Atomic Structure And Bonding Atomic Structure
Atoms are composed of protons, neutrons, and electrons. Protons are positively charged, electrons carry a negative charge, and neutrons are uncharged.
In a neutral atom, the nucleus of protons and neutrons is surrounded by electrons, the number of which is equal to the number of protons.
This number is also the same as the atomic number of the atom if the number of electrons and protons is not equal.
The atom or molecule containing the atom will necessarily carry a charge, which is called an ion. A negatively charged atom or molecule is termed an anion, and a positively charged species is called a cation.
The inert or noble gases, such as helium, neon, and argon, are particularly unreactive, and this has been related to the characteristic number of electrons they contain, 2 for helium, 10 for neon (2 + 8), and 18 for argon (2 + 8 + 8).
They are described as possessing ‘filled shells’ of electrons, which, except for helium, contain eight electrons, an octet.
Acquiring a noble gas-like complement of electrons governs the bonding together of atoms to produce molecules.
This is achieved by losing electrons, gaining electrons, or by sharing electrons associated with the unfilled shell, and leads to what we term ionic bonds or covalent bonds.
The unfilled shell involved in bonding is termed the valence shell, and the electrons in it are termed valence electrons.
Bonding And Valency
For many years now, these types of bonding have been represented in chemistry via a shorthand notation.
Ionic bonds have been shown as a simple electrostatic interaction of appropriate counter ions so that sodium chloride and magnesium chloride are conveniently drawn as Na+Cl– and Mg2+ 2Cl– respectively.
It becomes increasingly difficult to remove successive electrons from an atom, and ionic bonding is not usually encountered for some atoms, especially carbon.
Organic chemistry, the study of carbon compounds, is dominated by covalent bonding and the sharing of electrons.
A covalent bond between atoms involves the sharing of two electrons, one from each atom.
The sharing of two electrons is described as a single bond and is indicated in shorthand notation by a single line.
Depending upon the number of electrons an atom carries, it is able to form a certain number of bonds, and this number is called the valency of the atom.
The valency of hydrogen is 1, of oxygen 2, of nitrogen 3, and carbon 4.
This means that we can indicate the bonding in simple organic molecules such as methane, methanol, and methylamine via single bonds.
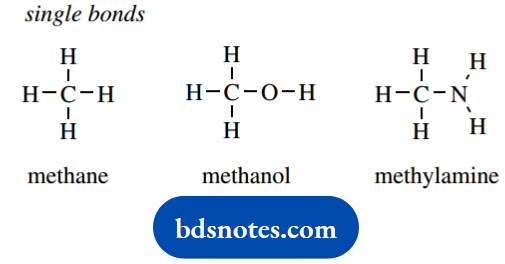
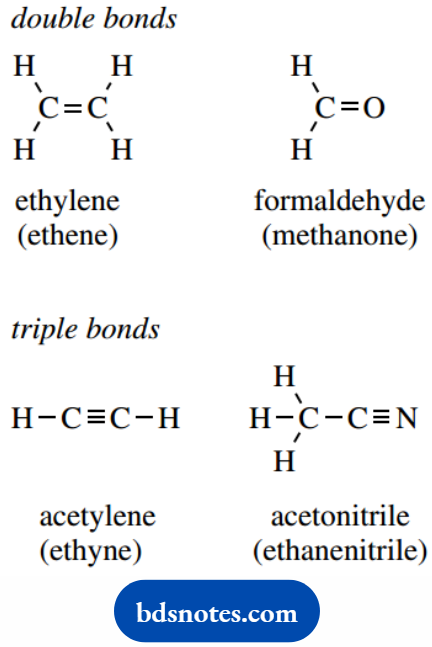
Carbon is particularly versatile, in that it can sometimes share two of its electrons with a second carbon, with nitrogen, or with oxygen.
It can even use three of its four valencies in bonding to another carbon, or to nitrogen. In this way, we generate double and triple bonds, indicated by two or three adjacent lines in our molecular representations.
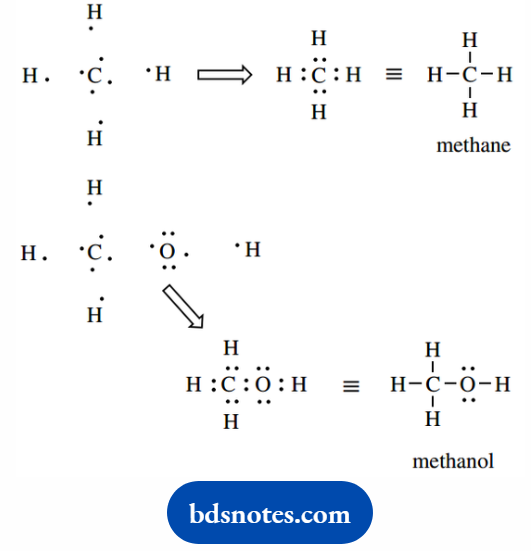
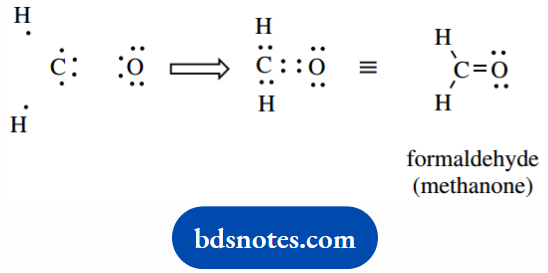
These are extensions of Lewis dot structures, where bonding electrons associated with each bond are shown as dots.
In our simple structures, bonding is associated with eight electrons in the valence shell of the atom, unless it is hydrogen when two electrons are required for bonding.
Whilst we have almost completely abandoned putting in electron dots for bonds.
We still routinely show some pairs of electrons not involved in bonding (lone pairs) because these help in our mechanistic rationalizations of chemical reactions.
This system has its merits and uses – indeed, we shall employ the line notation almost exclusively – but to understand how bonding occurs, and to explain molecular shape and chemical reactivity, we need to use orbital concepts.
Atomic Orbitals
The electrons in an atom surround the nucleus but are constrained within given spatial limits, defined by atomic orbitals. Atomic orbitals describe the probability of finding an electron within a given space.
We are unable to pinpoint the electron at any particular time, but we have an indication that it will be within certain spatial limits.
A farmer knows his cow is in a field, but, at any one time, he does not know precisely where it will be located.
Even this is not a good analogy, because electrons do not behave as nice, solid particles. Their behavior is in some respects like that of waves, and this can best be analyzed through mathematics.
Atomic orbitals are actually graphical representations of mathematical solutions to the Schrodinger wave equation. The equation provides not one, but a series of solutions termed wave functions ψ.
The square of the wave function, ψ2, is proportional to the electron density and thus provides us with the probability of finding an electron within a given space.
Calculations have allowed us to appreciate the shape of atomic orbitals for the simplest atom, i.e. hydrogen, and we make the assumption that these shapes also apply to heavier atoms, like carbon.
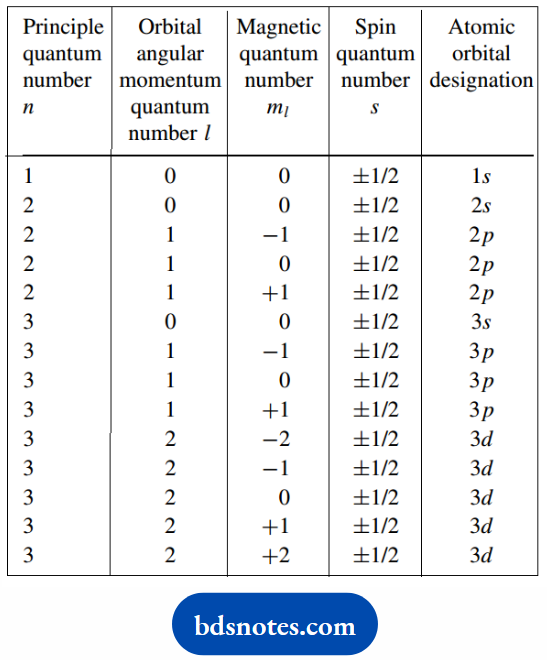
Each wave function is defined by a set of quantum numbers. The first quantum number.
The principal quantum number n, generally relates to the distance of the electron from the nucleus, and hence the energy of the electron.
It divides the orbitals into groups of similar energies called shells. The principal quantum number also defines the row occupied by the atom in the periodic table.
It has integral values, n = 1, 2, 3, 4, etc. The numerical values are used to describe the shell.
The second quantum number, the orbital angular momentum quantum number l, is generally related to the shape of the orbital and depends upon n, taking integral values from 0 to n − 1.
The different values are always referred to by letters: s for l = 0, p for l = 1, d for l = 2, and f for l = 3.
The third quantum number is related to the orbital orientation in space. It is called the magnetic quantum number ml, and depends upon l. It can take integral values from −l to +l.
For p orbitals, suffix letters are used to define the direction of the orbital along the x-, y-, or zaxes. Organic chemists seldom need to consider subdivisions relating to d orbitals.
Finally, there is the spin quantum number s, which may have only two values, i.e. ± 1/2. This relates to the angular momentum of an electron spinning on its own axis.
The magnitude of an electron’s spin is constant, but it can take two orientations. shows the possible combinations of quantum numbers for n = 1 to 3.
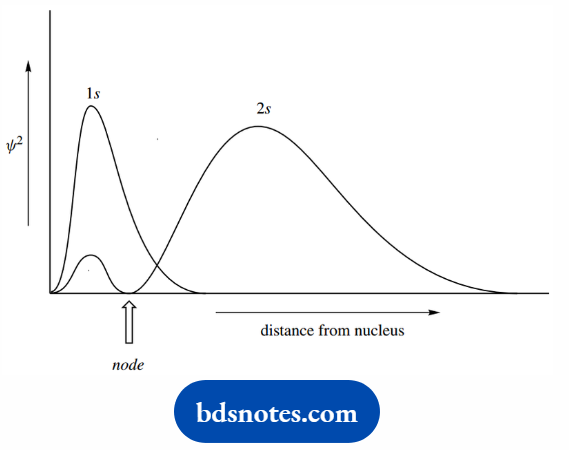
For a hydrogen atom, the lowest energy solution of the wave equation describes a spherical region about the nucleus, a 1 s atomic orbital.
When the wave equation is solved to provide the next higher energy level, we also get a spherical region of high probability, but this 2 s orbital is further away from the nucleus than the 1s orbital.
It also contains a node or point of zero probability within the sphere of high probability. Radial probability density plots.
Showing the probability of finding an electron at a particular distance from the nucleus is presented for the 1s and 2s orbitals, to illustrate the node in the 2s orbital.
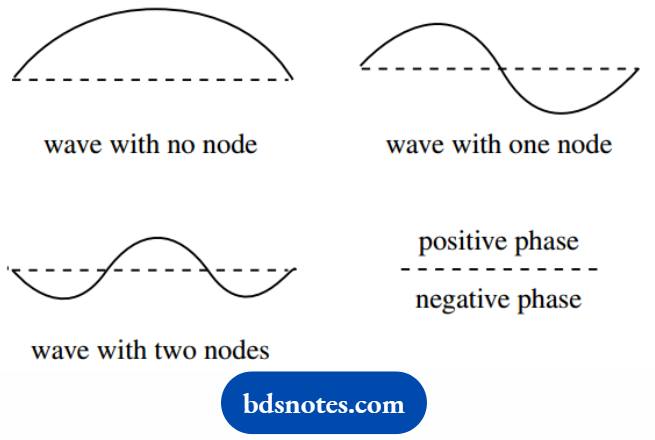
To appreciate the node concept, it is useful to think of wave analogies. Thus, a vibrating string might have no nodes, one node, or several nodes according to the frequency of vibration.
We can also realize that the wave has different phases, which we can label as positive or negative, according to whether the lobe is above or below the median line.
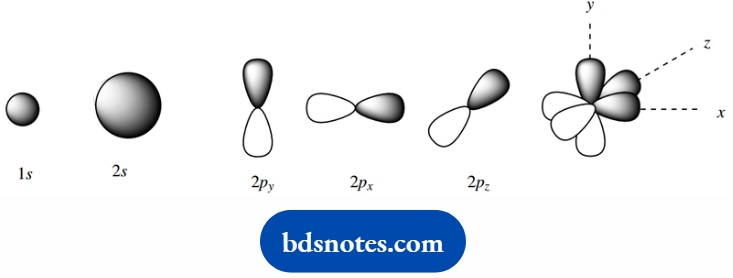
Then follow three additional atomic orbitals, which are roughly dumbbell or propeller-like in appearance. These are aligned along mutually perpendicular axes and are termed the 2 px, 2py, and 2pz orbitals.
These orbitals possess major probability regions on either side of the nucleus, but zero electron probability (a node) at the nucleus.
In one lobe of the orbital, the phase sign of the wave function is positive; in the other it is negative.
To avoid confusion with an electrical charge, the phase sign of the wave function is usually indicated by shading of the lobes; in everyday usage, we may draw them without either sign or shading.
These three orbitals are of equal energy, somewhat higher than that of the 2s orbital. We use the term degenerate to describe orbitals of identical energy. The general appearance of this orbital.
Consideration of 1s, 2s, and 2p orbitals will allow us to describe the electronic and bonding characteristics of most of the atoms encountered in organic molecules.
Atoms such as sulfur and phosphorus need 3s and 3p orbitals to be utilized, after which five more complex 3d orbitals come into play.
As the principal quantum number increases, so the average radius of the s orbitals or the length of the lobes of p orbitals increases.
And the electrons in the higher orbitals are thus located further from the nucleus. Each subsequent orbital is also at a higher energy level.
These energy levels can be calculated from the wave function. They may also be measured directly from atomic spectra, where lines correspond to electrons moving between different energy levels.
As the relative energy levels, 4s orbitals are actually of lower energy than 3d orbitals.
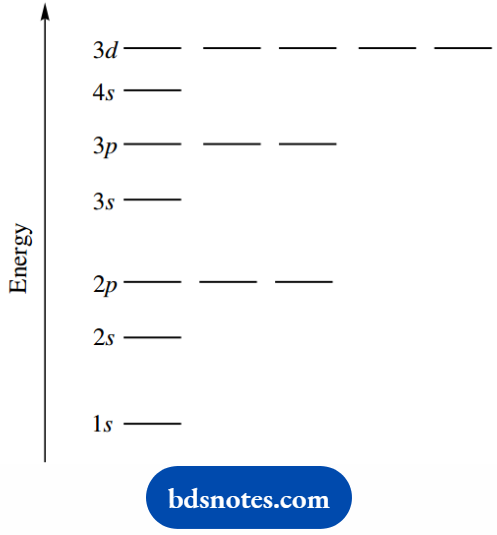
Electronic Configurations
Each atomic orbital can accommodate just two electrons, provided these can be paired by virtue of having opposite spin quantum numbers.
