacetal hydroxyl is also esterified, and thus any equilibration with an aldehyde form is now not possible. When this penta-acetate is treated with HBr, the anomeric acetate is preferentially lost under the acidic conditions, due to the stabilization conferred by the heterocyclic oxygen.
Note that this is the same type of intermediate we implicated in the conversion of hemiacetals into acetals. Acetobromoglucose then results from the nucleophilic attack of bromide onto the cationic system; in acetal formation, the nucleophile would be an alcohol. The anomeric effect is considerably larger when the substituent is halide than it is with alkoxy groups, so the product formed is almost exclusively the α-anomer.
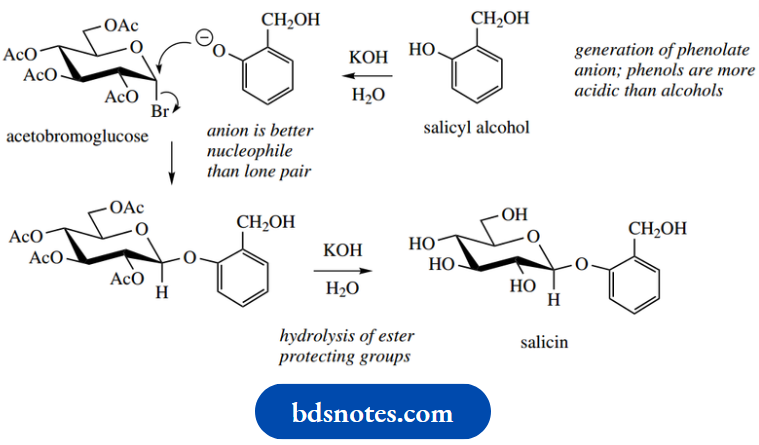
In the conversion of hemiacetals into acetals. Acetobromoglucose then results from the nucleophilic attack of bromide onto the cationic system; in acetal formation, the nucleophile would be an alcohol. The anomeric effect is considerably larger when the substituent is halide than it is with alkoxy groups, so the product formed is almost exclusively the α-anomer. The product is consequently the esterified β-glucoside derivative.
Further base treatment then hydrolyses the ester functions, liberating the glucoside salicin. As we shall, this type of substitution process is similar to the way glucosides (and polysaccharides) are produced in nature, though the enzymic reactions do not require any ester-protecting groups for the sugars.
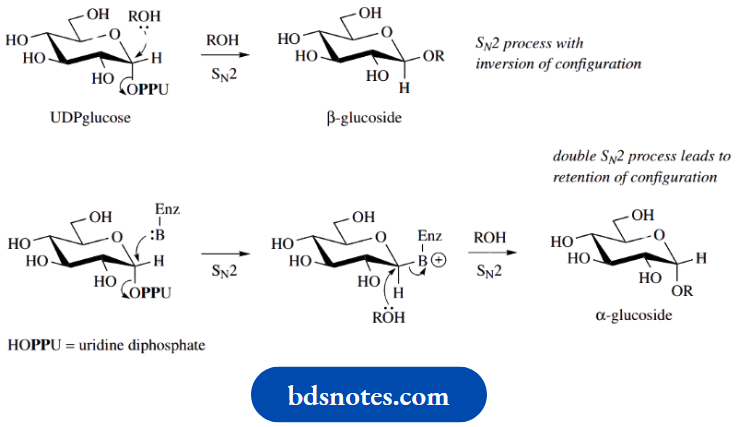
Biosynthesis of glycosides via U DPsugars The widespread occurrence of glycosides and polysaccharides in nature demonstrates there are processes for attaching sugar units to a suitable atom of an aglycone to give a glycoside, or to another sugar to give a polysaccharide. Linkages tend to be through oxygen, although they are not restricted to oxygen, since S-, N-, and C-glycosides are also well-known. The agent for glycosylation is a uridine diphosphosugar,
Example: UDP-glucose.
Of course, the uridine portion is itself a glycoside, an N-riboside of the pyrimidine base uracil. The glucosylation process can be envisaged as a simple SN2 nucleophilic displacement reaction, with an alcohol or phenol nucleophile, and a phosphate derivative as the leaving group. This SN2 displacement is analogous to that seen in the chemical synthesis of glycosides using acetobromoglucose

SN2 processes occur with inversion of configuration so since UDPglucose has its leaving group in the α-configuration, the product formed by the SN2 process has the β-configuration. This is the configuration most commonly found in natural O-glucosides. Some natural products do possess an α-linkage, however. It appears that such compounds originate via a double SN2 process, in which a nucleophilic group on the enzyme reacts first with the UDPglucose and then the hydroxy nucleophile displaces the enzymic group.
Other UDPsugars,
Example: UDP-galactose or UDPxylose, are utilized in the synthesis of glycosides containing different sugar units.
The S-, N-, and C-glycosides are formed by a similar process with the appropriate nucleophile. This type of reaction is also used in the biosynthesis of polysaccharides ), and in the metabolism of drugs and other foreign compounds via glucuronides.
Cyclic Acetals And Ketals Protecting Groups
We have just seen that intramolecular reactions between the carbonyl group and one or other of the hydroxyl functions readily lead to the formation of cyclic hemiacetal or hemiketal forms. Further, these products may then be converted into acetals or ketals by an intermolecular reaction with another alcohol molecule, giving us glycosides.
We could also form an acetal or ketal by supplying a carbonyl compound and exploiting the hydroxyl groups of the sugar. This provides a particularly useful means of protecting some of the hydroxyl groups whilst other reactions are carried out; the protecting group is then easily removed by effectively reversing the acetal/ketal reaction using hydrolytic conditions.
In principle, several different types of acetal or ketal might be produced. In this section, we want to exemplify a small number of useful reactions in which two of the hydroxyl groups on the sugar are bound up by forming a cyclic acetal or ketal with a suitable aldehyde or ketone reagent.
Aldehydes or ketones react with 1,2- or 1,3-diols under acidic conditions to form cyclic acetals or ketals. If the diol is itself cyclic, then the two hydroxyl groups need to be cis-oriented to allow the thermodynamically favourable fused-ring system to form.
Thus, cis-cyclohexanes-1,2- diol reacts with acetone to form a cyclic ketal, a 1,2- O-isopropylidene derivative usually termed, for convenience, an acetonide
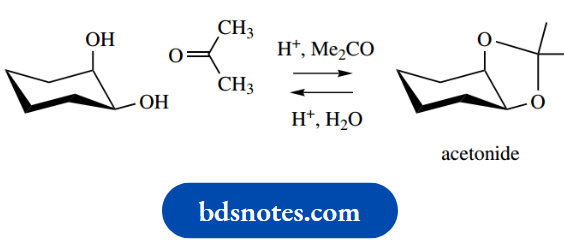
When required, the original diol may be regenerated by acid hydrolysis. Sugars are polyhydroxy compounds, and it is not always easy to predict which of the hydroxyls will react in this way. There are other complicating factors too.
