Acids And Bases Acid-Base Equilibria
A fundamental concept in chemistry is associated with proton loss and gain, i.e. acidity and basicity.
Acids produce positively charged hydrogen ions H+ (protons) in aqueous solution; the more acidic a compound is, the greater the concentration of protons it produces.
In water, protons do not have an independent existence but become strongly attached to a water molecule to give the stable hydronium ion H3O+. In the Bronsted–Lowry definition
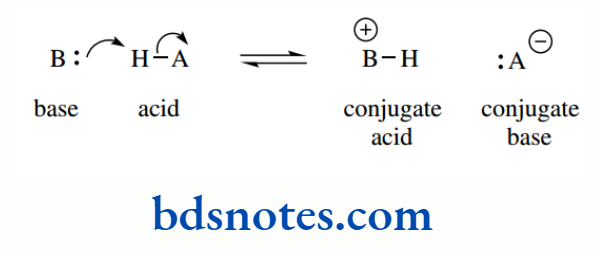
- An acid is a substance that will donate a proton;
- A base is a substance that will accept a proton.
Thus, the acid HCl ionizes in water to produce H3O+ and Cl− ions.
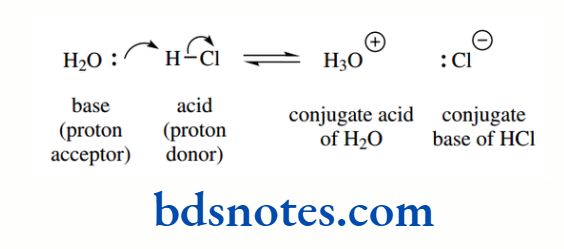
H3O+ is termed the conjugate acid (of the base H2O) and Cl− is termed the conjugate base (of the acid HCl).
In general terms, cleavage of the H–A bond in an acid HA is brought about by a base, generating the conjugate acid of the base, together with the conjugate base of the acid. You may wish to read that sentence again!
The Lewis definition of acids and bases is rather more general than the Brønsted–Lowry version (which refers to systems involving proton transfer) in that:
An acid is an electron-pair acceptor;
A base is an electron-pair donor.
Thus, Lewis acids include such species as boron trifluoride, which can react with trimethylamine to form a salt.
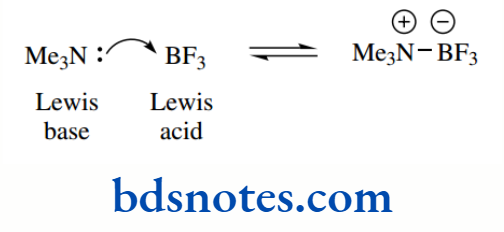
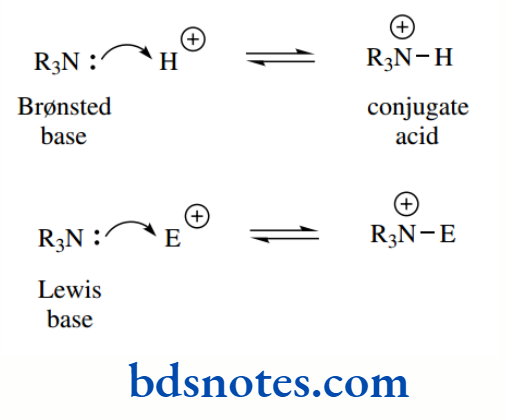
There is no fundamental difference between trimethylamine acting as a Brønsted base or as a Lewis base, except that in the Bronsted concept, it donates its electrons to a proton electrophile, whereas as a Lewis base, it donates its electrons to a Lewis acid electrophile.
Acidity And pKa Values
For the ionization of the acid HA in water
⇒ \(\mathrm{H}_2 \mathrm{O}+\mathrm{HA} \stackrel{K}{\rightleftharpoons} \mathrm{H}_3 \mathrm{O}^{\oplus}+\mathrm{A}^{\ominus}\) the equilibrium constant K is given by the formula
⇒ \(K=\frac{\left[\mathrm{A}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{[\mathrm{HA}]\left[\mathrm{H}_2 \mathrm{O}\right]}\)
where [HA] signifies the concentration of HA, etc. However, because the concentration of water is essentially constant in aqueous solution, a new equilibrium constant Ka is defined as
⇒ \(K_{\mathrm{a}}=\frac{\left[\mathrm{A}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{[\mathrm{HA}]}\)
K a is termed the acidity constant, and its magnitude allows us to classify acids as strong acids (a large value for Ka and, consequently, a high H3O+ concentration) or weak acids (a small value for Ka and, thus, a low H3O+ concentration). For example, the strong acid HCl has Ka = 107.