If the spins are the same, then the electrons must be located in different orbitals. We can now describe the electronic configuration for atoms of interest in the first two rows of the periodic table.
Electrons are allocated to atomic orbitals, one at a time so that orbitals of one energy level are filled before proceeding to the next higher level.
Where electrons are placed in orbitals of the same energy (degenerate orbitals, For Example. p orbitals) they are located singly in separate orbitals before two electrons are paired.
Further, the electronic configuration with the greatest number of parallel spins (same spin quantum number) results in the lowest energy overall.
The electronic configuration can be expressed as a list of those orbitals containing electrons, as shown below.
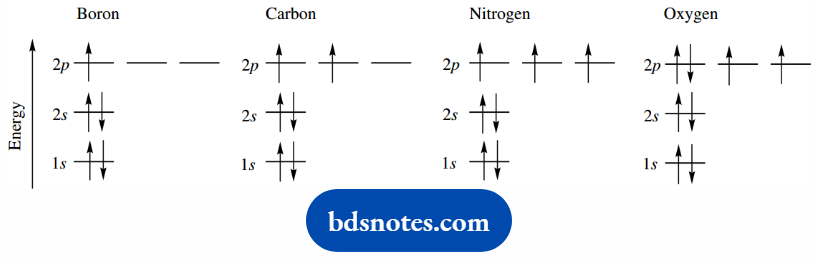
Although it is usual just to indicate the number of electrons in p orbitals, For Example. C: 1s22s22p2 as in the first column.
It is more informative to use the second column designation, i.e. C: 1s22s22px1 2py1, where the non-pairing of electrons is emphasized. As more 2p electrons are allocated, pairing becomes obligatory, For Example. O: 1s22s22px22py12pz1.
There is no hidden meaning in allocating electrons to 2px first. In any case, we are unable to identify which of these orbitals is filled first.

Alternatively, we can use the even more informative energy diagram. Electrons with different spin states are then designated by upward (↑) or downward (↓) pointing arrows.
Note particularly that the noble gas neon has enough electrons to fill the orbitals of the ‘2’ shell completely; it has a total of eight electrons in these orbitals, i.e. an octet.
In the case of helium, the ‘1’ shell orbital is filled with two electrons. The next most stable electronic configurations are those of argon, 1s22s22p63s23p6, and then krypton, 1s22s22p63s23p64s23d1046.
The filled electron shells are especially favorable and responsible for the lack of reactivity of these two elements. Attaining filled shells is also the driving force behind bonding.
Ionic Bonding
The simplest type of bonding to comprehend is ionic bonding. This involves the loss of an electron from one atom, and its transfer to another, with bonding resulting from the strong electrostatic attraction.
For this ionic bonding, the electron transfer is from an atom with a low ionization potential to an atom with high electron affinity.
The atomic objective is to mimic for each atom the nearest noble gas electronic configuration.
Let us consider sodium and chlorine. Sodium (1s22s22p63s1) has one electron more than neon (1s22s22p6), and chlorine (1s22s22p63s23p5) has one electron less than the noble gas argon (1s22s22p6 3s 23p6).
Chlorine has high electronegativity and acquires one electron to become a chloride anion Cl–. Sodium loses one electron to become the cation Na+.

The simplest of the cations we encounter is H+. This is the result of a hydrogen atom losing an electron, and simple arithmetic tells us that this entity now has no electrons.
Being composed of just a proton. We thus refer to H+ as a proton and combination with H+ as a protonation.
In favorable circumstances, we may see more than one electron being donated/acquired, For Ecample. Mg2+ O2-, though the more electrons involved the more difficult it is to achieve the necessary ionizations.
Molecules such as methane, CH4, are not obtained through ionic bonding, but through the covalent electron-sharing mechanism.
Covalent Bonding
Molecular orbitals: σ and π Bonds
We have used the electronic energy levels for atomic hydrogen to serve as a model for other atoms.
In a similar way, we can use the interaction of two hydrogen atoms giving the hydrogen molecule as a model for bonding between other atoms.
In its simplest form, we can consider the bond between two hydrogen atoms originates by bringing the two atoms together so that the atomic orbitals overlap.
Allowing the electrons from each atom to mingle and become associated with both atoms. This sharing of electrons effectively brings each atom up to the noble gas electronic configuration (He, two electrons).
Furthermore, it creates a new orbital spanning both atoms in which the two electrons are located; this is called a molecular orbital.
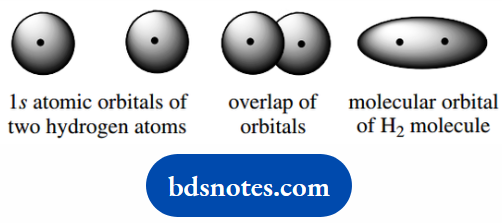
Graphically, we can represent. There must be some energy advantage by bonding, otherwise it would not occur.
The two atomic orbitals, therefore, are used to create a new molecular orbital of rather lower energy, the bonding molecular orbital.
However, since we are considering mathematical solutions to a wave equation, there is an alternative higher energy solution also possible.
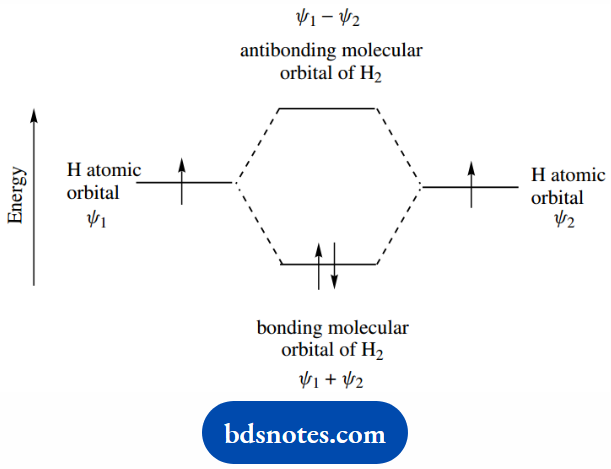
Remind yourself that the solution to x2 = 1 is x = +1 or −1. The higher energy solution is represented by the antibonding molecular orbital.
The bonding molecular orbital is where a combination of atomic orbitals leads to an increased probability of finding the electrons between the two atoms, i.e. bonding.
The antibonding molecular orbital is where a combination of atomic orbitals leads to a reduced or negligible probability of finding the electrons between the two atoms and does not produce bonding.
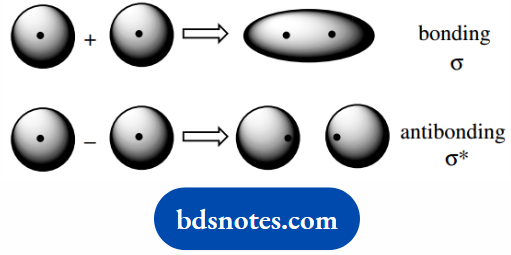
Combining the two atomic orbitals produces two molecular orbitals, here shown as ψ1 + ψ2, and ψ1 − ψ2: in the additive mode the electronic probability increases between the atoms.
Whereas in the subtractive mode, the electronic probability between the atoms decreases, i.e. the antibonding situation.
Bonding results where we have interaction of orbitals with the same phase sign of the wave function.
Whereas the antibonding orbital originates from the interaction of orbitals with different phase signs of the wave function.
This approach to molecular orbitals is called a linear combination of atomic orbitals: wave functions for the atomic orbitals are combined in a linear fashion, by simple addition or subtraction, to generate new wave functions for molecular orbitals.
The number of molecular orbitals formed is the same as the number of atomic orbitals combined. Electrons are allocated to the resultant molecular orbitals as with atomic orbitals.
We start with the lower energy orbital, putting one electron in each degenerate orbital before we add a second with spin pairing.
In the case of hydrogen, therefore, we have two spin-paired electrons in the bonding molecular orbital.
The antibonding orbital remains empty in the so-called ground state of the molecule, unless we input enough energy to promote one electron to the higher energy state, the excited state. This type of transfer gives rise to spectral absorption or emission.
The bonding in the hydrogen molecule formed by the overlap of s orbitals is called a sigma ( σ ) bond; the antibonding orbital is designated σ.
It is a term generally applied where orbital overlap gives a bond that is cylindrically symmetrical in cross-section when viewed along the bond axis. All single bonds are sigma bonds.
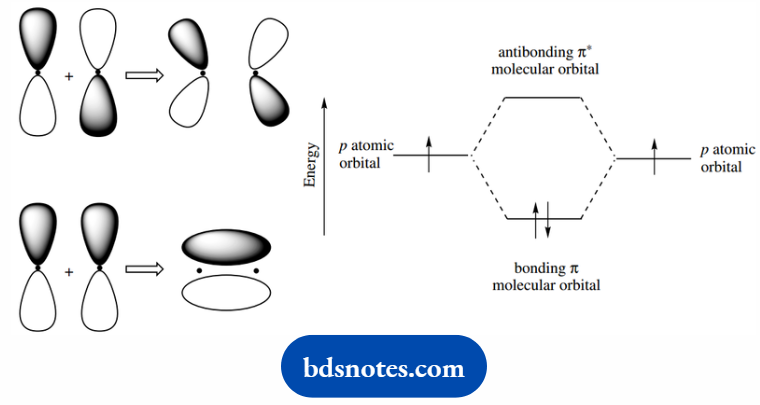
The other important type of bonding in organic molecules is the pi ( π ) bond, the result of side-to-side interaction of p orbitals.
Here, we consider the two lobes separately overlapping; the p orbitals have lobes of different phase signs, and for bonding, we require an overlap of lobes with the same phase sign.
This produces a bonding π molecular orbital with regions of greatest probability of finding electrons above and below the atomic axis. The π bond thus has a nodal plane passing through the bonded atoms.
The antibonding π orbital can be deduced in a similar manner. Double and triple bonds are characterized by π bonding. π bonds possess an enhanced reactivity not associated with σ bonds.
Hybrid Orbitals In Carbon
We start this section with a word of caution. Students frequently find hybridization a rather difficult concept to understand and appreciate. However, there is no particular reason why this should be so.
Chemistry is an experimental science, and to rationalize our observations we gradually develop and invoke a number of rules and principles.
Theories may have to change as scientific data increase, and as old principles cease to explain the facts.
All of the foregoing descriptions of atomic and molecular orbitals a hypotheses for atomic and molecular structure supported by experimental data.
So far, the description meets most of our needs and provides a good rationalization of chemical behavior.
However, it falls short in certain ways, and we have to invoke a further modification to explain the facts.
Here are three observations based upon sound experimental evidence, which are not accommodated by the above description of bonding:
- The hydrocarbon methane (CH4) is tetrahedral in shape with bond angles of about 109°, and the four C–H bonds are all equivalent and identical in reactivity.
- Ethylene (ethene, C2H4) is planar, with bond angles of about 120°, and it contains one π bond.
- Acetylene (ethyne, C2H2) is linear, i.e. bond angles 180°, and it contains two π bonds.
None of these observations follows immediately from the electronic configuration of carbon (1s22s22px12py1), which shows that carbon has two unpaired electrons, each in a 2p orbital.
From our study of bonding so far, we might predict that carbon will be able to bond to two other atoms, i.e. it should be divalent, though this would not lead to an octet of electrons.
Carbon is usually tetravalent and bonds to up to four other atoms. Therefore, we need to modify the model to explain this behavior. This modification is hybridization.
sp3 Hybrid Orbitals
Methane is a chemical combination of one carbon atom and four hydrogen atoms. Each hydrogen atom contributes one electron to a bond; so, logically, carbon needs to provide four unpaired electrons to allow the formation of four σ bonds.
The ability of carbon to bond to four other atoms requires unpairing of the 2s2 electrons. We might consider promoting one electron from a 2s orbital to the third, as yet unoccupied, 2p orbital.
This would produce an excited-state carbon; since the 2p orbital is of higher energy than the 2s orbital, the process would require the input of energy.
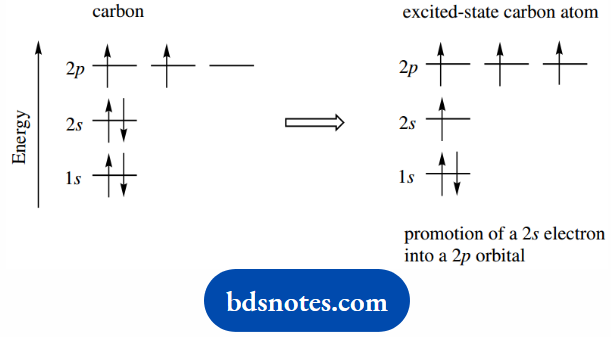
We could assume that the ability to form extra bonds would more than compensate for this proposed change.
We now have four unpaired electrons in separate orbitals, and the electronic configuration of carbon has become 1s22s2px1 2py12pz1.
Each electron can now form a bond by pairing with the electron of a hydrogen atom.
However, this does not explain why methane is tetrahedral and has four equivalent bonds.
The bond that utilizes the 2s electron would surely be different from those that involve 2p electrons.
The geometry of the molecule should somehow reflect that the p orbitals are positioned at right angles to each other, whilst the s orbital is spherical and might bond in any direction.
If all the bonds in methane turn out to be equivalent, they must be some sort of ‘hybrid’ version of those we have predicted.
We can explain many features of organic chemicals, including their reactivity and shape, by a mathematical model in which hybrid orbitals for carbon are derived by mixing the one 2s orbital and three 2p atomic orbitals.
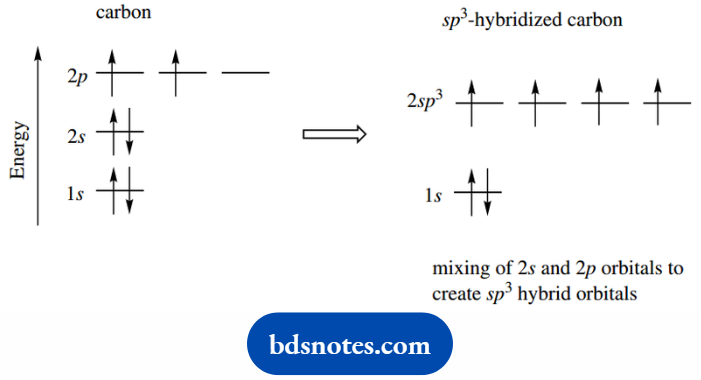
This generates four equivalent hybrid orbitals, which we designate sp3, since they are derived from one s orbital and three p orbitals.
The sp3 orbitals will be at an energy level intermediate between those of the 2s and 2p orbitals and will have properties intermediate between s and p, though with greater p character.
The mathematical model then provides us with the shape and orientation of these hybrid orbitals. For the convenience of drawing, we tend to omit the small lobes at the center of the array.