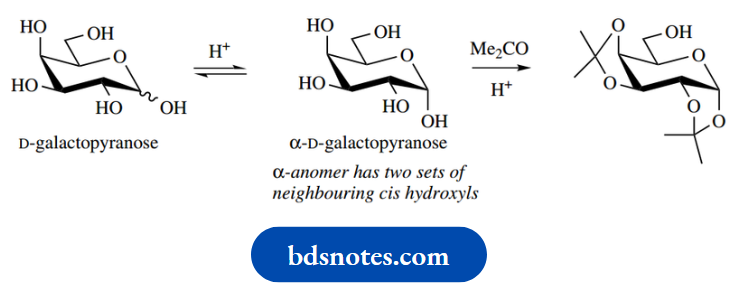
The ring size (pyranose/furanose) of the product may differ from that of the starting sugar. It may be that a more stable pyranose form does not have disoriented hydroxyl groups, whereas a less favoured furanose form does so that the latter can form cyclic acetals/ketals. The equilibration of pyranose/furanose forms allows this type of change to occur.
Thus, D-galactose reacts with acetone to give a diketal: the less-favoured α-form has two pairs of cis-oriented hydroxyls that can react. It thus yields a diacetonide. Only the primary alcohol group is left unprotected and is available for further modification if desired.
D-glucose provides a rather more complicated picture, unfortunately. Whilst the pyranose α-anomer could yield a mono-acetonide, there is no other pair of cis-hydroxyls that can react. However, it turns out that the furanose form has two sets of hydroxyls that can react; the product obtained is an acetonide of α-D-glucofuranose.
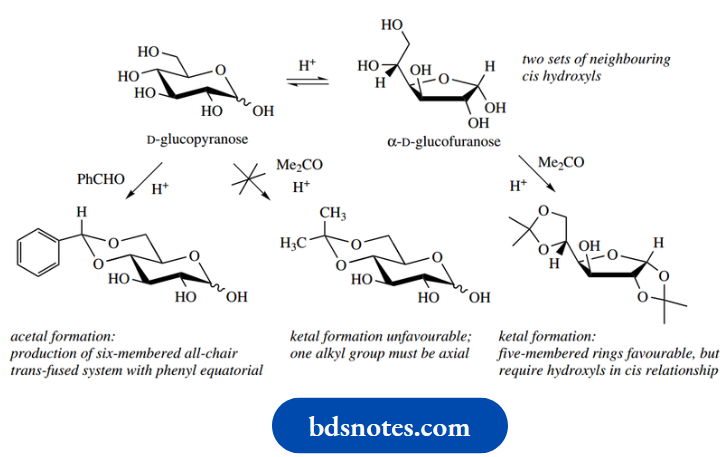
Note that a six-membered ketal ring involving the hydroxyls at 4 and 6 is not favoured; this is because such a ring would necessarily force one of the two methyls into an axial position. On the other hand, these two hydroxyls can be employed in forming a cyclic acetal with benzaldehyde. Benzaldehyde shows a tendency to form six-membered ring acetals, and because the two substituents are phenyl and hydrogen, we can have a favourable chair system with the phenyl equatorial.
It is not the intention to explain all such variations and add to potential confusion. The behaviour of most sugars for cyclic acetal and ketal formation is well documented for those wishing to work with these compounds. The objective here is merely to illustrate the potential for selective protection of the hydroxyl groups.
Oligosaccharides
The term oligosaccharide is frequently used to classify a small polysaccharide comprised of some two to five monomer units, a name derived from the Greek oligos, meaning a few. A pre-eminent example is the disaccharide sucrose, which we commonly call ‘sugar’ and is utilised widely as a sweetening agent and as the raw material for sweets and other confectionary. Other important disaccharides are maltose, a hydrolysis product from starch, and lactose, the main sugar component of cow’s milk.
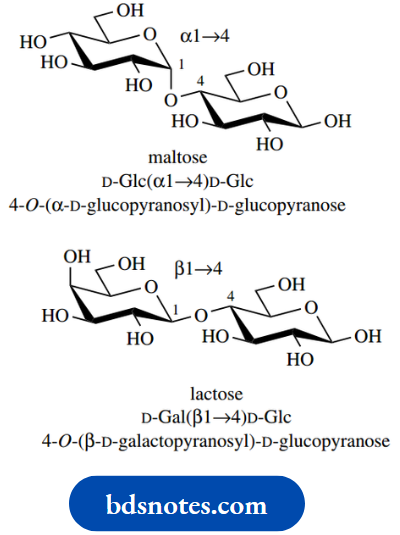
If we inspect these structures, we can see that they are acetals or ketals equivalent to the glycosides described above, though the alcohol portion is one of the hydroxyl groups of a second monosaccharide structure.
The linkages are conveniently defined by a shorthand system of nomenclature; this indicates the carbons that are joined by the acetal/ketal bond through the use of numbers and an arrow, together with the configuration α or β at the anomeric carbon. Note that each monosaccharide is numbered separately and there is no unique numbering system for the combined structure.
Thus, maltose becomes D-Glc( α1 → 4)D-Glc, which conveys the information that two molecules of D-glucose are bonded between carbon-1 of one molecule and carbon-4 of the second and that the configuration at the anomeric centre (C-1 of the first glucose residue) is α.
Similarly, lactose, a combination of D-galactose and D-glucose, is D-Gal( β1 → 4)D-Glc, the configuration at the anomeric centre of galactose being β. Note that the configuration at the hemiacetal anomeric centre in the second sugar (glucose) is not indicated; it could be α or β, as with a monosaccharide Longhand systematic nomenclature that treats one sugar as a substituent on the other can also be used.
In the systematic names, the ring size (pyranose or furanose) is also indicated. Thus maltose is 4- O-( α-D-glucopyranosyl)-D-glucopyranose, and lactose becomes 4- O-( β-D-galactopyranosyl)-Dglucopyranose.
Lactulose
Lactulose is a semi-synthetic disaccharide prepared from lactose and is composed of galactose linked β1 → 4 to fructose. Galactose is an aldose and exists as a six-membered pyranose ring, whereas fructose is a ketose and forms a five-membered furanose ring.
Systematically, lactulose is called 4- O-( β- D-galactopyranosyl)-D-fructofuranose; again, the configuration at the anomeric centre of fructose is unspecified. In abbreviated form, this becomes DGal( β1 → 4)D-Fru. Lactulose is widely employed as a laxative. It is not absorbed from the gastrointestinal tract, is predominantly excreted unchanged, and helps to retain fluid in the bowel by osmosis.
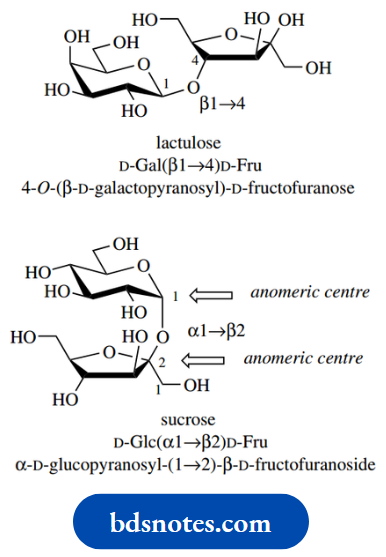
Sucrose
Sucrose is composed of glucose and fructose, and again we have a six-membered pyranose ring coupled to a five-membered furanose ring. However, there is a significant difference when we compare its structure with that of lactulose: in sucrose, the two sugars are both linked through their anomeric centres. In the shorthand representation, we thus have to indicate the configuration at each anomeric centre, so the linkage becomes α1 → β2. Sucrose is thus abbreviated to D-Glc(α1 → β2)D-Fru. The systematic nomenclature for sucrose is α-D-glucopyranosyl- (1 → 2)-β-D-fructofuranoside, which also includes the arrow to avoid confusion.
