Blood Disorders Questions And Answers
Question 1. What is anemia? Classify and describe clinical manifestations, laboratory diagnosis, and treatment planning of iron deficiency anemia.
Or
Define and classify anemia. Discuss clinical features, laboratory investigations, oral manifestations, and management of iron deficiency anemia.
Answer. Anemia is defined as an abnormal reduction in several circulating red blood cells, the quantity of hemoglobin, and the volume of packed red cells in a given unit of blood.
“Understanding blood disorders through FAQs: Q&A explained”
Classification of Anemia
- Anemia due to increased blood loss:
- Acute posthemorrhagic anemia
- Chronic posthemorrhagic anemia
- Anemia due to impaired red cell function:
- Cytoplasmic maturation defect:
- Deficient heme synthesis, i.e., iron deficiency anemia.
- Defective globin synthesis, i.e., thalassemic syndrome
- Nuclear maturation defect, i.e., vitamin B12 or folic acid deficiency, e.g., megaloblastic anemia
- Defect in stem cell proliferation and differentiation
- Aplastic anemia
- Pure red cell aplasia
- Anemia of chronic disorders
- Bone marrow infiltration
- Congenital anemia
- Cytoplasmic maturation defect:
“Importance of studying blood disorders for better diagnostic outcomes: Questions explained”
- Hemolytic anemia
- Acquired or extracorpuscular
- Immunohemolytic anemia
- Autoimmune hemolytic anemia
- Warm antibody autoimmune hemolytic anemia
- Cold antibody autoimmune hemolytic anemia
- Drug-induced immunohemolytic anemia.
- Isoimmune hemolytic anemia.
- Autoimmune hemolytic anemia
- Mechanical trauma: Microangiopathic hemolytic anemia
- Direct toxic effect: Malaria, bacteria, infection, and other agents.
- Acquired red cell membrane abnormalities: Paroxysmal nocturnal hemoglobinuria.
- Splenomegaly
- Hereditary or intracorpuscular
“Steps to explain causes of blood disorders: Genetic vs acquired factors: Q&A guide”
- Abnormalities of red cell membrane:
- Hereditary spherocytosis.
- Hereditary elliptocytosis.
- Hereditary stomatocytosis.
- Disorders of red cell interior:
- Red cell enzyme defect:
- Defects in HMP shunt: G6PD deficiency.
- Defects in the glycolytic pathway: Pyruvate kinase deficiency.
- Disorders of hemoglobin:
- Structurally abnormal hemoglobin, sickle syndrome, and other hemoglobinopathies.
- Reduced globin chain synthesis: Thalassemias.
- Red cell enzyme defect:
“Common challenges in diagnosing blood disorders effectively: FAQs provided”
- Morphologic:
- Microcytic hypochromic
- Normocytic normochromic
- Macrocytic normochromic.
Read And Learn More: Oral Medicine Question And Answers
Classification of Iron Deficiency Anemia
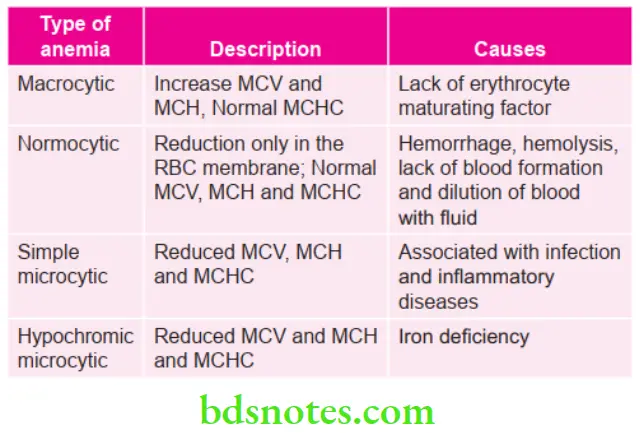
“Role of bone marrow dysfunction in causing blood disorders: Questions answered”
Anemia Clinical Manifestations
- It occurs in women in 4th to 5th decades of life.
- Patient experiences, tiredness, headache, paresthesia and lack of concentration.
- Koilonychia: Nails become flattened, brittle and spoon shaped.
- Neuropathy: There may be pin and needle sensation in extremities.
- Dysphagia: Some patients develop pharyngeal mucosal thickening.
- Gastrointestinal symptoms: Liver and spleen may be palpable. There may be gartrointestinal bleeding. Menorrhagia is present.
- Knuckle pigmentation can be appreciated.
“Early warning signs of issues addressed by understanding blood disorder pathogenesis: Common questions”
Anemia Oral Manifestations
- There is pallor of oral mucosa and gingiva.
- There is generalized atrophy of oral mucosa in tongue and buccal mucosa
- Tongue changes:
- There is redness, soreness or burning of tongue
- The filiform papilla over the anterior two-thirds of tongue is first to undergo atrophy
- In severe cases, fungiform papilla are affected leaving tongue smooth and waxy.
- Angular cheilitis: There is cracking and fissuring at the corners of mouth
- Softening of epithelium which leads to linear ulceration of skin, ascending upto and beyond the mucocutaneous junction. Bleeding from ulcerated tissues is appreciated
- Recurrent aphthous ulceration and candidal lesions can also occur in iron deficiency anemia
- Patient shows slow healing after oral surgical procedures.
“Asymptomatic vs symptomatic effects of ignoring blood disorder triggers: Q&A”
Anemia Laboratory Diagnosis
- Anemia is microcytic and hypochromic and the peripheral smear shows abnormal forms of RBCs.
- There is reduced hemoglobin level, as low as 4 g/100 mL.
- There is a normal or slightly reduced red cell count. MCV, MCH, MCHC all are reduced.
Understanding Blood Disorders: Q&A on Hemophilia, Thalassemia, and More
Anemia Treatment Planning/Management
- Iron supplement: Almost all patients are treated with oral supplements of iron by giving ferrous fumarate or ferrous sulfate. It is given in dose of 300 mg TDS or QDS for period of 6 months.
- The parental route of administration is suitable for few patients who are unable to take iron by mouth or who are unable to absorb iron by mouth.
- Recommended single dose of iron sorbitol is 1.5 mg/kg of body weight given daily.
- Parenteral iron supplements should be given at dose of 15 mg/kg body weight till maximum of 750 mg on two occasion atleast 7 days apart.
Leave a Reply