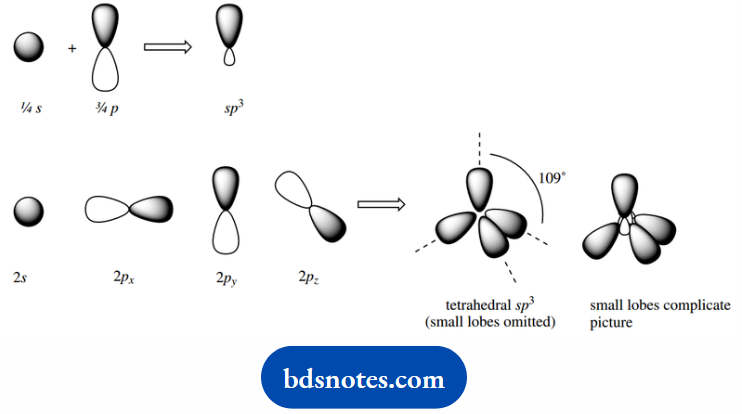
These new hybrid orbitals are then all equivalent, and spaced to minimize any interaction; this is a tetrahedral array.
The best way of arranging four groups around a central point. Each hybrid orbital can now accommodate one electron.
Now we can consider the bonding in methane. Using orbital overlap as in the hydrogen molecule as a model.
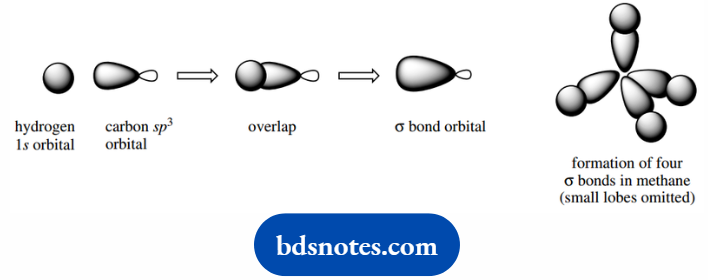
Each sp3 orbital of carbon can now overlap with a 1s orbital of a hydrogen atom, generating a bonding molecular orbital, i.e. a σ bond.
Four such bonds can be created, and they will be produced in a tetrahedral array.
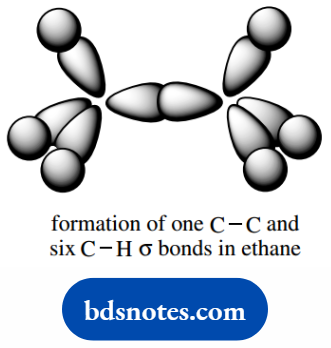
We can also consider C–C σ bonding, as in ethane (C2H6), by the overlap of two carbon sp3 orbitals. The three remaining sp3 orbitals of each carbon are used to make C–H σ bonds to hydrogen atoms.
It may be argued that we have actually started from the tetrahedral array in methane to propose a tetrahedral array of atomic orbitals in carbon.
This is undoubtedly true but is part of the process of refining the model as we need to explain new observations. We make models to describe nature; nature merely adopts a minimum energy situation.
We gain confidence in the approach by using a similar rationale to account for the second of the observations above, that ethylene is planar, with bond angles of about 120°, and contains one π bond.
sp2 Hybrid Orbitals
The sp3 hybrid orbitals of carbon were considered as a mix of the 2s orbital with three 2p orbitals.
To provide a model for ethylene, we now need to consider hybrid orbitals that are a mix of the 2s orbital with two 2p orbitals, giving three equivalent sp2 orbitals.
In this case, we use just three orbitals to create three new hybrid orbitals.
Accordingly, we find that the energy level associated with an sp2 orbital will be below that of the sp3 orbital: this time.
We have mixed just two high-energy p orbitals with the lower energy s orbital.
The four electrons are accommodated one in each hybrid orbital and one in the remaining 2p orbital.
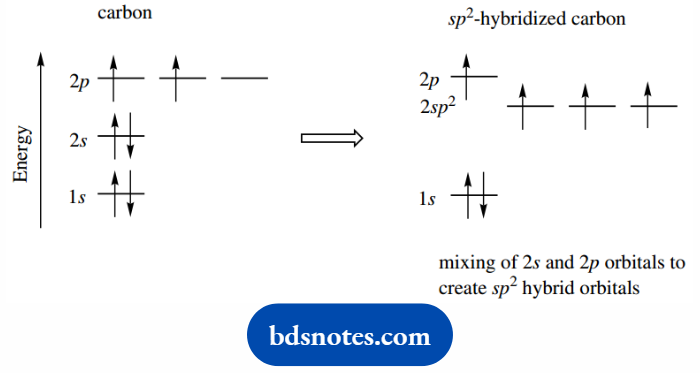
The sp2 hybrid orbitals are distributed in a planar array around the atom; this spacing minimizes any interactions. The 2p orbital is then located perpendicular to this plane.
Such information is again obtained from the mathematical analysis, but simple logic would lead us to predict that this is the most favorable arrangement to incorporate the components.
The sp2 orbital will be similar in shape to the sp3 orbital, but somewhat shorter and fatter, in that it has more s character and less p character.
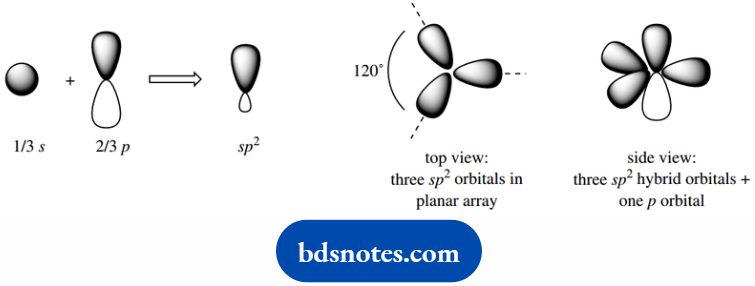
The bonding in ethylene is based initially on one C–C σ bond together with four C–H σ bonds, much as we have seen in ethane.
We are then left with a p orbital for each carbon, each carrying one electron, and these interact by side-to-side overlap to produce a π bond.
This makes the ethylene molecule planar, with bond angles of 120°, and the π bond has its electron density above and below this plane.
The combination of the C–C σ bond and the C–C π bond is what we refer to as a double bond; note that we cannot have π bond formation without the accompanying σ bond.

You will observe that it becomes progressively more difficult to draw a combination of σ and π molecular orbitals to illustrate the bonding that constitutes a double bond.
We often resort to a picture that illustrates the potential overlap of p orbitals by means of a dotted line or similar device. This cleans up the picture, but leaves rather more to the imagination.
The properties of an alkene (like ethylene) are special, in that the π bond is more reactive than the σ bond, so alkenes show a range of properties that alkanes (like ethane) do not.
We can only get an overlap of the p orbitals if their axes are parallel. If their axes were perpendicular, then there would be no overlap and, consequently, no bonding.
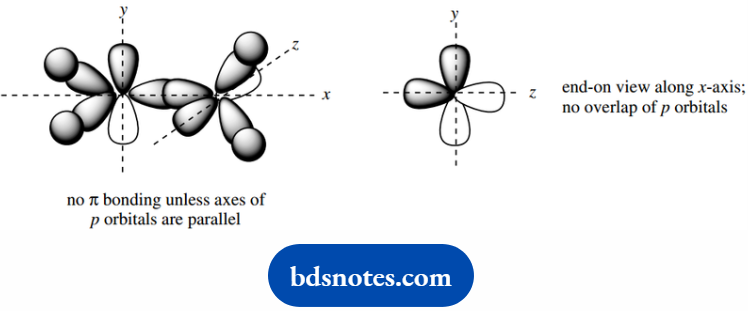
This situation might arise if we tried to twist the two parts of the ethylene molecule about the C–C link. This is not easily achieved and would require a lot of energy.
It can be achieved by absorbing sufficient energy to promote an electron to the antibonding π orbital.
This temporarily destroys the π bond, allows rotation about the remaining σ bond, and the π bond may reform as the electron is restored to the bonding orbital.
It accounts for a change in the configuration of the double bond, so-called cis–trans isomerism, and we shall see an example shortly.
Compounds with π bonds are said to be unsaturated, whereas compounds without π bonds containing only σ bonds are referred to as saturated.
sp hybrid orbitals
The third observation relates to acetylene (ethyne, C2H2), which is linear, i.e. bond angles of 180°, and contains two π bonds.
This introduces what we term triple bonds, actually a combination of one σ bond and two π bonds. In this molecule, we invoke another type of hybridization for carbon, that of sp hybrid orbitals.
These are a mix of the 2s orbital with one 2p orbital, giving two equivalent sp orbitals.
Each hybrid orbital takes one electron, whilst the remaining two electrons are accommodated in two different 2p orbitals.
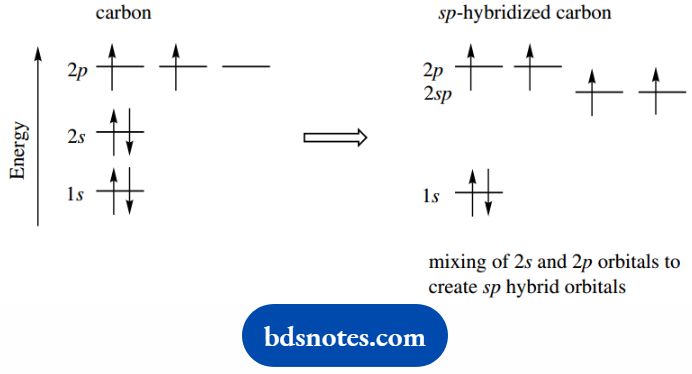
The sp hybrid orbitals can be visualized as a straight combination of an s and a p orbital, so that it will now be the shortest and fattest of the hybrid orbitals, with most s character and least p character.
Its energy will be above that of the s orbital, but below that of sp2 orbitals, since the p contribution is the higher energy component.
The atomic orbitals in sp-hybridized carbon are going to be two equivalent sp orbitals, arranged opposite each other to minimize interaction.
Plus the two remaining p orbitals, will be at right angles to each other, and also at right angles to the sp orbitals.
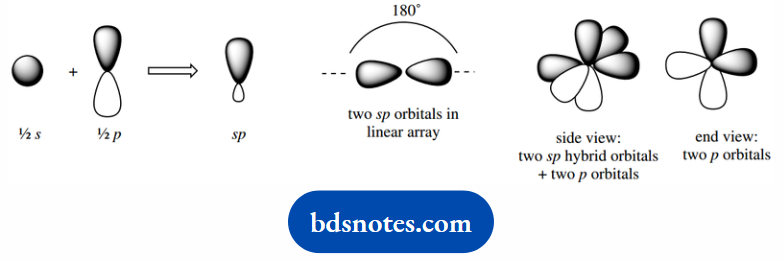
The bonding in acetylene has one C–C σ bond together with two C–H σ bonds; the p orbitals on each carbon, each carrying one electron, interact by side-to-side overlap to produce two π bonds.
Note again that the p orbitals can only overlap if their axes are parallel.

This makes the acetylene molecule linear, i.e. bond angles of 180°, and there are two π bonds with electron density on either side of this axis.
The properties of an alkyne, like acetylene, are also special in that the π bonds are again much more reactive than the σ bond.
Hybridization And Bond Lengths
We also note that there are significant differences in bond lengths for single, double, and triple bonds.
The carbon atoms in ethane are further apart (1.54 Å) than in ethylene (1.34 Å), and those in acetylene are even closer together (1.20 Å); Å refers to the Angstrom unit, 10-10 m.
This is primarily a consequence of the different nature of the σ bonds joining the two carbons.
Because sp2 hybrid orbitals have less p character than sp3 hybrid orbitals, they are less elongated; consequently, a σ bond formed from sp2 orbitals will be rather shorter than one involving sp3 orbitals.
By similar reasoning, sp hybrid orbitals will be shorter than sp2 orbitals, because they have even less p character, and will form even shorter C–C σ bonds.
There is a similar effect in the length of C–H bonds, but this is less dramatic, primarily because the hydrogen atomic orbital involved (1s) is considerably smaller than any of the hybrid orbitals we are considering.
Nevertheless, C–H bonds involving sphybridized carbon are shorter than those involving sp2-hybridized carbon, and those with sp3-hybridized carbon are the longest.
Note how we have resorted to another form of representation of the ethane, ethylene, and acetylene molecules here, representations that are probably familiar to you.
These line drawings are simpler, much easier to draw, and clearly show how the atoms are bonded – we use a line to indicate the bonding molecular orbital.
They do not show the difference between σ and π bonds, however. We also introduce here the way in which we can represent the tetrahedral array of bonds around carbon in a two-dimensional drawing.
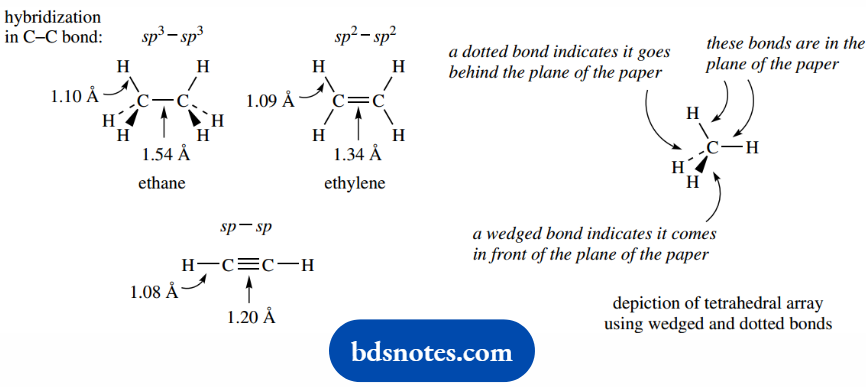
This is to use wedges and dots for bonds instead of lines. By convention, the wedge means the bond is coming towards you, out of the plane of the paper.
The dotted bond means it is going away from you, behind the plane of the paper. We shall discuss stereochemical representations in more detail later.
At the beginning of this section, we suggested that students often found hybridization a difficult concept to understand.
We should emphasize that hybridization is a model that helps us to appreciate molecular structure and predict chemical reactivity.
Do not think in terms of atomic orbitals merging to form hybrid orbitals, but consider that such orbitals already exist as the lowest energy arrangement.
Hybridization is our modification of the first model, which we saw had its limitations, to an improved model that provides a rationale for experimental observations.
As research progresses, we may have to apply even further modifications! At present, though, the concept of hybrid orbitals provides us with satisfactory explanations for many chemical features.
We have already seen that hybridization helps to define features such as bond angles and bond lengths that dictate molecular shape.
In later sections, we shall see that hybridization gives us good explanations for other aspects of chemistry, such as acidity and basicity.
The relative reactivity of nucleophiles, and the chemical behavior of compounds having conjugation or aromatic rings.
Carbanions, Carbocations, And Radicals
Before we move on from the hybrid orbitals of carbon.
We should take a look at the electronic structure of important reactive species that will figure prominently in our consideration of chemical reactions. First, let us consider carbanions and carbocations.
We shall consider the simplest examples, the methyl anion CH3_ and the methyl cation CH3+, though these are not going to be typical of carbanions and carbocations.