Since the two sugars in sucrose are both linked through their anomeric centres, this means that both the hemiacetal/hemiketal structures are prevented from opening; and, in contrast to maltose, lactose, and lactulose, there can be no open-chain form in equilibrium with the cyclic form. Therefore, sucrose does not display any of the properties usually associated with the masked carbonyl group. In nature, the formation of oligosaccharides, and also of polysaccharides, is dependent upon the generation of an activated sugar bound to a nucleoside diphosphate, typically a UDPsugar.
As outlined above ), nucleophilic displacement of the UDP leaving group by a suitable nucleophile generates the new sugar derivative. This will be a glycoside if the nucleophile is a suitable aglycone molecule, or an oligosaccharide if the nucleophile is another sugar molecule.
This reaction, mechanistically of SN2 type, should give an inversion of configuration at C-1 in the electrophile, generating a product with the β-configuration in the case of UDP-glucose, as shown. Many of the linkages formed between glucose monomers have the α-configuration, and it is believed that a double SN2 mechanism operates, which initially involves a nucleophilic group on the enzyme.
Polysaccharides
Structural Aspects
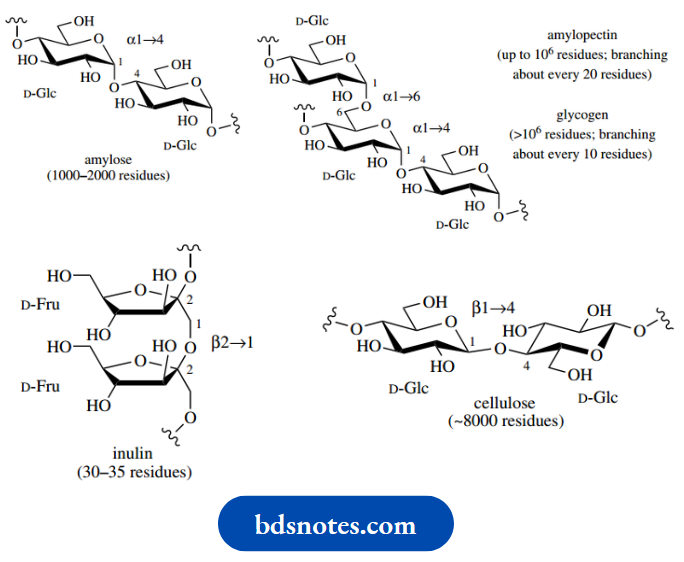
Polysaccharides fulfil two main functions in living organisms, as food reserves and as structural elements. Plants accumulate starch as their main food reserve, a material that is composed entirely of glucopyranose units, but in two different types of polymer, namely amylose and amylopectin. Amylose is a linear polymer containing some 1000–2000 glucopyranose units linked α1 → 4.
Amylopectin is a much larger molecule than amylose (the number of glucose residues varies widely, but may be as high as 106), and it is a branched-chain molecule. In addition to α1 → 4 linkages, amylopectin has branches at about every 20 units through α1 → 6 linkages. These branches then also continue with α1 → 4 linkages but may have subsidiary α1 → 6 branching, giving a tree-like structure.
The mammalian carbohydrate storage molecule glycogen is analogous to amylopectin in structure but is larger and contains more frequent branching, about every 10 residues. The branching in amylopectin and glycogen is achieved by the enzymic removal of a portion of the α1 → 4-linked straight chain containing several glucose residues, then transferring this short chain to a suitable 6-hydroxyl group. A less common storage polysaccharide found in certain plants is inulin, which is a relatively small polymer of fructofuranose, linked through β2 → 1 bonds.
Cellulose is reputedly the most abundant organic material on Earth, being the main constituent in plant cell walls. It is composed of glucopyranose units linked β1 → 4 in a linear chain. Alternate residues are ‘rotated’ in the structure, allowing hydrogen bonding between adjacent molecules, and construction of the strong fibres characteristic of cellulose, as in cotton.
Hydrolysis of polysaccharides
Hydrolysis of polysaccharides (and oligosaccharides) follows the comments under glycosides above. Thus, treatment of amylose, amylopectin, or cellulose with hot aqueous acid will result in the formation of glucose as the sole product, through hydrolysis of acetal linkages. Under milder, less forcing conditions, it is possible to isolate short-chain oligosaccharides as a result of random hydrolysis of linkages.
- More specific hydrolysis may be achieved by the use of enzymes. Thus, the enzyme α-amylase in saliva and the gut can catalyse hydrolysis of α1 → 4 bonds throughout the starch molecule to give mainly maltose, with some glucose and maltotriose, the trisaccharide of glucose.
- Amylose is hydrolysed completely by this enzyme, but the α1 → 6 bonds of amylopectin are not affected. Another digestive enzyme, α-1,6-glucosidase, is required for this reaction. Finally, pancreatic maltase completes the hydrolysis by hydrolysing maltose and maltotriose.
- The milk of mammals contains the disaccharide lactose as the predominant carbohydrate, to the extent of about 4–8%. Lactose, therefore, provides the basic carbohydrate nutrition for infants, who metabolize it via the hydrolytic enzyme lactase.
Lactase enzyme activity in adult humans is usually considerably lower than in infants. Lactose intolerance is a condition in certain adults who are unable to tolerate milk products in their diet.
- This is a consequence of very low lactase levels, such that ingestion of lactose can lead to adverse reactions, typically gastric upsets. Cellulose differs from amylose principally in the stereochemistry of the acetal linkages, which are α in amylose but β in cellulose.
- α-Amylase is specific for α1 → 4 bonds and is not able to hydrolyse β1 → 4 bonds. An alternative enzyme, termed cellulase, is required. Animals do not possess cellulase enzymes, and thus cannot digest wood and vegetable fibres that are predominantly composed of cellulose.
- Ruminants, such as cattle, are equipped to carry out cellulose hydrolysis, though this is dependent upon cellulase-producing bacteria in their digestive tracts.
Oxidation Of Sugars: Uronic Ccids
Sugars may be oxidized by a variety of reagents, and the most susceptible group in aldoses is the aldehyde. The use of aqueous bromine as a mild oxidizing agent achieves oxidation of the aldehyde group in D-glucose, and the product is the corresponding carboxylic acid D-gluconic acid.
The general term used for such a polyhydroxy carboxylic acid is an aldonic acid. These are named by substituting -one acid for -ose of the sugar. Polyhydroxy carboxylic acids have the potential to form lactones (cyclic esters,).
And D-gluconic acid readily forms a 1,4-lactone in solution. In principle, both five- and six-membered rings might be produced, but the five-membered system is favoured.
- More vigorous oxidation results in the oxidation of one or more hydroxy groups, with the primary alcohol group being attacked most readily.