However, for weak acids, the amount of ionization is much less and, consequently, the value of Ka is rather small. Thus, acetic acid CH3CO2H has Ka = 1.76 × 10−5. To avoid using such small numbers as these, Ka is usually expressed in the logarithmic form p Ka where pKa = − log10 Ka Accordingly, the pKa for acetic acid is 4.75:
⇒ \(K_{\mathrm{a}}=\frac{\left[\mathrm{A}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{[\mathrm{HA}]}\)
K a is termed the acidity constant, and its magnitude allows us to classify acids as strong acids (a large value for Ka and, consequently, a high H3O+ concentration) or weak acids (a small value for Ka and, thus, a low H3O+ concentration).
For example, the strong acid HCl has Ka = 107. However, for weak acids, the amount of ionization is much less and, consequently, the value of Ka is rather small. Thus, acetic acid CH3CO2H has Ka = 1.76 × 10−5. To avoid using such small numbers as these, Ka is usually expressed in the logarithmic form p Ka where pKa = − log10 Ka
Accordingly, the pKa for acetic acid is 4.75: pKa = − log(1.76 × 10−5) = −(−4.75) = 4.75
The pKa for hydrochloric acid can similarly be calculated to be −7: pKa = − log(107) = −7
This means there is an inverse relationship between the strength of an acid and pKa:
- A strong acid has a large Ka and, thus, a small pKa, i.e. A− is favoured over HA;
- A weak acid has a small K a and, thus, a large pKa, i.e. HA is favoured over A−
Or, put another way:
- The smaller the value of p Ka, the stronger the acid;
- The larger the value of p Ka, the weaker the acid.
We find that pKa values range from about −12 to 52, but it must be appreciated right from the start that a difference of one pKa unit represents a 10-fold difference in K a and, thus, a 10-fold difference in H3O+ concentration.
A twofold difference in acidity would be indicated by a pKa difference of just 0.3 units (log 2 = 0.3).
Accordingly, a difference of n pKa units indicates a 10n-fold difference in acidity, so the range −12 to 52 represents a huge factor of 1064. A compound with pKa < 5 is regarded as a reasonably strong acid, and those with pKa < 0 are very strong acids.
At first glance, negative pKa values seem rather strange, but this only means that the equilibrium lies heavily towards ionization; Ka is large and, therefore, pKa = − log Ka becomes negative.
⇒ \(\begin{gathered}
\mathrm{H}_2 \mathrm{O}+\mathrm{HA} \rightleftharpoons \mathrm{H}_3 \mathrm{O}+\mathrm{A}^{\ominus} \\
K_{\mathrm{a}}=\frac{\left[\mathrm{A}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{[\mathrm{HA}]} \quad \mathrm{p} K_{\mathrm{a}}=-\log _{10} K_{\mathrm{a}} \\
K_{\mathrm{a}}=0.01 \quad K_{\mathrm{a}}=0.1 \quad K_{\mathrm{a}}=1 \quad K_{\mathrm{a}}=10 \quad K_{\mathrm{a}}=100
\end{gathered}\)

As we use pKa values, we shall find that, in most cases, relative, rather than specific, values are all we need to consider to help us predict chemical behaviour and reactivity. Thus, from pKa values, we can see that acetic acid (pKa 4.75) is a weaker acid than hydrochloric acid (pKa − 7).
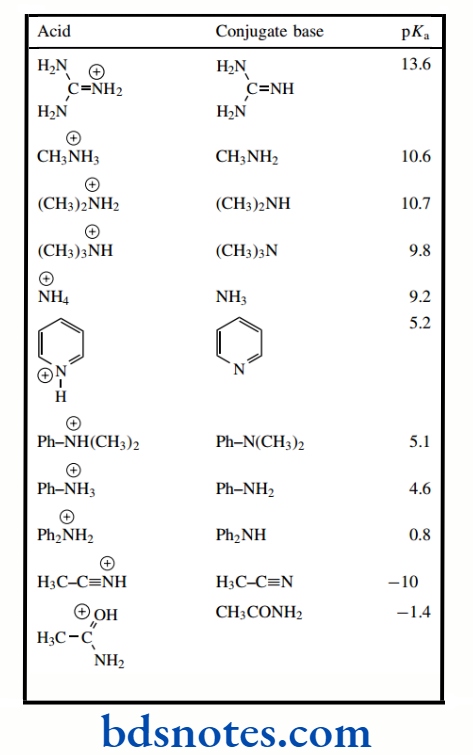
pKa values for a wide variety of different compounds. Compounds are listed in order of increasing acidity.
Although the pKa values included extend from about 52 to −10, values in the middle of the range are known most accurately.
This is because they can be measured readily in aqueous solution. Outside of the range from about 2 to 12, pKa values have to be determined in other solvents, or even by indirect methods; results are then extrapolated to give the value in water.
Have been intentionally rounded to stress that a high level of accuracy is usually inappropriate. The range of pKa values that can be measured in water is determined by the ionization of water itself, i.e. −1.74 (the pKa of H3O+) to 15.74 (the pKa of H2O); Acids that are stronger than H3O+ simply protonate water, whereas bases that are stronger than HO− remove protons from water.