We shall be meeting. In that, they lack features to enhance their stability and utility.
The methyl anion is what would arise if we removed H+ (a proton) from methane by fission of the C–H bond so that the two electrons are left with carbon.
We can immediately deduce that carbon has its full octet of electrons and that we shall have a tetrahedral array of three bonds and a lone pair of electrons in sp3 orbitals.
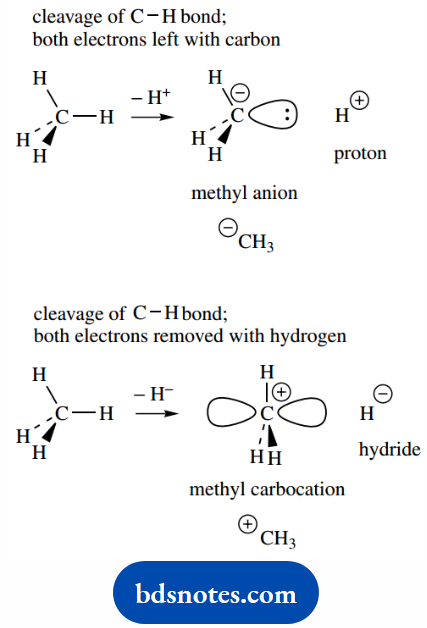
On the other hand, methyl carbocation is the result of removing a hydride anion (a hydrogen atom and an electron) from methane by fission of the C–H bond so that the two electrons are removed with hydrogen.
We can now deduce that carbon has only six electrons in its outer shell. This arrangement is best accommodated by sp2 hybridization and a vacant p orbital.
The alternative of four sp3 hybrid orbitals with one unfilled does not minimize repulsion between the filled orbitals, and is also a higher energy arrangement.
To deduce this, we need to go back to the energy diagrams for sp3 and sp2 hybrid orbitals.
The lower p character of sp2 hybrid orbitals means they are of lower energy than sp3 orbitals; this is because the 2p orbitals are of higher energy than the 2s orbital.
Consequently, we can work out that six electrons in sp2 orbitals will have a lower energy than six electrons in sp3 orbitals.
Hence, the methyl carbocation is of a planar sp2 nature with an unoccupied p orbital at right angles to this plane. The consequences of this will be developed.
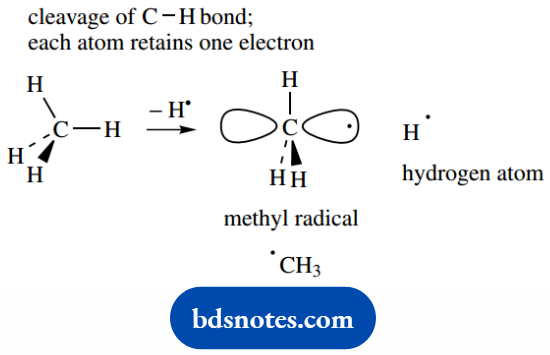
There is also a third type of reactive species that we shall, namely radicals. Briefly, radicals are uncharged entities that carry an unpaired electron.
A methyl radical CH3 results from the fission of a C–H bond in methane so that each atom retains one of the electrons.
In the methyl radical, carbon is sp2 hybridized and forms three σ C–H bonds, whilst a single unpaired electron is held in a 2p orbital oriented at right angles to the plane containing the σ bonds.
The unpaired electron is always shown as a dot. The simplest of the radical species is the other fission product, a hydrogen atom.
Hybrid Orbitals In Oxygen And Nitrogen
Hybridization concepts can also be applied to atoms other than carbon. Here, we look at how we can understand the properties of oxygen and nitrogen compounds by considering hybrid orbitals for these atoms.
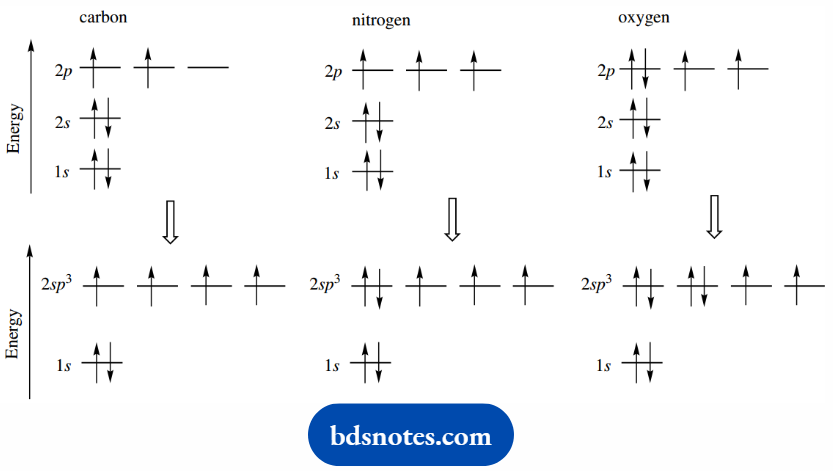
Let us recap on the electronic configurations of oxygen and nitrogen. Nitrogen has one more electron than carbon, and oxygen has two more.
For each atom, we can consider hybrid sp3 orbitals derived from the 2s and 2p orbitals as we have seen with carbon.
We shall then obtain electronic configurations in which nitrogen has two paired electrons in one of these orbitals, whilst the remaining three orbitals each have a single unpaired electron available for bonding.
Oxygen has two sets of paired electrons and has two unpaired electrons available for bonding.
σ bonds
The simplest compounds to consider here are ammonia and water.
It is apparent from the above electronic configurations that nitrogen will be able to bond to three hydrogen atoms, whereas oxygen can only bond to two.
Both compounds share part of the tetrahedral shape we saw with sp3-hybridized carbon.
Those orbitals not involved in bonding already have their full complement of electrons, and these occupy the remaining part of the tetrahedral array.
These electrons are not inert, but play a major role in chemical reactions; we refer to them as lone-pair electrons.
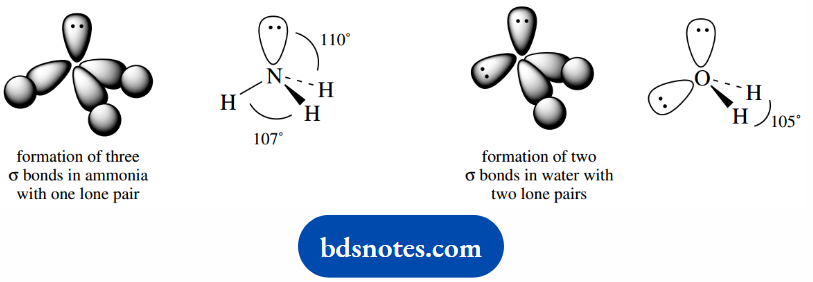
These orbital pictures tend to get a little confusing, in that we really need to put in the elemental symbol to distinguish it from carbon, and we usually wish to show the lone pair electrons.
We accordingly use a compromise representation that employs the cleaner line drawings for part of the structure.
And shows the all-important orbital with its lone pair of electrons. These are duly shown for ammonia and water.
The tetrahedral geometry resultant from these sp3-hybridized nitrogen and oxygen atoms is found to exist in both ammonia and water.
Bond angles in these molecules are not quite the 109° of the perfect tetrahedron, because the electrons in the lone pair atomic orbital are not involved in bonding.
They are, therefore, closer to the nucleus than the electrons in the N–H or O–H bond σ molecular orbitals.
Lone pairs thus tend to exert a greater electronic repulsive force between themselves, and also towards the bonding electrons, than the σ bonding electrons do to each other.
The net result is that bond angles between lone pairs, or between lone pairs and σ bonds, are somewhat greater than between σ bonds, a distortion of the perfect tetrahedral array.
Lone pair electrons may be used in bonding. Since they already have a complement of two electrons, bonds will need to be made to an atom that is electron deficient, For Example. a proton.
Thus, the ammonium cation and the hydronium cation also share tetrahedral geometry, and each possesses a σ bond formed from lone pair electrons.
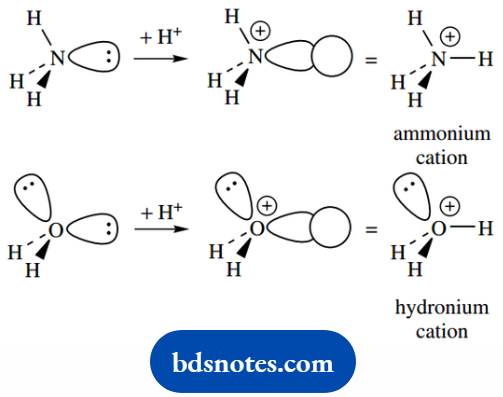
The hydronium cation still possesses a lone pair of electrons. It does not bond to a second proton for the simple reason that the cation would then be required to take on an unfavorable double positive charge.
π bonds
When we consider double bonds to oxygen, as in carbonyl groups (C=O), or to nitrogen, as in imine functions (C=N).
We find that experimental data are best accommodated by the premise that these atoms are sp2 hybridized. This effectively follows the pattern for carbon-carbon double bonds.
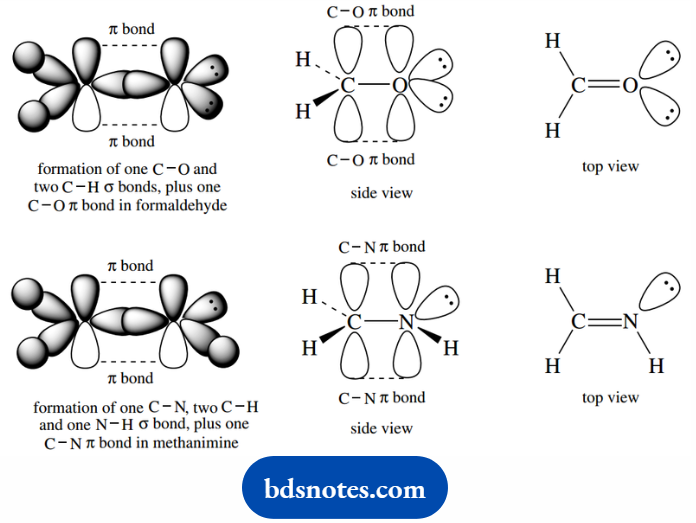
The double bond is again a combination of a σ bond plus a π bond resulting from the overlap of p atomic orbitals. The carbonyl oxygen carries two lone pairs in sp2 orbitals, whereas nitrogen carries one.
Thus, the main difference from the alkene structure, apart from the atoms involved, is that lone pairs in atomic orbitals replace one or more of the σ molecular orbitals that constituted the C–H bonds.
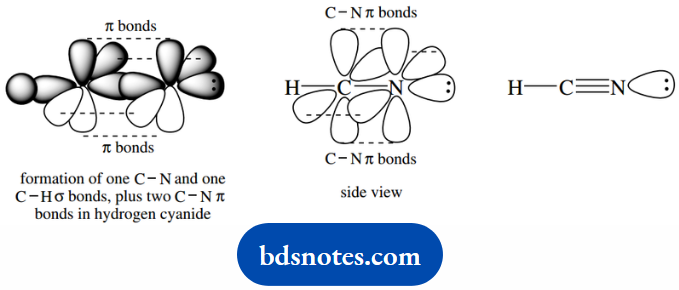
The atoms around the double bond are in a planar array, just as in an alkene. Triple bonds are also encountered in cyanides or nitriles.
We can compare these with alkynes in much the same way. With sp-hybridized nitrogen, we can form one C–N σ bond and two C–N π bonds.
Leaving a lone pair of electrons on nitrogen in an sp atomic orbital. The cyanide or nitrile system is linear, just like an alkyne.
Bond Polarity
The nucleus of each atom has a certain ability to attract electrons. This is termed its electronegativity.
This means that, when it is bonded to another atom, the bonding electrons are not shared equally between the two atoms.
Covalent bonds may, therefore, possess a charge imbalance, with one of the atoms taking more than its share of the electrons.
This is referred to as bond polarity. An atom that is more electronegative than carbon will thus polarize the bond, and we can consider the atoms as being partially charged.
This is indicated in a structure by putting partial charges ( δ+ and δ−) above the atoms.
It can also be represented by putting an arrowhead on the bond, in the direction of electron imbalance. Alternatively, we use a specific dipole arrow at the side of the bond.

In general, electronegativities increase from left to right across the periodic table and decrease going down a particular column of the periodic table.
The relative electronegativities of those atoms most likely to be found in typical organic molecules are included.
The numbers (Pauling electronegativity values) are on an arbitrary scale from Li = 1 to F = 4.
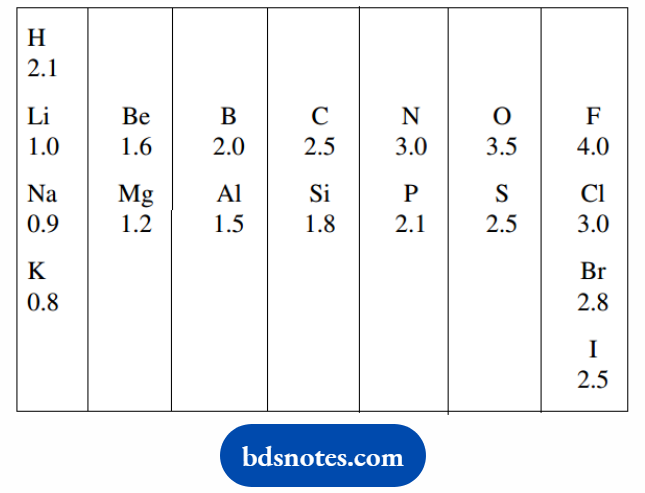
From the sequence shown, it is readily seen that hydrogen and carbon are among the least electronegative atoms we are likely to encounter in organic molecules.
The relatively small difference in electronegativities between hydrogen and carbon also means there is not going to be much polarity associated with a C–H bond.
Most atoms other than hydrogen and carbon when bonded to carbon are going to be electron-rich; therefore, bonds may display considerable polarity.
This polarity helps us to predict chemical behavior, and it is crucial to our prediction of chemical mechanisms.
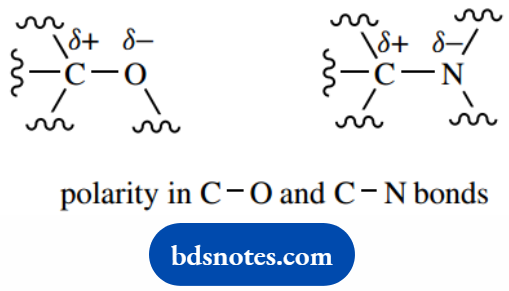
We must also modify our thinking of bonding as being simply ionic (where there is the transfer of electrons between atoms) or covalent (where there is equal sharing of electrons).
These represent two extremes, but bond polarity now provides a middle ground where there is a sharing of electrons, but an unequal sharing.