- Thus, oxidizing either D-glucose or D-gluconic acid with aqueous nitric acid leads to a dicarboxylic acid, D-glucaric acid.
- Dicarboxylic acids of this type are termed aldaric acids. Again, aldaric acids readily form five-membered lactones, which may be the 1,4- or 3,6- lactones, or the dilactone.
Determination of blood glucose levels
The peptide hormone insulin is produced by the pancreas and plays a key role in the regulation of carbohydrate, fat, and protein metabolism. In particular, it has a hypoglycaemic effect, lowering the levels of glucose in the blood.
A malfunctioning pancreas may produce a deficiency in insulin synthesis or secretion, leading to the condition known as diabetes mellitus.
- This results in increased amounts of glucose in the blood and urine, diuresis, depletion of carbohydrate stores, and subsequent breakdown of fat and protein. Incomplete breakdown of fat leads to the accumulation of ketones in the blood, severe acidosis, coma, and death.
- Where the pancreas is still functioning, albeit less efficiently, the condition is known as type 2 diabetes (non-insulin-dependent diabetes, NIDDM)and can be managed satisfactorily by a controlled diet or oral antidiabetic drugs.
- In type 1 diabetes (insulin-dependent diabetes, IDDM), pancreatic cells no longer function, and injections of insulin are necessary, one to four times daily, depending on the severity of the condition.
- These treatments need to be combined with a controlled diet and regular monitoring of glucose levels but do not cure the disease, so treatment is lifelong. Quick and easy methods have been developed so that patients can monitor their blood glucose levels regularly. One such method depends upon the oxidation of glucose to gluconic acid in a reaction catalysed by the enzyme glucose oxidase.
This enzyme can be obtained from several microorganisms,
Example: Aspergillus and Penicillium species, and for convenience are usually immobilized onto a suitable support. The microbial enzyme converts glucose into gluconic acid utilizing molecular oxygen as an oxidant, but detection of the process is dependent upon the simultaneous production of hydrogen peroxide.

Hydrogen peroxide may be detected by exploiting a secondary chemical reaction that produces a coloured product; this is compared with a standard colour chart to indicate the colour intensity and, therefore, give a measure of the glucose concentration.
Alternatively, it may be scanned in a colourimeter to give a more accurate assay. Even more accuracy can be obtained by using a voltammetric sensor, in which the hydrogen peroxide is oxidized to oxygen on an electrode surface, thus generating an electrical current that is directly proportional to the glucose concentration
⇒ \(\mathrm{H}_2 \mathrm{O}_2+2 \mathrm{HO}^{-} \rightleftharpoons \mathrm{O}_2+\mathrm{H}_2 \mathrm{O}+2 \mathrm{e}^{-}\)
The method is highly specific for glucose. Related sugars, such as mannose, xylose and galactose, are not oxidized by this enzyme or react only in trace amounts.
Uronic acids:
Uronic acids are produced from aldoses when just the terminal –CH2OH group has been oxidized to a carboxylic acid. They are named after the parent sugar, substituting -uronic acid for -use; thus, D-glucuronic acid is the 6-carboxylic acid analogue of D-glucose. It should be apparent from the preceding comments that it will not be possible to oxidize the primary alcohol function selectively in the presence of the more reactive aldehyde group, so it becomes necessary to protect the aldehyde by an appropriate means.
It may also be desirable to protect other hydroxyls.
For example:
The formation of the acetonide of galactose protects the aldehyde and all hydroxyls except that at position 6 . It now remains to oxidize the primary alcohol and remove the protecting groups.
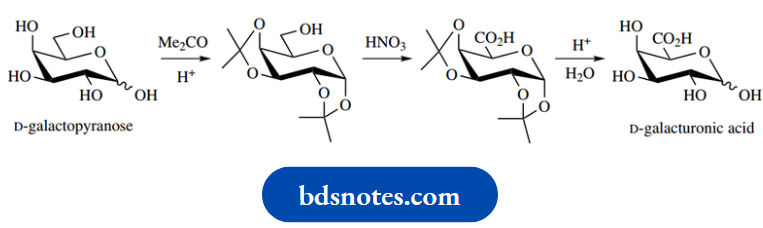
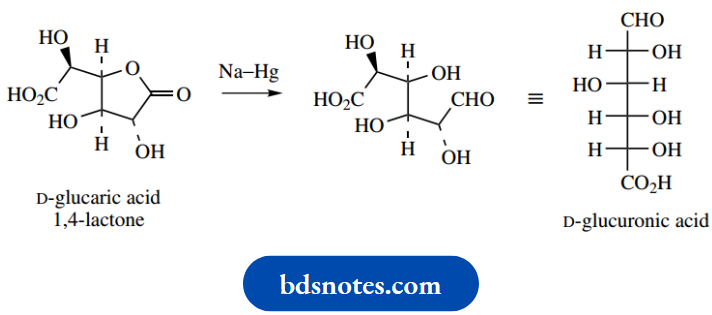
An alternative approach is to oxidize both the carbonyl and primary alcohol functions to carboxylic acids, then selectively reduce that corresponding to the required aldehyde. This may be achieved by reducing the 1,4-lactone of D-glucaric acid, using the same reaction as where it was employed in the synthesis of labelled glucose.
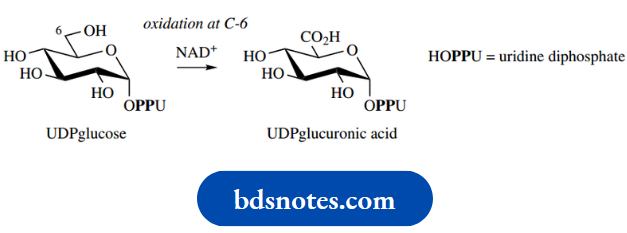
Uronic acids are found in nature, but they are formed enzymatically by selective oxidation of the primary alcohol function of a sugar. Oxidation takes place not on the free sugar, but on UDPsuga derivatives, as utilized in glycoside biosynthesis. UDP-glucuronic acid is an important carrier in the metabolism of drug molecules.
Some examples of natural uronic a cid d derivatives
Polymers of uronic acids are encountered in nature in structures known as pectins, which are essentially chains of D-galacturonic acid residues linked α1 → 4, though some of the carboxyl groups are present as methyl esters. These materials are present in the cell walls of the fruit, and the property that aqueous solutions under acid conditions form gels is the basis of jam-making.
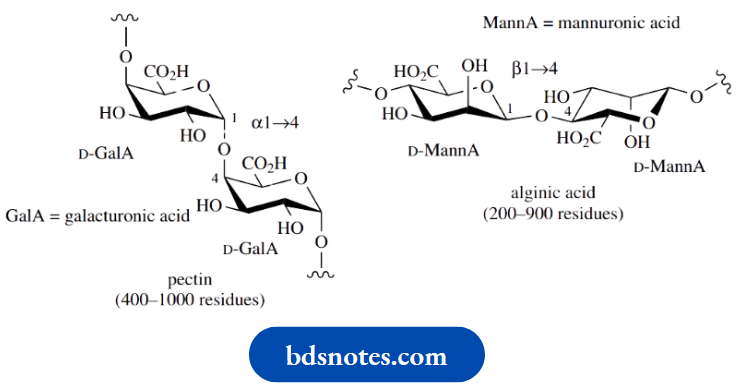
Alginic acid is a polymer of D-mannuronic acid residues joined by β1 → 4 linkages. It is the main cell wall constituent of brown algae (seaweed). Salts of alginic acid are valuable thickening agents in the food industry, and the insoluble calcium salt is the basis of absorbable alginate surgical dressings.