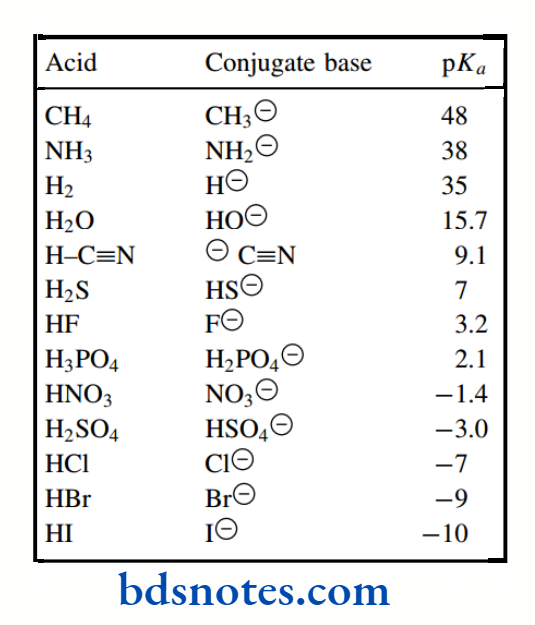
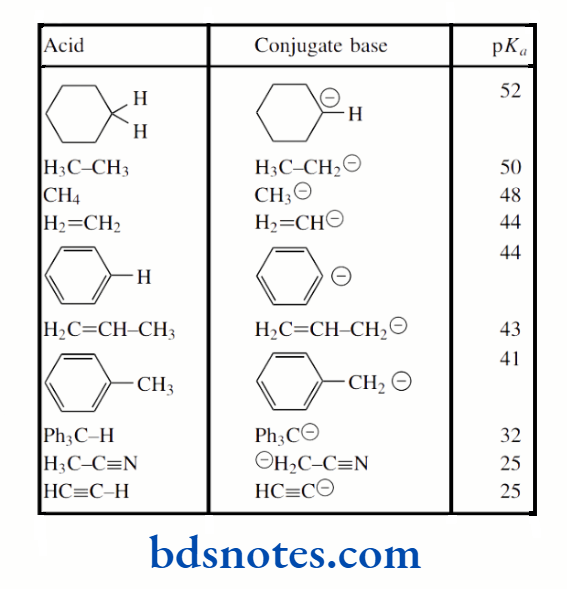
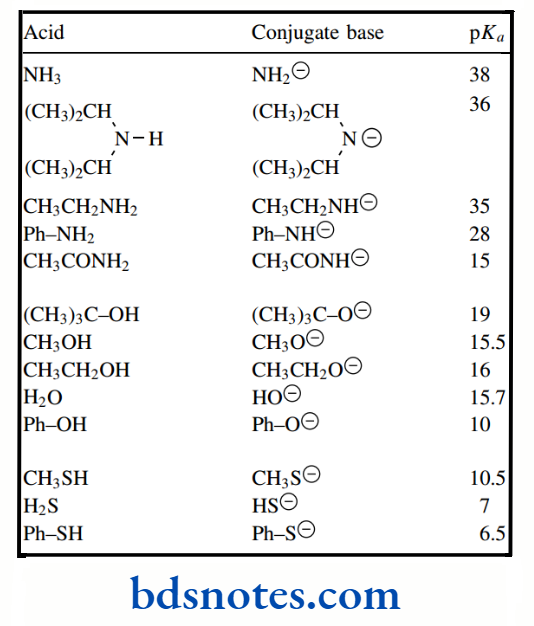
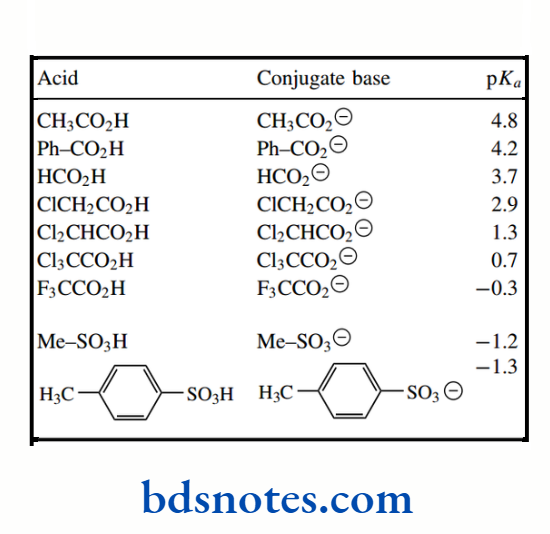
However, the fact that they do have to be measured means that as you look in the literature for the pKa of a particular compound you may find slightly different values can be presented.
Do not let this confuse you. As mentioned above, relative, rather than specific, values are our main concern. We have chosen to present the pKa values as a series of tables, rather than in a single one.
This should help you to locate a particular compound according to its functional group, and we hope that this will also emphasize similarities and differences in related structures.
It also means that you may find some examples turning up in more than one table. As we consider different aspects of chemical reactivity in subsequent chapters, we shall see how pKa values can be used to predict whether a reagent is a good or a poor nucleophile, whether it can function as a good leaving group, and how easy it is to generate anionic nucleophiles.