Bond polarity in a molecule can often be measured by a dipole moment, expressed in Debye units (D). However, the physical measurement provides only the overall dipole moment, i.e. the sum of the individual dipoles.
A molecule might possess bond polarity without displaying an overall dipole if two or more polar bonds are aligned so that they cancel each other out.
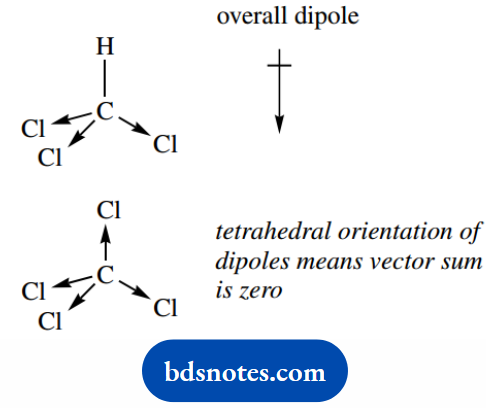
The C–Cl bond is polar, but although chloroform (CHCl3) has a dipole moment (1.02 D), carbon tetrachloride (CCl4) has no overall dipole.
Because of the tetrahedral orientation of the dipoles in carbon tetrachloride, the vector sum is zero.
Polarization in one bond can also influence the polarity of an adjacent bond.
Thus, in ethyl chloride, the polarity of the C–Cl bond makes the carbon more positive ( δ+); consequently, electrons in the C–C bond are drawn towards this partial positive charge.
The terminal carbon thus also experiences a partial positive charge, somewhat smaller than δ+ and so depicted as δ δ+.
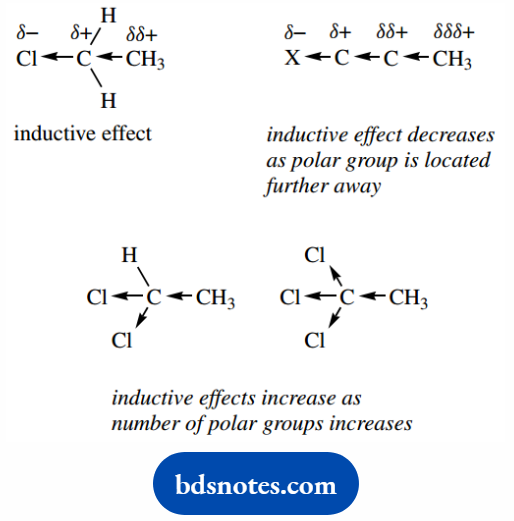
This transmission of polarity through the σ bonds is termed an inductive effect. It is relatively short range, decreasing rapidly as the original dipole is located further away.
It becomes unimportant after about the third carbon atom. However, the effects will increase with the number of polar groups.
So we see increasing polarization effects with 1,1-dichloroethane and 1,1,1-trichloroethane. We shall often need to consider inductive effects when attempting to predict chemical reactivity.
Conjugation
Double bonds, whether they be C=C, C=O, or C=N, are sites of special reactivity in a molecule.
This reactivity may take on different characteristics if we have two or more double bonds in the same molecule, depending on whether the double bonds are isolated or conjugated.
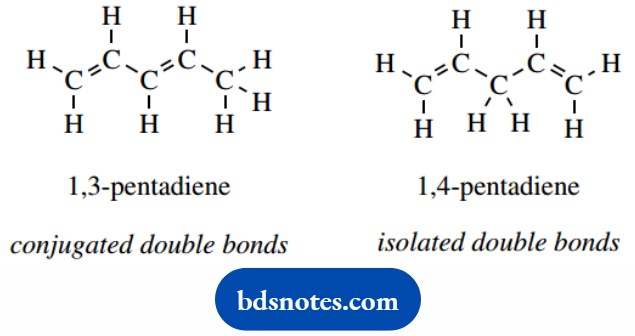
We use the term conjugated to describe an arrangement in which double bonds are separated by a single bond.
Thus, in 1,3-pentadiene the double bonds are conjugated, whereas in 1,4-pentadiene they are isolated or nonconjugated.
The nomenclature ‘diene’ indicates two C=C double bonds, the numbers the position in the molecule.
Conjugated dienes usually display rather different chemical reactivity and spectral properties from non-conjugated dienes.
The differences arise from the nature of the π orbitals in the double bond system. Consider 1,4- pentadiene first.
We may draw this to show an overlap of p orbitals to create two separate π bonds, and effectively that is all there is that is worthy of note.
The double bonds are isolated entities that do not interact.
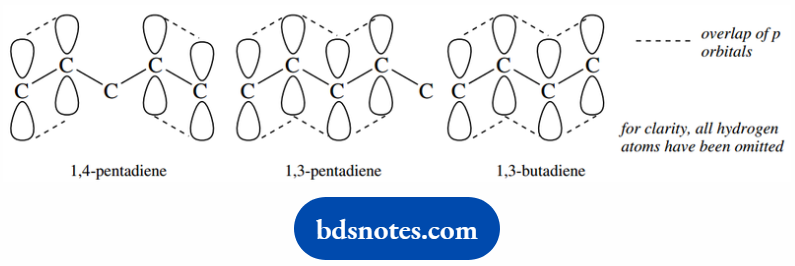
In 1,3-pentadiene, however, the p orbitals are all able to overlap in such a way that a lower energy molecular orbital can be formed.
We have more physical data available for 1,3-butadiene, so let us consider this slightly simpler conjugated system instead.
We have four 2p orbitals on four adjacent carbon atoms, and these can overlap to produce four π molecular orbitals.
These are as shown, and their relative energies can be visualized from the bonding interactions possible.
Remember, bonding results from the overlap of orbitals that have the same phase sign of the wave function, whereas antibonding orbitals originate from the interaction of orbitals with different phase signs of the wave function.
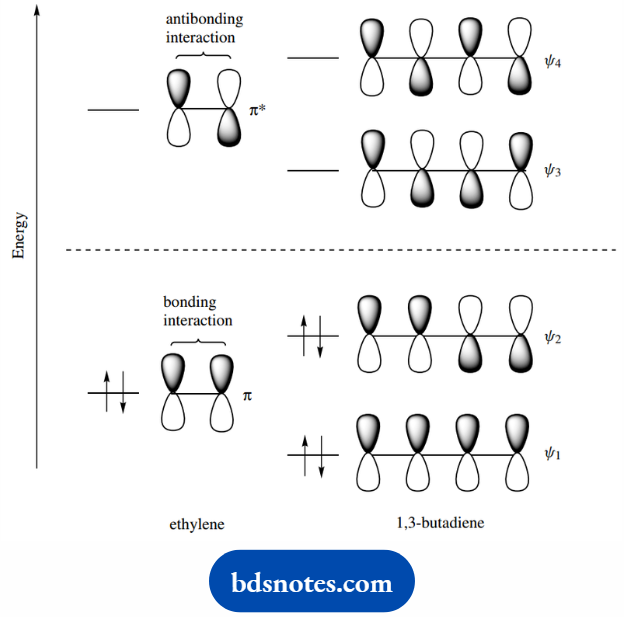
Thus, ψ1 has three bonding interactions and no antibonding interactions, ψ2 has two bonding interactions and one antibonding interaction.
ψ3 has one bonding interaction and two antibonding interactions, and ψ4 has no bonding interaction and three antibonding interactions. The four electrons will be allocated to ψ1 and ψ2.
Conjugation introduces a number of features. We can consider that the π electrons in a conjugated system are no longer associated with specific bonds.
But are delocalized over those atoms constituting the conjugated system. This has energy implications. The overall energy associated with butadiene is actually less than we might expect.
It is lower than that of non-conjugated dienes, For Example. 1,4-pentadiene, and less than what we might estimate from figures for the monounsaturated but-1-ene.
Thus, compounds with two conjugated double bonds are thermodynamically more stable (less reactive) than compounds with two isolated double bonds.
In due course, we shall see that the double bond reactivity of butadiene is also influenced by conjugation: butadiene behaves differently from compounds with isolated double bonds.
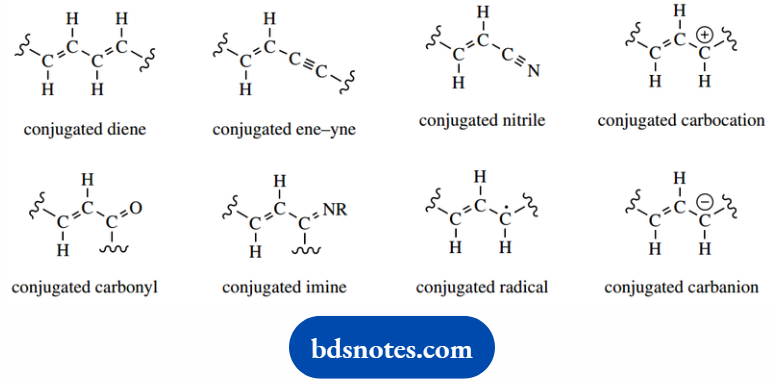
We also need to appreciate that conjugation, and its influence on reactivity is not restricted to alkenes.
Any system containing two or more π bonds may be conjugated, so that we can include triple bonds (alkynes), carbonyl groups, imines, and nitriles in this description.
In its broadest sense, conjugation refers to a system that has a p orbital adjacent to a π bond allowing the delocalization of electrons.
The adjacent p orbital may be a vacant one, as in a carbocation, one that contains a single electron. As in a radical, or maybe part of another π bond, as in a conjugated diene.
At first glance, a conjugated anion does not fit the broad definition of conjugation, since we would expect the carbanion center to be sp3 hybridized.
Nevertheless, there is delocalization of electrons and this system is considered to be conjugated.
In this system, delocalization results from accommodating the negative charge in a p orbital rather than an sp3 orbital, so that we again achieve p orbital overlap.
Conjugated systems also give characteristic spectral absorptions, especially in the UV–visible regions.
As the extent of conjugation increases, i.e. more than two double bonds separated by single bonds, these compounds have more intense absorptions at longer wavelengths (lower energies).
This is because the energy difference between bonding and antibonding molecular orbitals becomes smaller with increasing conjugation.
The spectral data arise from the transition of an electron between these energy levels.
This means that, with increasing conjugation, the characteristic absorption moves from the UV to the visible region and, typically, the compound becomes colored.
A compound appears colored to the human eye when it is removed by absorption of some of the wavelengths from white light.
Carotenoids, vitamin A, and vision
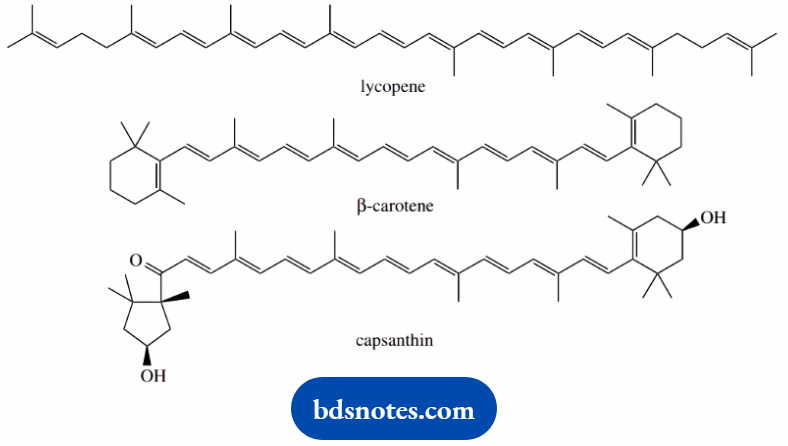
Carotenoids are a group of natural products found predominantly in plants. They are characterized by an extended chain of conjugated double bonds, giving an extended π electron system.
They are highly colored and contribute to yellow, orange, and red pigmentations in plants.
Lycopene is the characteristic carotenoid pigment in ripe tomato fruits, and the orange color of carrots is caused by β-carotene. Capsanthin is the brilliant red pigment of capsicum peppers.
Carotenoids function along with chlorophylls in photosynthesis as accessory light-harvesting pigments, effectively extending the range of light that can be absorbed by the photosynthetic apparatus.
The absorption maximum of carotenoids is typically between 450 and 500 nm, which indicates that the energy difference between bonding and antibonding molecular orbitals is quite small.
This absorption maximum corresponds to blue light so with blue light absorbed, the overall impression to the human eye is of a bright yellow-orange coloration.
Recent research suggests that carotenoids are important antioxidant molecules for humans, helping to remove toxic oxygen-derived radicals, and thus minimizing cell damage.
The most beneficial dietary carotenoid in this respect is lycopene, with tomatoes as the predominant source.
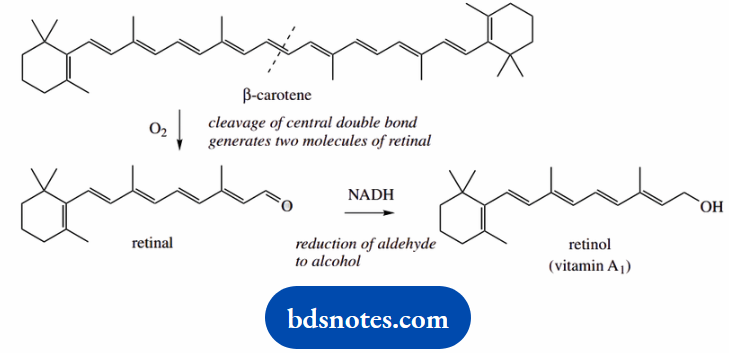
Vitamin A1 (retinol) is derived in mammals by oxidative metabolism of plant-derived dietary carotenoids in the liver, especially β-carotene.
Green vegetables and rich plant sources such as carrots help to provide us with adequate levels.
Oxidative cleavage of the central double bond of β-carotene provides two molecules of the aldehyde retinal, which is subsequently reduced to the alcohol retinol.
Vitamin A1 is also found in a number of foodstuffs of animal origin, especially eggs and dairy products. Some structurally related compounds, including retinal, are also included in the A group of vitamins.
A deficiency of vitamin A leads to vision defects, including a visual impairment at low light levels, termed night blindness.
For the processes of vision, retinol needs to be converted first by oxidation into the aldehyde retinal, and then by enzymic isomerization to cis-retinal.
Cis-retinal is then bound to the protein opsin in the retina via an imine linkage to give the red visual pigment rhodopsin.