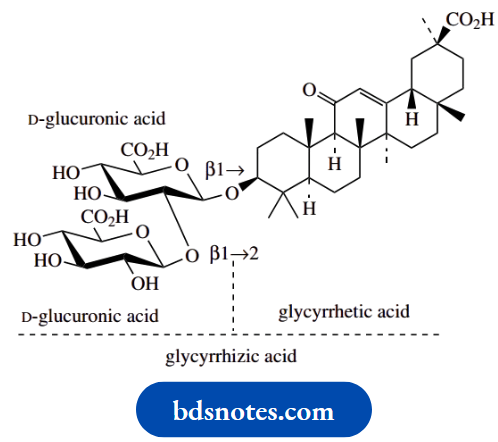
The intensely sweet constituent in the root of liquorice (Glycyrrhiza glabra) is glycyrrhizin, a mixture of potassium and calcium salts of glycyrrhizic acid. It is said to be 50–150 times as sweet as sucrose. Glycyrrhizic acid is a glycoside of the triterpene aglycone glycyrrhizic acid. The sugar portion is a disaccharide comprised of two molecules of D-glucuronic acid, so is termed a diglucuronide. Liquorice is used in confectionery and as a flavouring agent for beers and stouts. It also finds considerable use in drug formulations to mask the taste of bitter drugs and for its emulsifying surfactant properties.
Glucuronides in drug metabolism
One of the principal ways by which foreign compounds are removed from the body is to conjugate them into glucuronic acid. This conjugation process not only binds the unwanted compound but also converts it into a highly polar material that is water soluble and can be excreted in aqueous solution, typically via the kidneys. The polarity is provided both by the hydroxyl groups and by the ionizable carboxylic acid group. Typical chemicals that may become conjugated with glucuronic acid include alcohols, phenols, carboxylic acids, amines, and thiols.
Drugs must also be considered as foreign compounds, and an essential part of drug treatment is to understand how they are removed from the body after their work is completed. Glucuronide formation is the most important of so-called phase II metabolism reactions. Aspirin, paracetamol, morphine, and chloramphenicol are examples of drugs excreted as glucuronides.
Glucuronides
Glucuronides are formed in mammals by reaction with uridine diphosphoglucuronic acid (UDPglucuronic acid; UDP-GA) in processes catalysed by uridine diphosphoglucuronyltransferase enzymes. This reaction is entirely analogous to the enzymic glycosylation process we looked at above. The reaction with UDP-GA can be envisaged as a simple SN2 nucleophilic displacement reaction, with an appropriate nucleophile,
Example: An alcohol or amine, and a phosphate derivative as the leaving group.
UDP glucuronyltransferase enzymes have very broad substrate specificity and can catalyse reactions with a wide variety of foreign molecules and drugs.
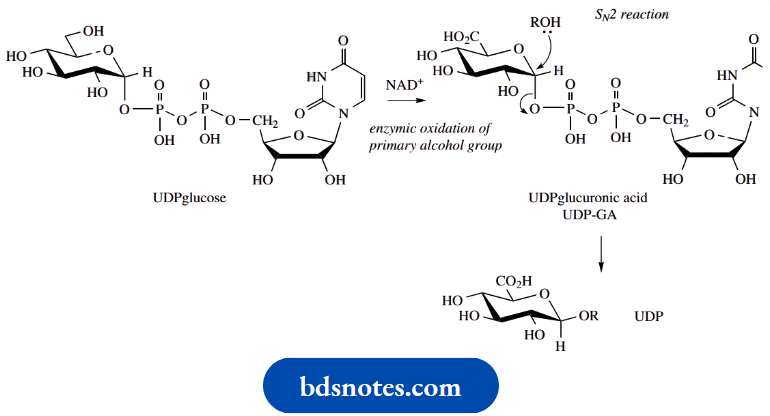
UDP-GA is formed from UDP-glucose by enzymic oxidation of the primary alcohol group. We have already noted that UDP-glucose is also the biochemical precursor of glucose-containing polysaccharides,
Example: Starch and glycogen
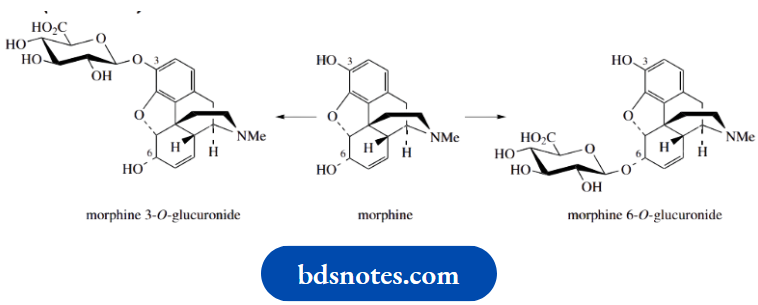
The opium alkaloid morphine is one of the most valuable analgesics for the relief of severe pain. It is known to be metabolized in the body to O-glucuronides, by reaction at the phenolic and alcoholic hydroxyls.
The glucuronides formed are water soluble and readily excreted. An interesting feature is that the two monoglucuronides have significantly different pharmacological activities. Although morphine 3-O-glucuronide is antagonistic to the analgesic effects of morphine, morphine 6-O-glucuronide is a more effective and longer-lasting analgesic than morphine itself, and with fewer side effects.
Vitamin C
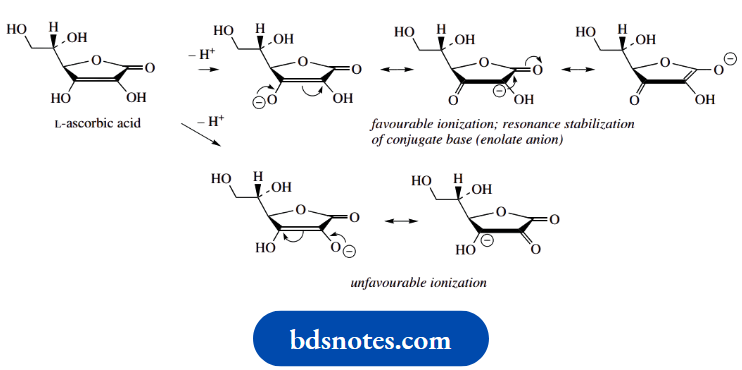
Vitamin C, also known as L-ascorbic acid, clearly appears to be of a carbohydrate nature. Its most obvious functional group is the lactone ring system, and, although termed ascorbic acid, it is certainly not a carboxylic acid. Nevertheless, it shows acidic properties, since it is an enol, in fact, an enediol. It is easy to predict which enol hydroxyl group is going to ionize more readily.