We shall also find that pKa values can tell us how much of a compound or a drug is ionized under particular conditions and, therefore, whether or not it can be produced in a soluble form.
It is now appropriate to consider some of the electronic and structural features that influence pKa so that we can rationalize and predict relative.
Electronic And Structural Features That Influence Acidity
Electronegativity
The more electronegative an element is, the more it helps to stabilize the negative charge of the conjugate base.
For example, the acidities of compounds of second-row elements in the periodic table increase as the atom to which hydrogen is attached becomes more electronegative:
pKa values for CH4, NH3, H2O and HF are about 48, 38, 16 and 3, respectively, i.e. we have increasing acidity left to right as the electronegativity of the atom attached to hydrogen increases.
Bond energies
Within a single column of the periodic table, acidities increase as one descends the column: pKa values for HF, HCl, HBr, and HI are about 3, −7, −9, and −10 respectively, i.e. we have increasing acidity on descending the group.
This is the reverse of what might be expected simply based on electronegativities but relates to the increasing size of the atom and the corresponding improved ability to disperse the negative charge over the atom.
We are seeing a weakening in bond strengths on descending the group. Similarly, although sulfur is less electronegative than oxygen, thiols (RSH) are more acidic than alcohols (ROH). For example, pKa values for methanethiol and methanol are 10.5 and 16 respectively.
Inductive effects
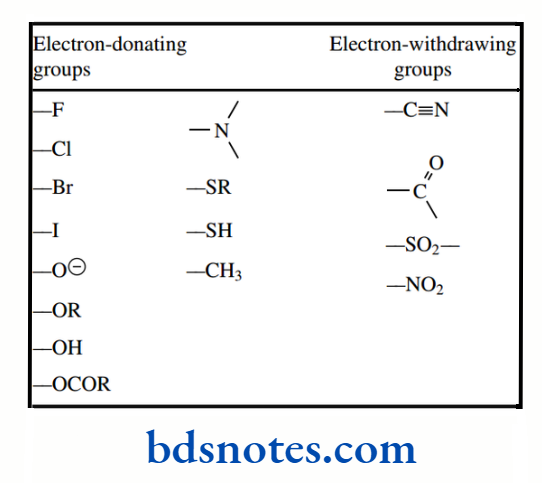
Electron-donating and electron-withdrawing groups influence acidity by respectively destabilizing or stabilizing the conjugate base.
his inductive effect, a charge polarization transmitted through σ bonds, causes a shift in electron density, and its influence may easily be predicted.
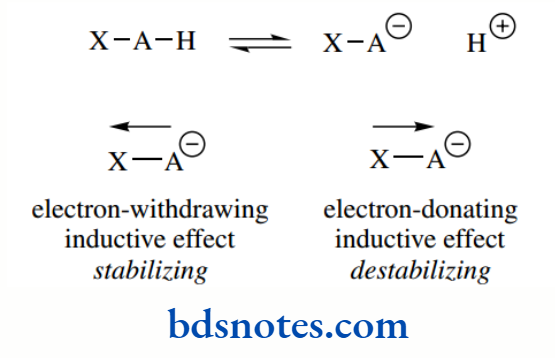
Thus, electron-withdrawing groups increase acidity:
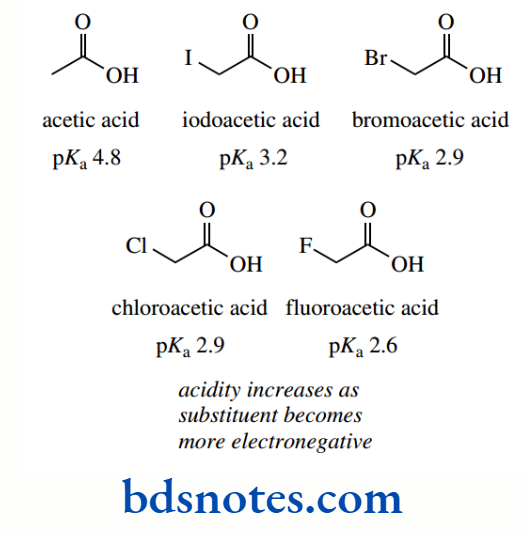
pKa values for the simple carboxylic acid acetic acid and its halogenated derivatives chloroacetic acid, dichloroacetic acid, and trichloroacetic acid are about 4.8, 2.9, 1.3, and 0.7 respectively, the inductive effects of the chlorine atoms spreading the charge of the conjugate base and thus stabilizing it.
Increasing the number of halogen atoms increases this effect, with a consequent increase in acidity. Note that the introduction of one chlorine atom increases acidity by a factor of almost 100, and trichloroacetic acid is a strong acid.
Because of the different electronegativities of the various halogens, we can also predict that fluorine will have a greater effect than chlorine, which in turn will increase acidity more than bromine or iodine.