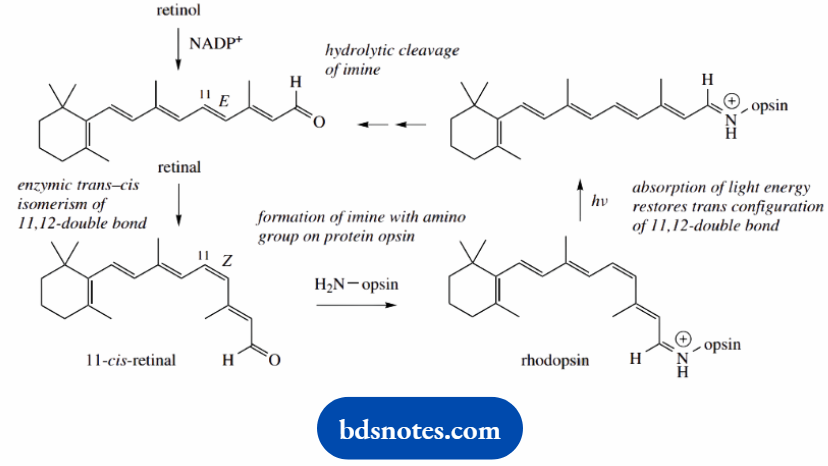
Rhodopsin is sensitive to light by a process that involves the isomerization of the cis-retinal portion back to the trans form, thus translating the light energy into a molecular change that then triggers a nerve impulse to the brain.
The absorption of light energy promotes an electron from a π to a π* orbital, thus temporarily destroying the double bond character and allowing rotation.
Trans-retinal is then subsequently released from the protein by hydrolysis, and the process can continue.
Aromaticity
Aromatic compounds constitute a special group of conjugated molecules; these are cyclic unsaturated molecules with unusual stability and characteristic properties.
The term aromatic originates from the odor displayed by many of the simple examples.
Benzene
The parent compound is benzene. Benzene, C6H6, contains an array of six sp2-hybridized carbons, each attached by a σ bond to the adjacent carbons, and by a third σ bond to a hydrogen atom.
The six p atomic orbitals from carbon are all aligned so that they can overlap to form molecular orbitals, and this is most favorable when the carbons are all in one plane.
The lowest energy molecular orbital can be considered as an extended ring-like system with a high electron probability above and below the plane of the ring.
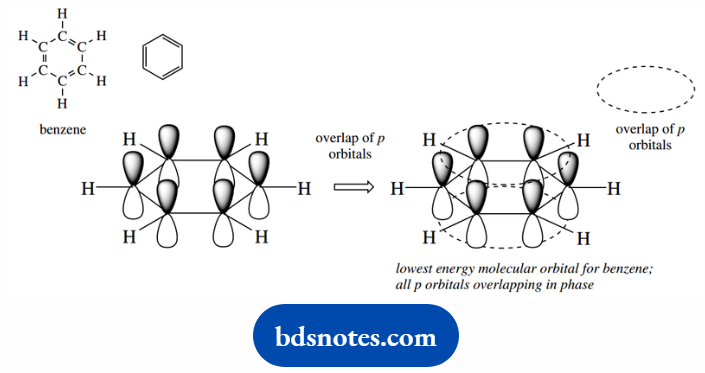
This is a bonding π molecular orbital in a conjugated system extending over all six atoms. The electrons will be distributed evenly, or delocalized, over the whole molecule.
The six p atomic orbitals combine to give six molecular orbitals for the π system. The relative energies for these are shown.
There is one low-energy bonding molecular orbital and two degenerate bonding orbitals at higher energy. There will be an analogous array of antibonding orbitals at higher energy.
The six electrons are assigned to these orbitals as we have seen previously, beginning with the lowest energy level.
This leads to the six electrons completely filling the bonding molecular orbitals and providing an extremely favorable arrangement.
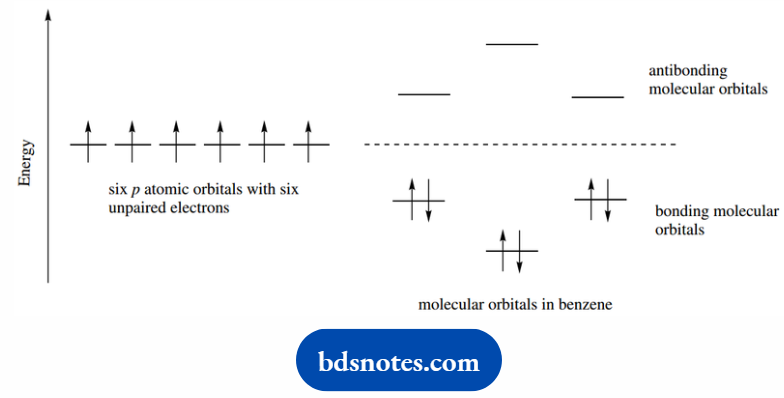
In that, the overall energy is significantly below that of six electrons in the contributing p atomic orbitals.
The energy stabilization is considerable, and also much more than could be accounted for by simple conjugation. The special stability afforded by this planar cyclic array is what we understand by aromaticity.
The chemical reactivity associated with aromatic systems will be covered.
Cyclooctatetraene
Let us consider the origins of benzene’s aromatic stabilization. Another cyclic hydrocarbon, cyclooctatetraene (pronounced cyclo-octa-tetra-ene), certainly looks conjugated according to our criteria.
But chemical evidence shows that it is very much more reactive than benzene, and does not undergo the same types of reaction. It does not possess the enhanced aromatic stability characteristic of benzene.
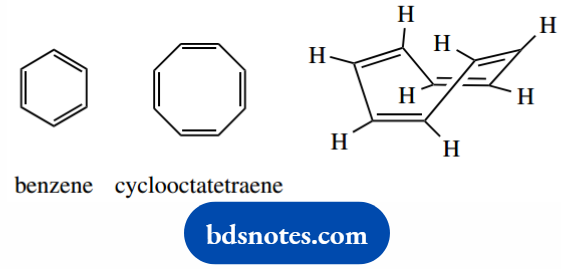
Further, cyclooctatetraene has been shown to be non-planar; it adopts a tub shape. This originates from bond angles.
A regular octagon has internal bond angles of 135°, quite far from the optimum angle of 120° for sp2 hybridization. In benzene’s hexagon, the internal angle is 120°, a perfect fit for sp2 geometry.
Cyclooctatetraene thus distorts from the planar to relieve this strain. A careful consideration of this shape may then suggest the immediate consequences.
This are because none of the double bonds are in the same plane; therefore, there is going to be no overlap of p orbitals between the double bonds.
We cannot get any enhanced stability associated with conjugation.
Huckel’s Rule
Cyclooctatetraene has eight π electrons and benzene has six. The number of π electrons that confer aromaticity is given by Huckel’s rule:
A planar cyclic conjugated system will be particularly stable if the number of π electrons is 4n + 2, where n is an integer (0, 1, 2, 3, etc).
Although the significance of this will not become apparent until later (see below), we must stress that 4n + 2 refers to the number of π electrons and not the number of atoms in the ring.
Benzene, therefore, with six π electrons (n = 1, 4n + 2 = 6), is aromatic; however, cyclooctatetraene, with eight π electrons, is not aromatic.
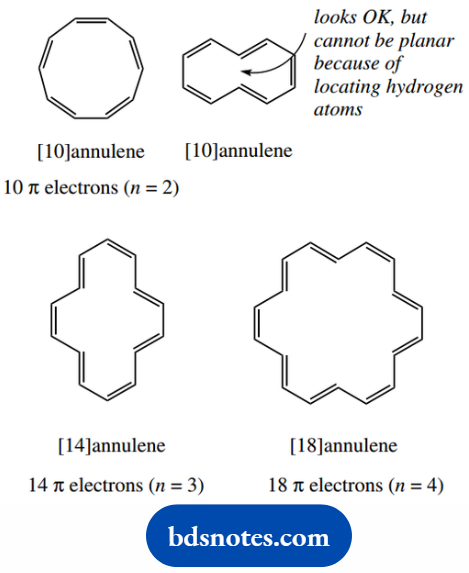
Also aromatic would be a system with 10 π electrons (n = 2), or 14 π electrons (n = 3). The first of these would be the compound [10]annulene, and the second [14]annulene.
Annulene is a general term for a carbon ring system with alternating single and double bonds; the number in brackets is the number of carbons in the ring.
For example, we could call benzene [6]annulene, though in practice, nobody ever does.
Whereas [14]annulene shows aromatic properties, [10]annulene, unfortunately, does not, but we know this is a consequence of the molecule adopting a non-planar shape.
The interior angle for a planar 10-carbon system would have to be 144°, and this is too far removed from the sp2-hybridized angle of 120° to be feasible.
As ring sizes get larger, it becomes possible to have a cyclic system where all bond angles can be the ideal 120°.
There is a way of drawing a 10-carbon ring system with angles of 120°, but we must realize that this attempts to place two hydrogens in the same space.
This is clearly not feasible; as the hydrogens are pushed away from each other, therefore, this must lead to a non-planar molecule.
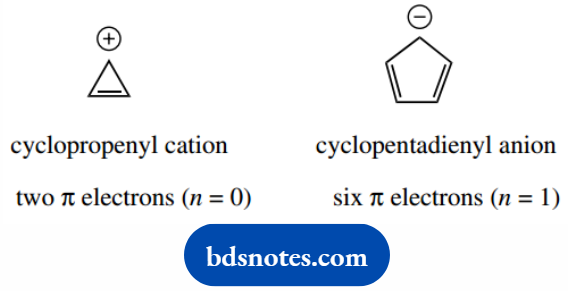
Structures that are also aromatic are the cyclopropenyl cation (2 π electrons; n = 0) and the cyclopentadienyl anion (6 π electrons; n = 1).
Although we do not wish to pursue these examples further, they are representative of systems where the number of π electrons is not the same as the number of carbon atoms in the ring.
The stabilization conferred by aromaticity results primarily from the much lower energy associated with a set of electrons in molecular orbitals compared with them being in atomic orbitals.
We have seen how this originates in benzene by allocating electrons to the bonding orbitals.
We can apply the same procedure to other annulene compounds, and there exists a very neat way of finding the relative energies of molecular orbitals without resources for mathematical calculations.
This device, the Frost circle, inscribes the appropriate polygon in a circle, with one vertex pointing vertically downwards.
The intersections of other vertices with the circle then mark the positions of the molecular orbitals.
The position of the horizontal diameter represents the energy of the carbon p orbital; intersections below this are bonding.
Those above are antibonding, and nonbonding orbitals are on the diameter line. Frost circles for benzene and cyclooctatetraene are drawn.
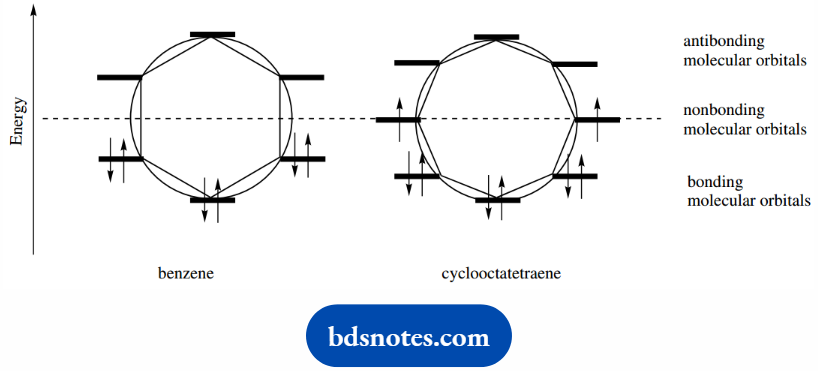
We can immediately see that allocating six electrons into the benzene molecular orbitals fills all three bonding orbitals (a closed shell structure) and there is substantial aromatic stabilization.
In that, the energy associated with electrons in the molecular orbitals is greatly reduced compared with that of electrons in the six atomic orbitals.
For cyclooctatetraene, allocating eight electrons to the molecular orbitals leads to three filled orbitals.
But then the remaining two electrons are put singly into each of the degenerate nonbonding orbitals.
Cyclooctatetraene does not have a filled shell structure like benzene, but has two nonbonding electrons; it does not have the special stability we see in benzene.
As we have seen, cyclooctatetraene also adopts a non-planar shape, lacks the stabilization associated with conjugation, and behaves like four separate normal alkenes.
Kekule Structures
Benzene is usually drawn as a structure with alternating single and double bonds. We can draw it in two ways.
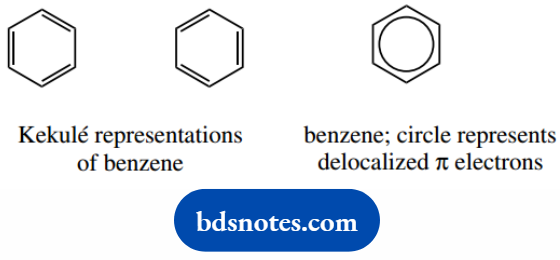
These two forms are so-called Kekule structures; but neither is correct, in that benzene does not have single and double bonds. This immediately follows from a measurement of C–C bond lengths.
For sp2– hybridized carbons, we expect the C=C bonds to be about 1.34 Å, whereas the C–C bond length would be about 1.47 Å.
Measurements show that all˚ of the carbon-carbon bond lengths are the same, at 1.40 Å. This length is between that of single and double bonds.
And suggests that we have C–C bonds that are somewhat between single and double bonds in character. From the point of stability.
And now also bond lengths, we must view benzene as quite different from cyclohexatriene. To emphasize this, a different representation for the benzene ring has been proposed, i.e. a circle within a hexagon.
The circle represents the six π-electron system, and this, therefore, highlights the special nature of the aromatic ring.
As we shall see in due course, this representation has considerable limitations, and most chemists, ourselves included, do not use it.
Aromaticity And Ring Currents
One can demonstrate the particular stability of aromatic compounds by their characteristic chemical reactions.
For example, benzene reacts with bromine only with difficulty and gives bromobenzene, a substitution product.
This leaves the aromatic ring intact. By contrast, a typical alkene reacts readily with bromine by an addition process to give a dibromo product.
This reaction destroys the π bond. When it comes to compounds such as annulenes, it is not always easy to synthesize sufficient material to demonstrate typical chemical reactivity.
And a simple spectroscopic analysis for aromaticity is infinitely preferable. Nuclear magnetic resonance (NMR) spectroscopy has provided such a probe.
The proton NMR signals for hydrogens on a double bond are found in the region δ 5–6 ppm. In contrast, those in benzene are detected at δ 7.27 ppm.
This substantial difference is ascribed to the presence of a ring current in benzene and other aromatic compounds.
A ring current is the result of circulating electrons in the π system of the aromatic compound. Without entering into any discussion on the origins of NMR signals.
The ring current creates its own magnetic field that opposes the applied magnetic field, and this affects the chemical shift of protons bonded to the periphery of the ring.
Signals are shifted downfield (greater δ) relative to protons in alkenes. Proton NMR spectroscopy can, therefore, be used as a test for aromaticity.
In this way, [14]annulene and [18]annulene have been confirmed as aromatic.
Aromatic Heterocycles
In due course, we shall see that unsaturated cyclic compounds contain atoms other than carbon, For Example. nitrogen, oxygen, or sulfur, can also be aromatic.