It must be the one β to the carbonyl, the ionization of which produces a conjugate base that is nicely resonance stabilized. Indeed, note that these resonance forms correspond to those of an enolate anion derived from a 1,3-dicarbonyl compound. Ionization of the α-hydroxyl provides less favourable resonance, and the remaining hydroxyls are typical non-acidic alcohols. Thus, the pKa of vitamin C is 4.0 and is comparable to that of carboxylic acid.
Vitamin C is essential for the formation of collagen, the principal structural protein in skin, bone, tendons, and ligaments, being a cofactor in the hydroxylation of the amino acids proline to 4-hydroxyproline, and of lysine to 5-hydroxylysine. These hydroxy amino acids account for up to 25% of the collagen structure. Vitamin C is also associated with some other hydroxylation reactions,
Example:
The hydroxylation of tyrosine to dopa (dihydroxyphenylalanine) in the pathway to catecholamine Deficiency leads to scurvy, a condition characterized by muscular pain, skin lesions, fragile blood vessels, bleeding gums, and tooth loss. Vitamin C also has valuable antioxidant properties, and these are exploited commercially in the food industries.
Most animals can synthesize vitamin C, though humans and primates cannot and must obtain it via their diet. Citrus fruits, peppers, guavas, rose hips, and blackcurrants are especially rich sources, but it is present in most fresh fruits and vegetables.
In animals, ascorbic acid is synthesized in the liver from D-glucose, by a pathway that initially involves specific enzymic oxidation of the primary alcohol function, giving D-glucuronic acid. This is followed by a reduction to L-gluonic acid, which is effectively a reduction of the carbonyl function in the ring-opened hemiacetal.
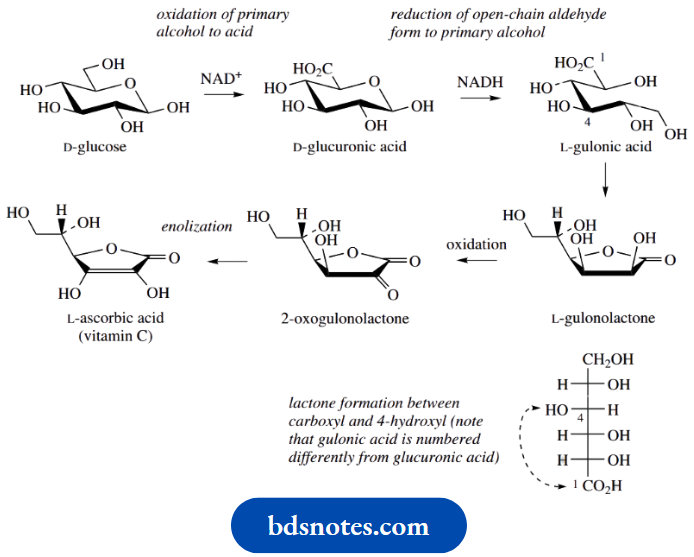
Lactone formation in gluonic acid leads to the favourable five-membered system, and then oxidation of the secondary alcohol to a carbonyl effectively gives ascorbic acid. However, the more favourable structure of ascorbic acid is the enol tautomer with the conjugated α, β-unsaturated lactone.
Ascorbic acid formation in plants follows an analogous pathway, starting from either D-glucose or D-galactose. Man and other primates appear deficient in the enzyme that oxidizes gluconolactone to keto-lactone, and we are thus dependent on a dietary source of vitamin C.
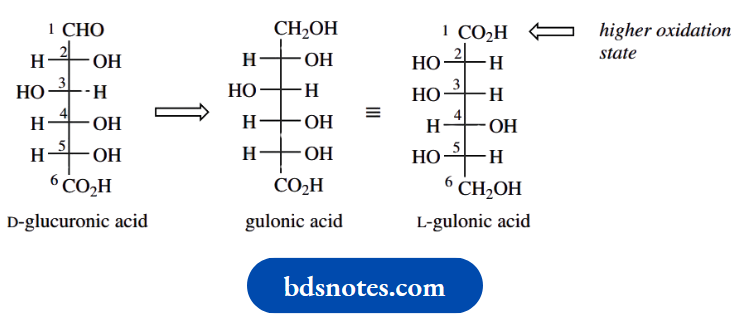
An unfortunate twist is the apparent configurational change from D to L in going from glucuronic to gluconic acid. This is a consequence of renumbering. In gluonic acid, the carboxylic acid group has the higher oxidation state, becomes the topmost substituent in the Fischer projection, and is numbered carbon-1.
As a result, the D descriptors for glucuronic acid and the L descriptors for gluconic acid now refer to two different chiral centres. You can see why we were rather unenthusiastic about the value of D and L. We are also uncomfortable that the –CH2OH → –CO2H change inferred in the glucose → glucuronic acid by convention maintains the same configuration in the two compounds. From the gluonic acid example, we might reasonably expect to apply the same type of renumbering in glucuronic acid.
Aminosugars
Aminosugars are the result of the replacement of one or more hydroxyl groups in sugar by amino groups. They are formed in nature by transamination processes on appropriate keto sugars, which are themselves the product of regiospecific enzymic oxidation processes.
Thus, D-glucosamine (2-amino-2-deoxy-D-glucose) is readily appreciated as a metabolic product from D-glucose. Note here the convenient way we can name an aminosugar by relating it to a normal sugar via the removal of a hydroxyl (2-deoxy) and then the addition of an amino (2-amino)
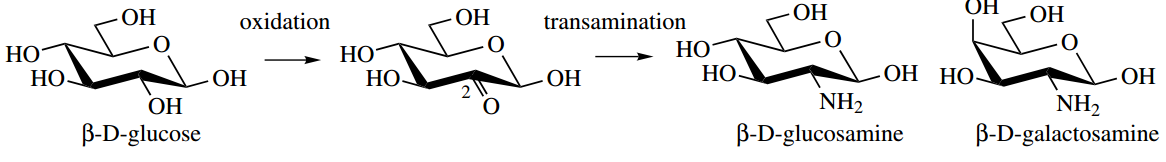
D-glucosamine and D-galactosamine, usually as N-acetyl derivatives, are part of the structures of several natural polysaccharides, whilst other uncommon aminosugars are components of the aminoglycoside antibiotics. We have also noted the occurrence of N-glycosides, where the nitrogen substitution is at the anomeric centre.