This is reflected in the observed acidities of monohalogenated acetic acids, though the increased acidity of chloroacetic acid (pKa 2.87) over bromoacetic acid (pKa 2.90) is not apparent because of the rounding-up process.
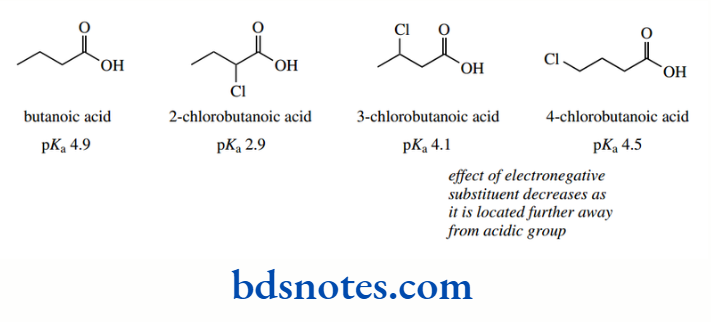
The inductive effect is a rather short-range effect, and its influence decreases rapidly as the substituent in question is located further away from the site of the negative charge because it has to be transmitted through more bonds.
Thus, the effect on the acidity in butanoic acid derivatives can be seen to diminish with distance. 2- Chlorobutanoic acid (pKa 2.9) shows a significant enhancement in acidity over butanoic acid (pKa 4.9), whereas 3-chlorobutanoic acid (pKa 4.1) and 4-chlorobutanoic acid (pKa 4.5) show rather more modest changes.
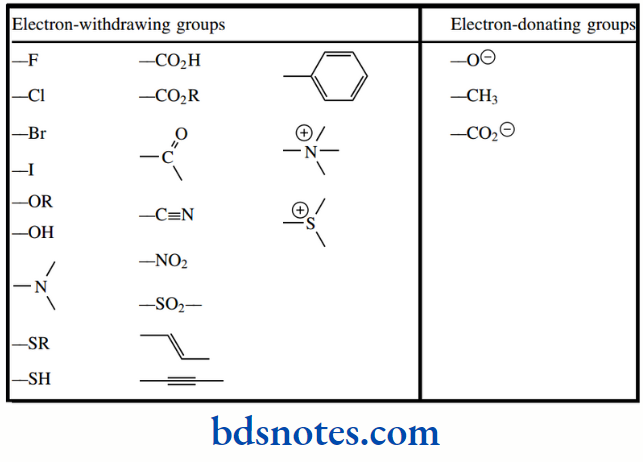
Other electron-withdrawing groups that increase the acidity of acids include, listed in decreasing order of their effect: –NO2, –N+R3, –CN, –CO2R, –CO–, –OR and –OH.
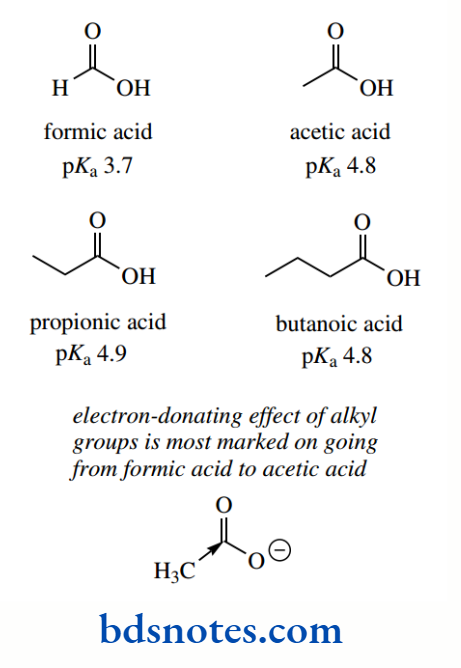
Electron-donating groups will have the opposite effect, destabilizing the conjugate base by increasing electron density, and thus producing weaker acids. The most common electron-donating groups encountered are going to be alkyl groups, though the effect from alkyl groups is rather small.
Indeed, it is not immediately apparent why there should be any inductive effect at all since the substitution of hydrogen by alkyl should not lead to any bond polarization. At this point, we should merely note that alkyl groups have a weak electron-donating effect – it may not be strictly an inductive effect.
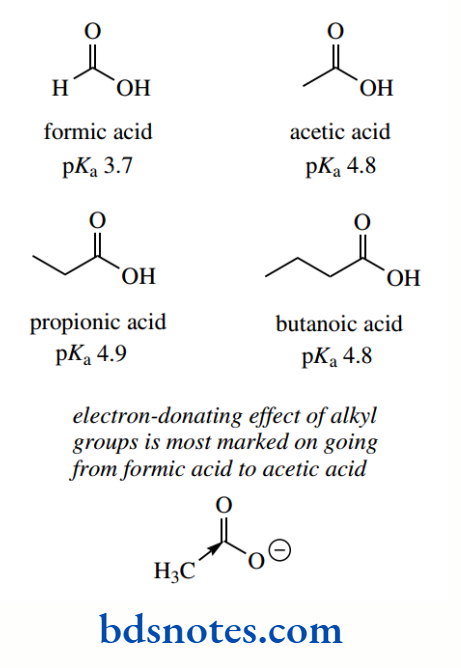
The pKa value for formic acid (pKa 3.7) makes it more acidic than acetic acid. The electron-donating effect of the methyl group is most marked on going from formic acid to acetic acid since the acidity of propionic acid (pKa 4.9) and butanoic acid vary little from that of acetic acid.