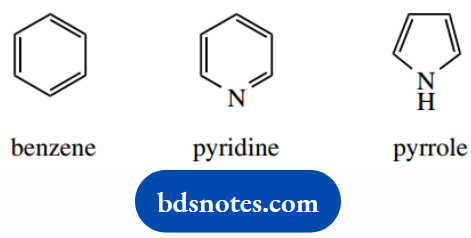
For example, pyridine can be viewed as a benzene ring in which one CH has been replaced by a nitrogen. It is aromatic and, like benzene, displays enhanced stability.
Pyrrole is a five-membered heterocycle but also displays aromaticity. Like the cyclopentadienyl anion, the number of π electrons is not the same as the number of atoms.
In pyrrole, nitrogen provides two of the six π electrons. Examples of these molecules are discussed under heterocycles.
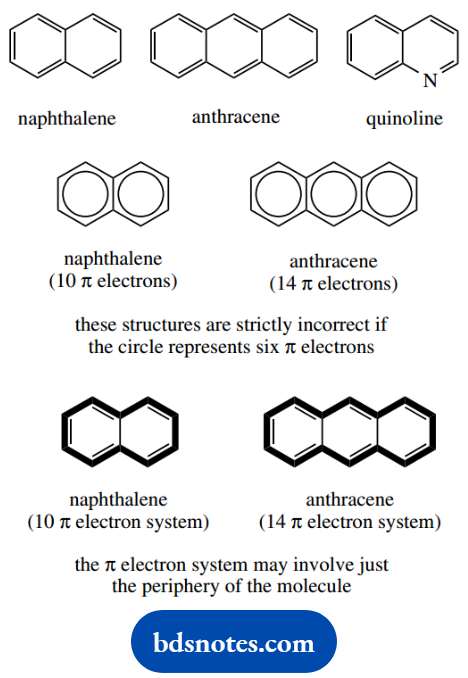
We may also encounter aromatic hydrocarbons that feature fused rings. Thus, naphthalene is effectively two benzene rings fused together, and anthracene has three fused rings.
The heterocycle quinoline is a fusion of benzene and pyridine. These ring systems are undoubtedly aromatic, and they display the enhanced stability and reactivity associated with simple aromatic compounds like benzene.
Molecular orbital calculations suggest that the π electrons in naphthalene are delocalized over the two rings and this results in substantial stabilization.
These molecules are planar, and all p orbitals are suitably aligned for overlap to form π bonding molecular orbitals.
Although we can draw Kekule´s structures for these compounds, it is strictly incorrect to use the circle in hexagon notation since the circle represents six π electrons.
Naphthalene has 10 carbons, and therefore 10 π electrons and anthracene has 14 π electrons. The circle notation suggests 12 or 18 π electrons.
Note that Huckel’s rule applies only to monocyclic compounds, although 10 π electrons (naphthalene) and 14 π electrons (anthracene) seem to meet the criteria for aromaticity.
There is good evidence to suggest we should consider the aromatic system not as a combination of benzene rings, but as a single ring involving the periphery of the molecule.
Resonance Structures And Curly Arrows
The molecular orbital picture of benzene proposes that the six π electrons are no longer associated with particular bonds.
But are effectively delocalized over the whole molecule, spread out via orbitals that span all six carbons.
This picture allows us to appreciate the enhanced stability of an aromatic ring, and also, in due course, to understand the reactivity of aromatic systems.
There is an alternative approach based on Lewis structures that is also of particular value in helping us to understand chemical behavior.
Because this method is simple and easy to apply, it is an approach we shall use frequently. This approach is based on what we term resonance structures.
Let us go back to the two Kekule representations´ for benzene. The Lewis structure for benzene has alternating single and double bonds, but there are two ways of writing this.
In one form, a particular bond is single; in the other form, this bond has become double.
Resonance theory suggests that these two structures are both valid representations and that each contributes to the structure of benzene.
But the true structure is something in between, a lower energy hybrid of the two Kekule forms, a resonance hybrid.
If this is the case, then each bond is neither single nor double, but, again, something in between.
As we have already seen, all C–C bond lengths in benzene are 1.40 Å, which is in between the bond lengths for single (1.47 Å) and double (1.34 Å) bonds.
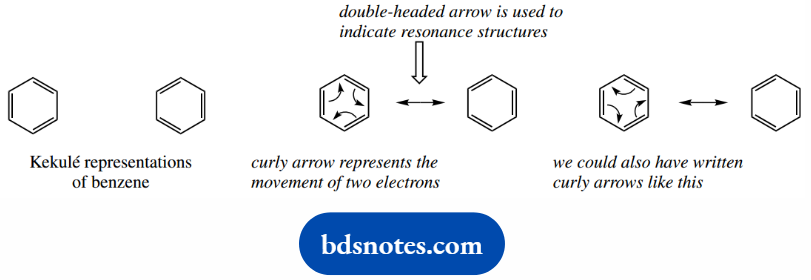
To indicate resonance forms, we use a double-headed arrow between the contributing structures. This arrow is reserved for resonance structures and is never used elsewhere.
The difference between the two structures is that the electrons in the π bonds have been redistributed, and we can illustrate this by use of another type of arrow, a curly arrow.
This arrow is used throughout chemistry to represent the movement of two electrons.
In the benzene case, a cyclic movement of electrons accounts for the apparent relocation of double bonds, though there are two ways we might show this process; both are equally satisfactory.
Benzene is a nice example to choose to illustrate the concept of resonance. It is not the best example for explaining the rules governing the use of curly arrows, so we must move to some simpler compounds.
Being able to draw curly arrows is an essential skill for an organic chemist, and you will see from a cursory glance at the following chapters just how frequently they are employed.
We shall use the same curly arrows and precisely the same principles for predicting the outcome of chemical reactions.
They allow us to follow bond-making and bond-breaking processes, and provide us with a device we can use to keep track of the electrons.
- The curly arrow represents the movement of two electrons.
- The tail of the arrow indicates where the electrons are coming from, and the arrowhead where they are going.
- Curly arrows must start from an electron-rich species. This can be a negative charge, a lone pair, or a bond.
- Arrowheads must be directed towards an electron-deficient species. This can be a positive charge, the positive end of a polarized bond, or a suitable atom capable of accepting electrons, i.e. an electronegative atom.
In our brief introduction to Lewis structures, we paid particular attention to valency, the number of bonds an atom could make to other atoms via the sharing of electrons.
We must now broaden this idea to consider atoms in a molecule that are no longer neutral, but which carry a formal positive or negative charge.
This means we are considering cations and anions, as in ionic bonding, but the atom involved is still part of a molecule, and the molecule consequently also carries a formal charge.
We have already met a few such entities in this chapter, For Example. the ammonium and hydronium cations, looking specifically at the molecular orbital descriptions.
As indicated above, the use of curly arrows may involve species with positive or negative charges.
As simple examples, ammonia and water are neutral molecules. Nitrogen has five valence electrons, and it acquires a stable octet of electrons in making three bonds to hydrogen atoms.
Each hydrogen has its stable arrangement of two electrons. The nitrogen in ammonia also carries a lone pair of electrons.
Oxygen, with six valence electrons, makes two bonds to hydrogen atoms. Its octet of electrons will carry two lone pairs.
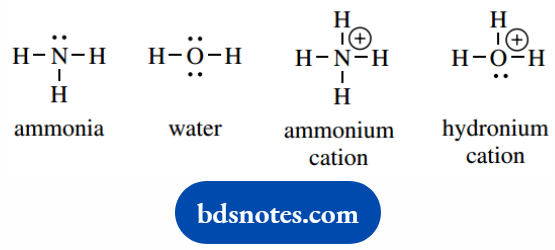
We can deduce the charge associated with ammonia and water by simply considering that the component atoms are neutral, and that all we have done is share the electrons, so the molecules must also be neutral.
The formal charge on an individual atom can be assessed more rigorously by subtracting the number of valence electrons assigned to an atom in its bonded state from the number of valence electrons it has as a neutral-free atom.
Electrons in bonds are considered as shared equally between the atoms, whereas unshared lone pairs are assigned to the atom that possesses them.
Formal charge = Number of valence electrons as neutral free atom – Number of valence electrons assigned in bounded state
Hence, for nitrogen, the number of valence electrons in a free atom is five. In ammonia, the number of assigned electrons is also five (three in bonds plus a lone pair).
Therefore, the formal charge on nitrogen is zero. For hydrogen, the formal charge is also zero, since the number of valence electrons is one, and the number of assigned electrons is one.
For oxygen, the number of valence electrons in a free atom is six. In water, the number of assigned electrons is also six (two in bonds plus two lone pairs).
Therefore, the formal charge on oxygen is zero. The hydrogens are also uncharged, as in ammonia.
Now consider the ammonium and hydronium cations. In the ammonium system, for nitrogen, the formal charge is now +1.
This follows from the number of valence electrons, i.e. five, minus the number of assigned electrons, i.e. four (four in bonds).
In the hydronium system, the formal charge on oxygen is also +1. This is assessed from the number of valence electrons, i.e. six, minus the number of assigned electrons, i.e. five (three in bonds plus a lone pair).
Of course, we already knew that ammonium and hydronium cations were the result of bonding neutral ammonia or water with a proton (charge +1), so an overall charge of +1 comes as no particular surprise.
Other systems are less familiar, and will therefore have to be assessed carefully. For example, what charge is associated with the structure shown on the left below?
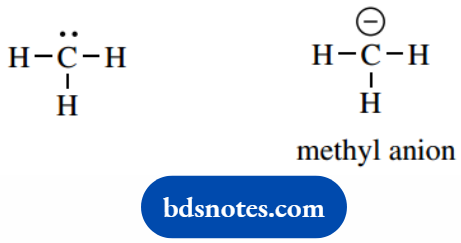
This is the methyl anion and carries one negative charge. Carbon has four valence electrons, and in this structure, the number of assigned electrons is five (three bonds plus a lone pair).
Therefore, the formal charge on carbon is 4 − 5 = −1. We must always indicate the charge in structures pictured as shown in the right-hand representation.
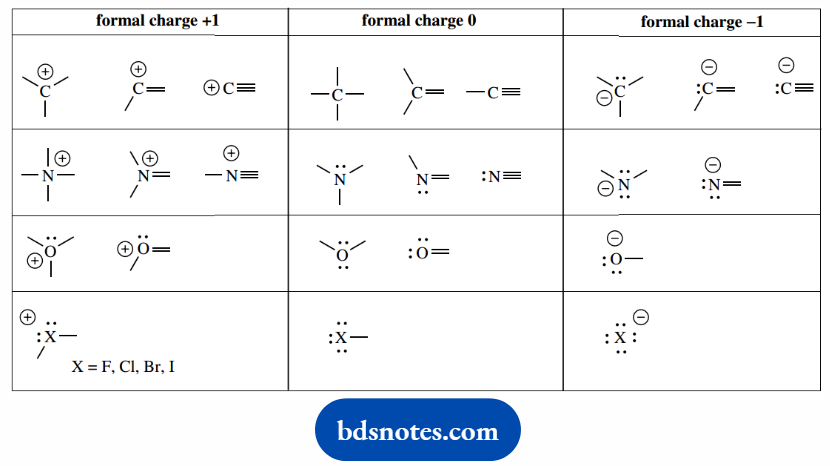
The left-hand representation is incomplete and, therefore, wrong. The most common formal charges we shall meet are summarized.
Now let us return to curly arrows and resonance structures.
- Resonance structures differ only in the position of the electrons; the positions of the atoms do not change.
- Resonance structures can be interconverted by the movement of electrons indicated by curly arrows.
- Three main types of electron movement can be implicated:
Bonding To Nonbonding
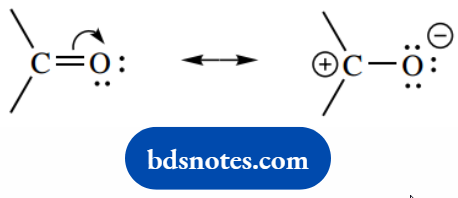
Nonbonding To Bonding
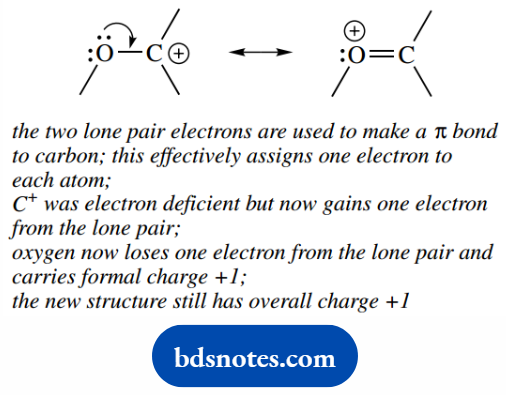
Bonding To New Bonding
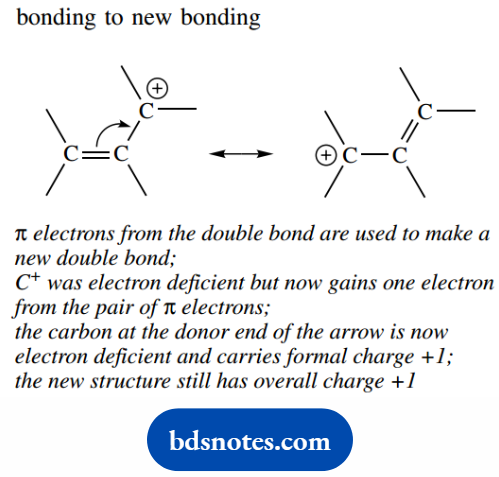
- All structures must be valid Lewis structures. An atom may become electron deficient, but, on the other hand.
- It must never be shown with more valence electrons than it can accommodate. For example, it is not possible to have pentavalent carbon.
- The overall charge must remain the same.
- There should be the same number of unpaired electrons in each structure.
This redistribution of electrons provides us with one or more new resonance structures (also called canonical structures, limiting structures, or monomers). However, some structures are more realistic than others.
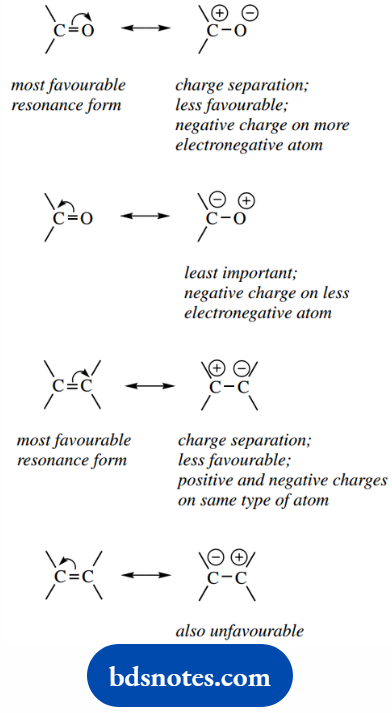
- The more covalent bonds a structure has, the more stable it is.
- A structure in which all the atoms have the noble gas structure is particularly stable.
- Separation of charge in a resonance structure decreases stability.
- Structures with charge separation are more stable if the negative charge is located on an electronegative atom.
- Structures with adjacent-like charges are disfavoured, as are those with multiple isolated charges.