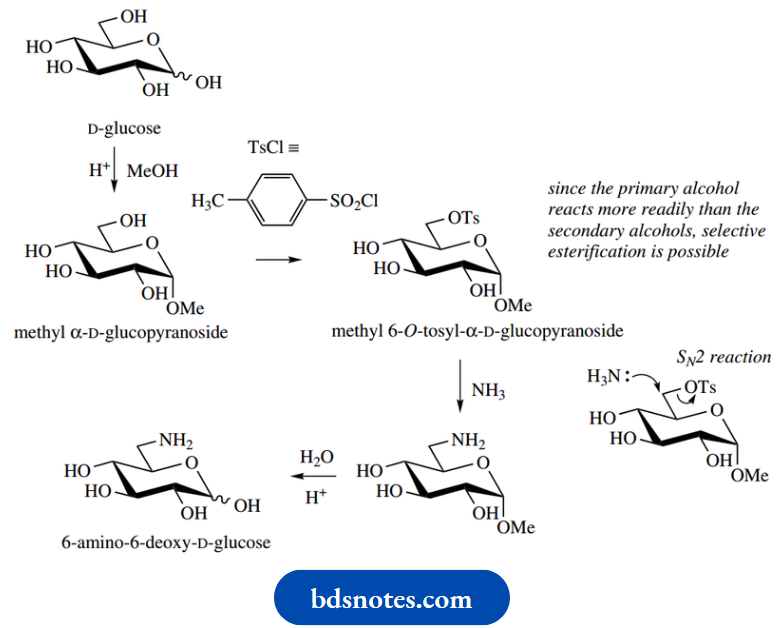
A simple chemical approach to aminosugars is to use SN2 displacement by ammonia of a suitable leaving group, such as a tosylate (toluene p-sulfonate, see
This process can be made selective for position 6 since the less-hindered primary alcohol group is more readily esterified than the secondary alcohol.
2-Aminosugars such as glucosamine may be synthesized by a modified Kiliani–Fischer process. The starting aldose, here D-arabinose, is treated with ammonia, producing an imine, and then with HCN to yield epimeric 2-aminonitriles.
The remaining steps lead to a mixture of D-glucosamine and D-mannosamine, which will need to be separated. Protecting groups such as cyclic acetals and ketals may also be employed to achieve selective reaction.
Aminosugars and aminoglycoside antibiotics
The aminoglycosides form an important group of antibiotic agents and are immediately recognizable as modified carbohydrate molecules. Typically, they have two or three uncommon sugars attached through glycoside linkages to an aminocyclitol, i.e. an amino-substituted hydroxycyclohexane system.
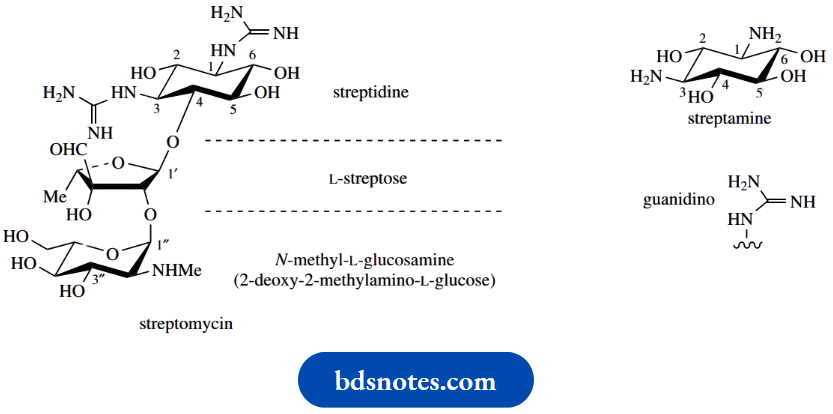
The first of these agents to be discovered was streptomycin from Streptomyces griseus. Its structure contains the aminocyclitol streptamine, though both amino groups are bound as guanidino substituents in the derivative streptidine.
Other medicinally useful aminoglycoside antibiotics are based on the aminocyclitol 2-deoxystreptamine,
Example:
Gentamicin C1 from Micromonospora purpurea. Although streaming and 2-deoxystreptamine are cyclohexane derivatives, they are both of carbohydrate origin and derived naturally from glucose
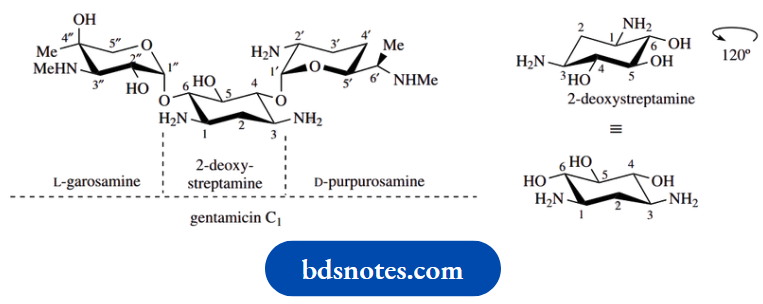
The other parts of streptomycin, namely L-streptose and the aminosugar 2-deoxy-2-methylaminoL-glucose ( N-methyl-L-glucosamine), are also ultimately derived from D-glucose. Gentamicin C1 contains two aminosugars, L-garosamine and D-purpurosamine.
The aminoglycoside antibiotics have a wide spectrum of activity, including activity against some Gram-positive and many Gram-negative bacteria. However, their widespread use is limited by nephrotoxicity, which results in impaired kidney function, and by ototoxicity, which is a serious side-effect that can lead to irreversible loss of hearing.
These antibiotics are thus reserved for the treatment of serious infections where less-toxic drugs have proved ineffective. The aminoglycoside antibiotics interfere with protein biosynthesis by acting on the smaller 30S subunit of the bacterial ribosome.
Aminosugars are also components of many macrolide antibiotics. These are macrocyclic lactones with a ring size typically of 12–16 atoms. Two or more sugar units are attached through glycoside linkages, these sugars tending to be unusual 6-deoxy structures often not found outside of this class of compounds,
Example: L-cladinose.
At least one sugar is an amino sugar, e.g. D-desosamine. These antibiotics have a narrow spectrum of antibacterial activity, principally against Gram-positive microorganisms. Their antibacterial spectrum resembles but is not identical to, that of the penicillins, so they provide a valuable alternative for patients allergic to the penicillins.
Erythromycin produced by cultures of Saccharopolyspora erythraea is the principal macrolide antibacterial currently used in medicine. It exerts its antibacterial action by inhibiting protein biosynthesis, binding to the larger 50S subunit of bacterial ribosomes and blocking the translocation step.
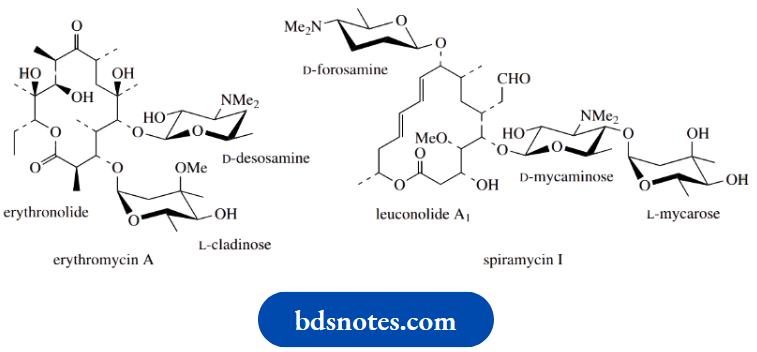
Spiramycin is another macrolide, recently introduced into medicine for the treatment of toxoplasmosis, infections caused by the protozoan Toxoplasma gondii. This contains a 16-membered lactone ring (erythromycin has a 14-membered ring), and two aminosugars, D-mycaminose and D-forosamine. D-Forosamine is remarkable in having only one hydroxyl group, and that is bound up in the hemiacetal ring system.