The electron-donating effect from alkyl substituents is relatively small, being considerably smaller than inductive effects from most electron-withdrawing groups, and also rapidly diminishes along a carbon chain.
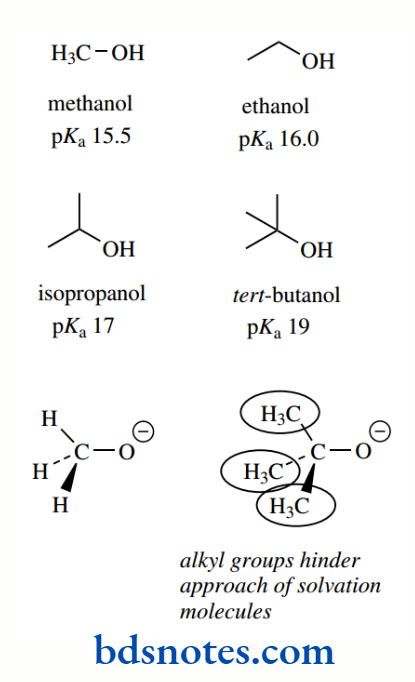
Alcohols are much less acidic than carboxylic acids; but, as one progresses through the sequence of methanol, ethanol, isopropanol, and tert-butanol, pKa values gradually increase from 15.5 to 19, a substantial decrease in acidity.
Although this was originally thought to be caused by the inductive effects of methyl groups, it is now known to be primarily related to solvation effects. In solution, the conjugate base anion is surrounded by polar solvent molecules.
This solvation helps to stabilize the conjugate base and thus increases the acidity of the alcohol. As we get more alkyl groups, the solvation of the anion is diminished because of the increased steric hindrance they cause and observed acidity also decreases.
Hybridization effects
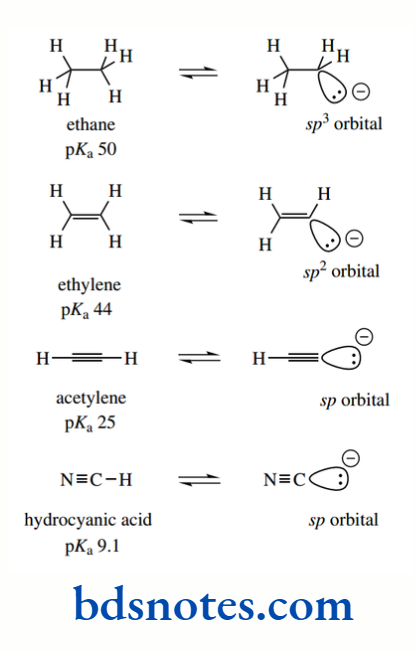
The acidity of a C–H bond is influenced by the hybridization state of the carbon atom attached to the acidic hydrogen. Dissociation of the acid generates an anion whose lone pair of electrons is held in a hybridized orbital.
We can consider sp orbitals to have more s character than sp² orbitals, and similarly, sp² orbitals to have more s character than sp³ orbitals.
Since s orbitals are closer to the nucleus than p orbitals, it follows that electrons in an sp-hybridized orbital are held closer to the nucleus than those in an sp² orbital; those in an sp² orbital are similarly closer to the nucleus than those in an sp3 orbital.
It is more favourable for the electrons to be held close to the positively charged nucleus, and thus a sp-hybridized anion is more stable than a sp²-hydridized anion, which is more stable than a sp3³-hybridized anion. Thus, the acidity of a C–H bond decreases as the s character of the bond decreases.
The pKa of the hydrocarbon ethane is about 50, that of ethylene is about 44, and that of acetylene is about 25. The hybridization of the C–H bond in ethane is sp³ (25% s character), in ethylene it is sp² (33% s character), and in acetylene it is sp (50% s character).
This makes alkynes (acetylenes) relatively acidic for hydrocarbons. It is also a contributing factor in the acidity of HCN (pKa 9.1), where the conjugate base cyanide is a hybridized anion, though additional stabilization comes from the electronegative nitrogen atom.
So far we have considered the hybridization state of the orbital associated with the anionic charge. However, the hybridization state elsewhere in the molecule may also affect acidity.
The more character an orbital has, the closer the electrons are held to the nucleus, and this effectively makes the atom more electronegative.
This may be explained in terms of hybridization modifying inductive effects, such that sp-hybridized carbons are effectively more electronegative than sp²-hybridized carbons, and similarly, sp²-hybridized carbons are more electronegative than sp³-hybridized carbons.