- The σ bond framework and steric factors must permit a planar relationship between contributory resonance structures.
This is illustrated for a carbonyl compound and an alkene.
We then consider the potential relative importance of the resonance structures we have drawn.
- Equivalent resonance structures contribute equally to the hybrid.
- Structures that are not equivalent do not contribute equally; the more stable a structure is, the more it is likely to contribute.
- Highly unstable structures make little contribution and may be ignored.
Acceptable resonance structures can then be imagined as contributing to the overall electronic distribution in the molecule.
By considering the properties of contributing structures, we can also predict some of the properties of the molecule.
We imagine that the molecule is not fully represented by a single structure, but is better represented as a hybrid of its contributing resonance forms.
It is likely that the energy associated with the molecule is actually lower than that of any contributing resonance form.
Therefore, the delocalization of electrons that resonance represents is a stabilizing feature.
The larger the number of stable resonance structures we can draw, the greater the extent of delocalization.
The difference in energy between the actual molecule and that suggested by the best of the resonance structures is termed the resonance energy or resonance stabilization energy. This can usually only be an estimated amount.
The resonance terminology and the double-headed arrow may give the impression that the structures are rapidly interconverting. This is not true.
We must appreciate right from the start that resonance structures are entirely hypothetical.
They are our (sometimes clumsy) attempt to write down on paper what the bonding in the molecule might be like, and they may depict only the extreme possibilities.
The molecule is presumably happily going about its business in a form that we cannot easily depict. Nevertheless, resonance structures are extremely useful and do help us to explain chemical behavior.
Let us look again at the simple examples shown above and the consequences of our hypothetical resonance structures.
We shall see that most of the reactions of simple carbonyl compounds, like formaldehyde, are a consequence of the presence of an electron-deficient carbon atom.
This is accounted for in resonance theory by a contribution from the resonance structure with charge separation.
The second example shows the so-called conjugate acid of acetone, formed to some extent by treating acetone with acid.
Protonation in this way typically activates acetone towards reaction, and we find that electron-rich reagents (nucleophiles) attack the carbon atom.
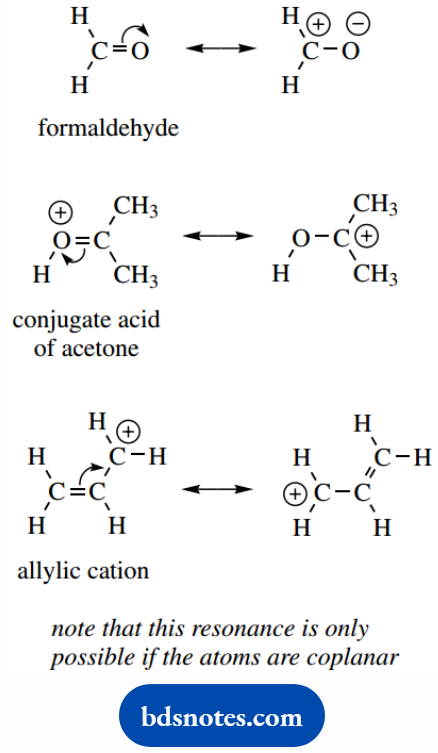
This is reasonable since we can show this carbon as positively charged in the right-hand resonance structure. The third example is the allylic cation.
This is a reasonably stable carbocation, and we attribute this to resonance stabilization; this is particularly favorable in this case since both contributing resonance forms are identical.
We can visualize the allylic cation as an entity in which the positive charge is delocalized over the whole structure (strictly, it is the electrons that are delocalized.
But we are one short of a full complement and it is the positive charge that dominates the representation).
Hydrogen Bonding
Hydrogen bonds (H-bonds) describe the weak attraction of a hydrogen atom bonded to an electronegative atom, such as oxygen or nitrogen, to the lone pair electrons of another electronegative atom.
These bonds are different in nature from the covalent bonds we have described; they are considerably weaker than covalent bonds, but turn out to be surprisingly important in chemistry and biochemistry.
Let us consider a molecule possessing an O–H σ bond. This bond is polar because hydrogen is less electronegative than oxygen.
And this allows the partially positive hydrogen atom to associate with a center in another molecule carrying a partial negative charge. This is likely to be the oxygen atom in another molecule.
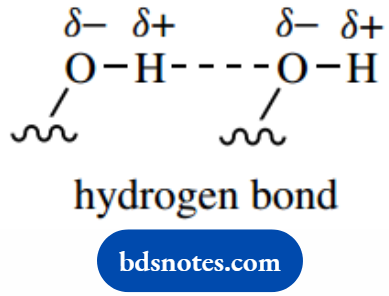
If we consider the interaction between two water molecules, then the partial positive charge on hydrogen induced by the electronegativity of oxygen is attracted to the high electron density of the oxygen lone pair in another molecule.
This hydrogen is now linked to its original oxygen by a σ bond, and to another oxygen by an electrostatic attraction.
The bond length of the H–O hydrogen bond is typically about twice the length of the H–O covalent bond.
The hydrogen bond is very much weaker than the covalent bond, though stronger than other interactions between molecules.
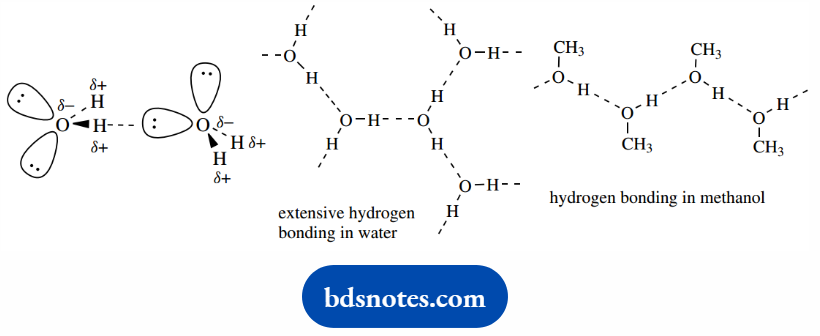
In water, further hydrogen bonds involving other molecules are formed, leading to a network throughout the entire sample.
The extensive hydrogen bonding in water is responsible for some of water’s unusual properties, its relatively high boiling point.
And its high polarity makes it a particularly good solvent for ionic compounds. Alcohols also exhibit hydrogen bonding; but, with only a single O–H, the network of bonds cannot be as extensive as in water.
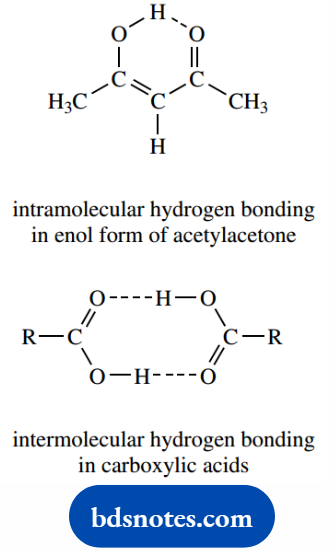
Hydrogen bonds connecting different molecules are termed intermolecular, and when they connect groups within the same molecule they are called intramolecular.
A simple example of an intramolecular hydrogen bond is seen in the enol form of acetylacetone.
Carboxylic acids are known to form hydrogen-bonded ‘dimers’ in solution through two quite strong intermolecular hydrogen bonds.
Hydrogen bonds involving N–H are also common, and we can also meet hydrogen bonding between alcohols and amines.
Hydrogen Bonds And DNA
The nucleic acids known as deoxyribonucleic acid (DNA) are the molecules that store genetic information. This information is carried as a sequence of bases in the polymeric molecule.
Remarkably, the interpretation of this sequence depends upon simple hydrogen bonding interactions between base pairs.
Hydrogen bonding is fundamental to the double helix arrangement of the DNA molecule, and the translation and transcription via ribonucleic acid (RNA) of the genetic information present in the DNA molecule.
In DNA, the base pairs are adenine–thymine and guanine–cytosine. Adenine and guanine are purine bases, and thymine and cytosine are pyrimidines.
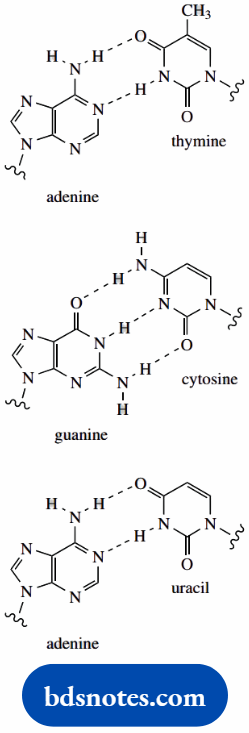
Thus, each purine residue is specifically linked by hydrogen bonding to a pyrimidine residue. This may involve either two or three hydrogen bonds, with hydrogen of N–H groups bonding to oxygen or to nitrogen.
The result of these interactions is that each base can hydrogen bond only with its complementary partner. The specific base pairing means that the two strands in the DNA double helix are complementary.
Wherever adenine appears in one strand, thymine appears opposite it in the other; wherever cytosine appears in one strand, guanine appears opposite it in the other.
Another pyrimidine base, uracil, is found in RNA instead of thymine. Base pairing between adenine and uracil involves two hydrogen bonds and resembles the adenine–thymine interaction.
This type of base pairing is of importance in transcription, the synthesis of messenger RNA.
Molecular Models
We soon come to realize that molecules are not two-dimensional objects as we draw on paper.
They are three-dimensional and their overall size and shape can have a profound effect on some of their properties, especially biological properties.
We have seen that four single bonds to carbon are distributed in a tetrahedral array, an arrangement that minimizes any steric or electrostatic interactions.
Atoms around double bonds are in a planar array, and angles are 120◦. Again, this trigonal arrangement minimizes interactions. A triple bond creates a linear array of atoms.
Now, careful measurements of bond angles and also bond lengths in a wide variety of molecules have convinced us that these features are sufficiently constant that we can use them to predict the shape and size of other molecules.
Bond lengths in molecules usually correlate with one of five kinds, and their typical measurements are shown. The five types of bonds are:
- Single bonds between atoms, one of which is hydrogen;
- Single bonds between atoms, neither of which is hydrogen;
- Double bonds;
- Triple bonds;
- Bonds in aromatic rings.
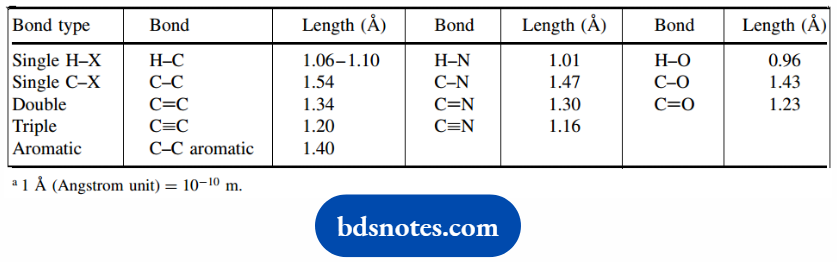
Bond angles can be related to hybridization, and so can bond lengths.
Thus, electrons in sp hybrid orbitals are held closer to the nucleus than electrons in sp2 orbitals, which are correspondingly closer than electrons in sp3 orbitals.
The bond lengths below follow this generalization. The shortest bond lengths involve bonds to hydrogen, the smallest atom that utilizes an s orbital in bonding.
Note also that aromatic carbon-carbon bonds have a bond length between that of single and double bonds, a feature of aromatic bonding.
With these five typical bond lengths, and the typical bond angles for tetrahedral, trigonal, and linear arrays, it becomes possible to construct molecular models to predict a molecule’s size and shape.
This may be achieved via a molecular model kit, or by computer graphics.
Many different molecular model kits have been produced over the years, each varying in their approach to atoms and bonds, and also in their cost.
However, there are three main types, which can provide us with three main types of information. These are the framework, ball-and-stick, and space-filling versions.
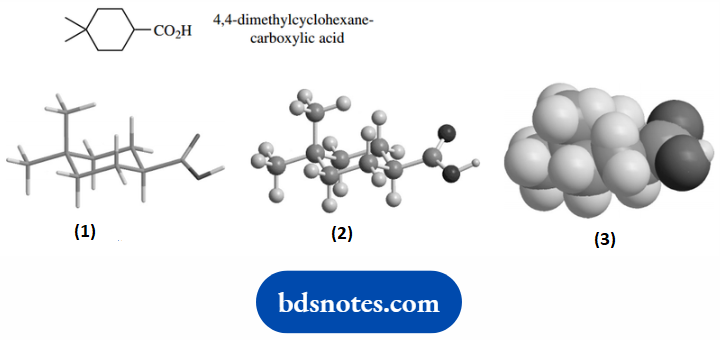
Framework models concentrate on the bonds in the molecule. In the least expensive kits, these are represented by narrow tubes, joined by linker pieces that signify the atom position.
The linkers have four, three, or two stubs to fit into the tubes, according to the number of bonds required, and these are also arranged at appropriate angles (tetrahedral 109◦, trigonal 120°, linear 180°).
The linkers are also colored differently to show the atom they represent, typically black for carbon, white for hydrogen, red for oxygen, and blue for nitrogen.
The resultant model tells us about the bonding, and the overall size and shape of the molecule, but gives us little indication about the volume taken up by the atoms and electrons.
Nevertheless, this type of model is probably the most popular, and it provides a lot of information. It is the three-dimensional equivalent of our two-dimensional line drawings of structures.
In ball-and-stick models, balls with holes drilled at appropriate angles are used to represent the atoms, and sticks or springs are used for the bonds to link them.
The resultant model is rather like the framework model, but has representations of the atoms. Models tend to look better than the framework type, but in practice tend to be bigger and less user friendly.
Space-filling model kits are even less user friendly. They employ specially shaped atomic pieces that clip together, each representing the volume taken up by the atom and its bonding electrons.
This system produces a rather globular model that indicates the whole bulk of the molecule, including the electron clouds that are involved in bonding.
The value of this type of model is that it shows just how big the molecule really is, and not just the atoms and or bonds.
When one is faced with interpreting how a molecule might interact with, say, a receptor protein, then space-filling models are essential.
There is a downside, however, because it becomes very difficult indeed to visualize the structure and bonding in the molecule.
These three types of models are illustrated, though we have employed computer graphics to generate these pictures.
Significantly, computer modeling programs allow the generation of images according to the three main types of models; each provides us with subtly different information, so they are not alternatives but are actually complementary.
Whilst many people still like to handle and view a model, computer programs have allowed us to create and manipulate representations of three-dimensional molecules rapidly with varying degrees of accuracy and sophistication.
We can easily view the image from any angle, and can see the effect that any molecular modifications might have.
Further, although interactions of groups in a molecule can lead to changes in bond angles, and to a lesser extent in bond lengths, ordinary models may not show this.
Computer graphics programs are able to carry out quite sophisticated energy calculations to show the most favorable arrangement of atoms, the interatomic distances, and the bond angles.
Leave a Reply