Polymers Containing Aminosugars
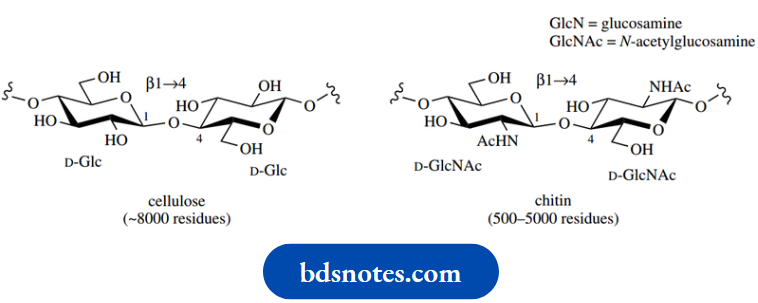
The structure of chitin is rather similar to that of cellulose, though it is composed of β1 → 4- linked N-acetylglucosamine residues. Chitin is a major constituent in insect skeletons and the shells of crustaceans,
Example: Crabs and lobsters; as with cellulose, its strength again depends on hydrogen bonding between adjacent molecules, producing rigid sheets. Chemical deacetylation of chitin provides chitosan, a valuable industrial material used for water purification because of its chelating properties, and in wound-healing preparations
Bacterial cell walls contain peptidoglycan structures in which the carbohydrate chains are composed of alternating β1 → 4-linked N-acetylglucosamine and O-lactyl-N-acetylglucosamine (also called N-acetylmuramic acid) residues. These chains are cross-linked via peptide structures. Part of the peptidoglycan of Staphylococcus aureus is shown here
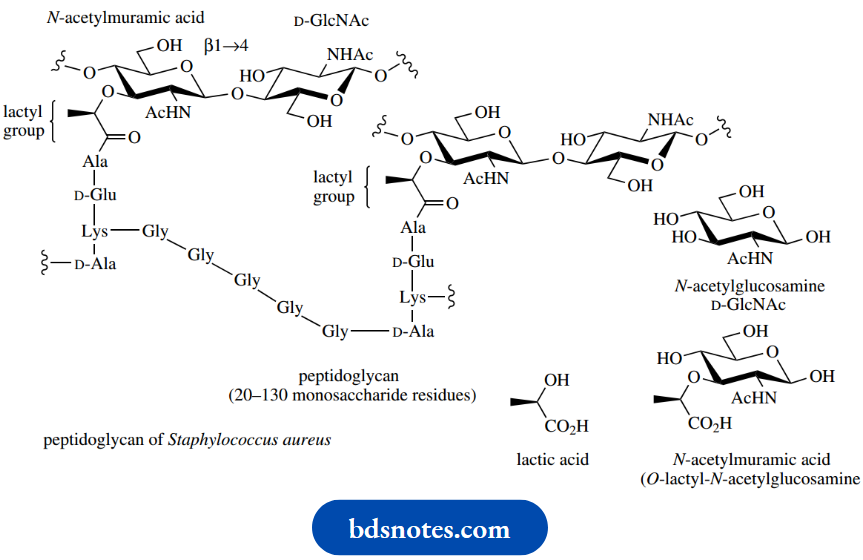
This shows the involvement of the lactyl group of the N-acetylmuramic acid in linking the peptide with the carbohydrate via an amide/peptide bond. The biological activities of the β-lactam antibiotics,
Example:
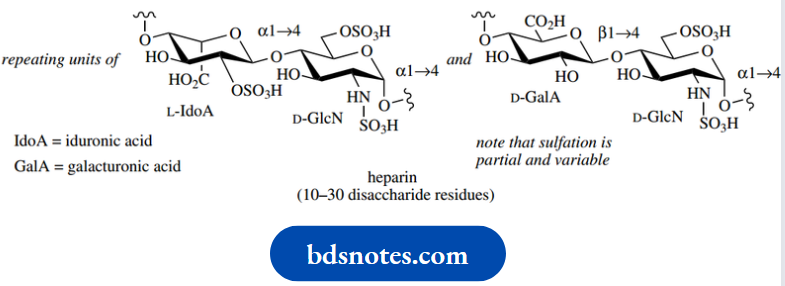
Penicillins and cephalosporins stem from an inhibition of the cross-linking mechanism during the biosynthesis of the bacterial cell wall. The mammalian blood anticoagulant heparin is also a carbohydrate polymer in which amino sugars (glucosamine) alternate with uronic acid residues.
Polymers of this kind are known as anionic mucopolysaccharides or glycosaminoglycans. Heparin consists of two repeating disaccharide units, in which the amino functions and some of the hydroxyls are sulfated, producing a heterogeneous polymer. The carboxyls and sulfates together make heparin a strongly acidic water-soluble material.
Carbohydrate determinants of blood groups
Most people are aware that blood is classified into several types, the blood groups. These are termed A, B, O, etc. It is essential in blood transfusions that the donor blood matches that of the recipient, otherwise, antibodies are produced in the new blood. This leads to aggregation of red blood cells, with potentially fatal results through blockage of blood vessels.
The blood group antigens are glycoproteins, carbohydrates having an attached protein chain, and the various blood groups can be correlated with a single monosaccharide residue in the carbohydrate portion. At the end of the carbohydrate section in type O blood antigens, there is a D-galactopyranose ring to which is attached an L-fucopyranose sugar through an α1 → 2 linkage.
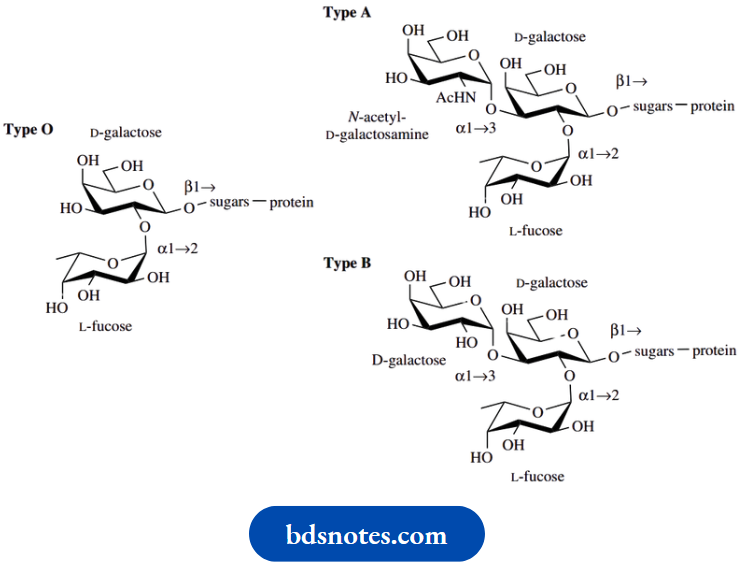
In type B blood antigens, the galactose residue has a second D-galactose residue attached, through an α1 → 3 linkage. In type A blood antigens, the second sugar residue is now N-acetyl-D-galactosamine, again attached through an α1 → 3 linkage. It has been found that enzymic removal of the terminal galactose residue from type B or of the N-acetyl-galactosamine residue from type A converts the B or A antigens into O antigens. It is also known that individuals with type B or type A antigens possess additional enzyme systems that specifically add the extra terminal carbohydrate unit to the type O antigen.
Leave a Reply