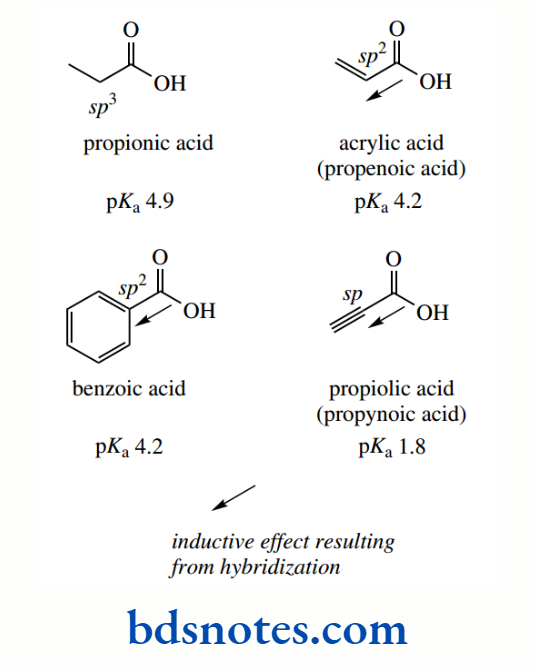
The pKa values for the following acids illustrate that, as the carbon atom adjacent to the carboxylic acid group changes from sp³ to sp³ to sp hybridization, the acidity increases, in accord with the electronegativity explanation above.
Note that benzoic acid (sp² hybridization) has a similar pKa to acrylic acid (propenoic acid), which also has sp² hybridization.
Resonance delocalizes zation effects
Delocalization of charge in the conjugate base anion through resonance is a stabilizing factor and will be reflected by an increase in acidity.
Drawing resonance structures allows us to rationalize that the negative charge is not permanently localized on a particular atom, but may be dispersed to other areas of the structure. We should appreciate that a better interpretation is that the electrons are contained in a molecular orbital that spans several atoms.
However, drawing resonance structures provides a simple and convenient way of predicting stability through delocalization. The pKa of ethanol is 16, and that of acetic acid is 4.8.
The increased acidity of acetic acid relative to ethanol can be rationalized in terms of the delocalization of charge in the acetate anion, whereas in ethoxide the charge is localized on oxygen. Even more delocalization is possible in the methanesulfonate anion, and this is reflected in the increased acidity of methanesulfonic acid (pKa − 1.2)
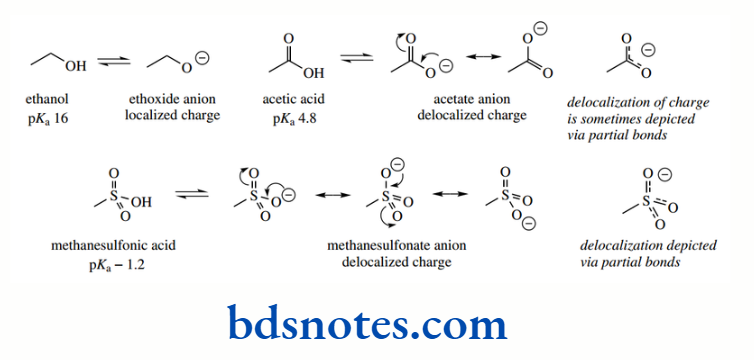
We have also shown some representations of acetate and methanesulfonate anions that have been devised to emphasize resonance delocalization; these include partial bonds rather than double/single bonds.
Although these representations are valuable, they can lead to some confusion in interpretation. It is important to remember that there is a double bond in these systems. Therefore, we prefer to draw out the contributing resonance structures.
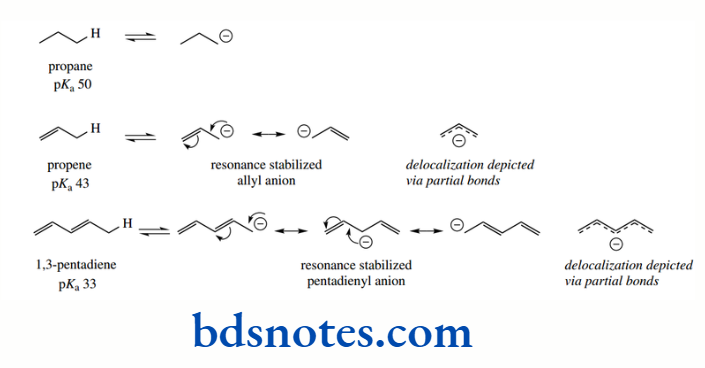
The alkane propane has pKa 50, yet the presence of the double bond in propene means the methyl protons in this alkene have pKa 43; this value is similar to that of ethylene (pKa 44), where increased acidity was rationalized through sp² hybridization effects.
1,3-Pentadiene is yet more acidic, having pKa 33 for the methyl protons. In each case, increased acidity in the unsaturated compounds may be ascribed to the delocalization of charge in the conjugate base. Note that we use the term allyl for the propenyl group.
Resonance stabilization is also responsible for the increased acidity of a C–H group situated adjacent to a carbonyl group.
The anion is stabilized through delocalization of charge, similar to that seen with the allyl anion derived from propene; but this system is even more favourable, in that delocalization allows the charge to be transferred to the electronegative oxygen atom.
As a result, acetone (pKa 19) is significantly more acidic than propene (pKa 43). Anions of this type, termed enolate anions, are some of the most important reactive species used in organic chemistry
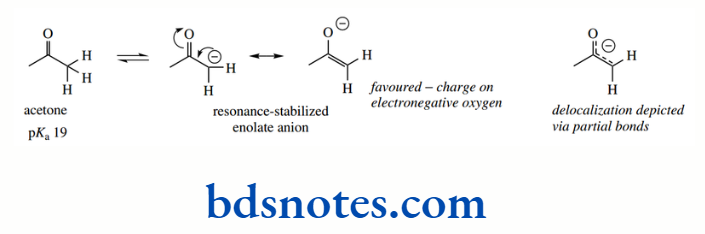
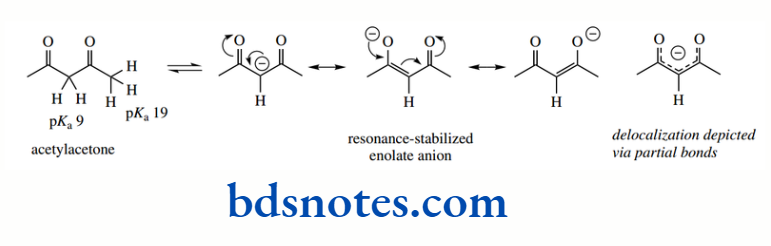
The acidity of a C–H is further enhanced if it is adjacent to two carbonyl groups, as in the 1,3- diketone acetylacetone. The enolate anion is stabilized by delocalization, and both carbonyl oxygens can participate in the process.
This is reflected in the pKa 9 for the protons between the two carbonyls, whereas the terminal protons adjacent to just a single carbonyl have pKa 19, similar to acetone above.
Increased delocalization has a profound effect on the acidity. These two values should be compared with that of the hydrocarbon propane (pKa 50)
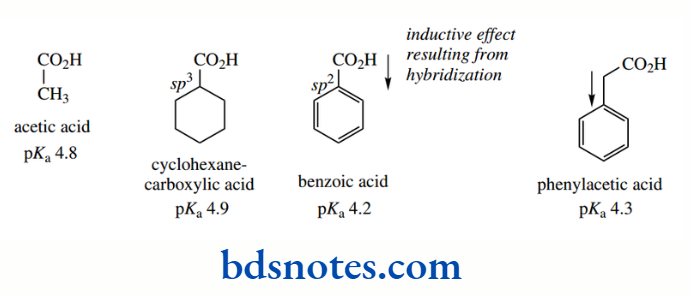
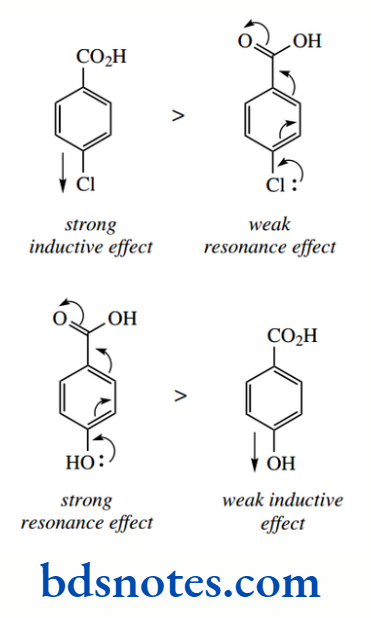
Aromatic rings are themselves excellent examples of resonance and delocalization of electrons They also influence the acidity of appropriate substituent groups, as seen in benzoic acids. Benzoic acid (pKa 4.2) is a stronger acid than acetic acid (pKa 4.8), and it is also stronger than its saturated analogue cyclohexane carboxylic acid (pKa 4.9).
The phenyl group exerts an electron-withdrawing effect because the hybridization of the ring carbons is sp²; consequently, electrons are held closer to the carbon atom than in a sp³-hybridized orbital. This polarizes the bond between the aromatic ring and the carboxyl. The pKa of phenylacetic acid (pKa 4.3), compared with acetic acid (pKa 4.8), demonstrates the inductive effect of a benzene ring.
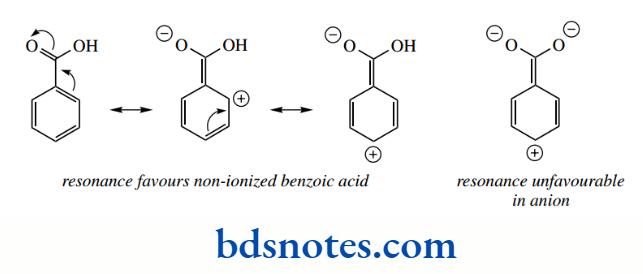
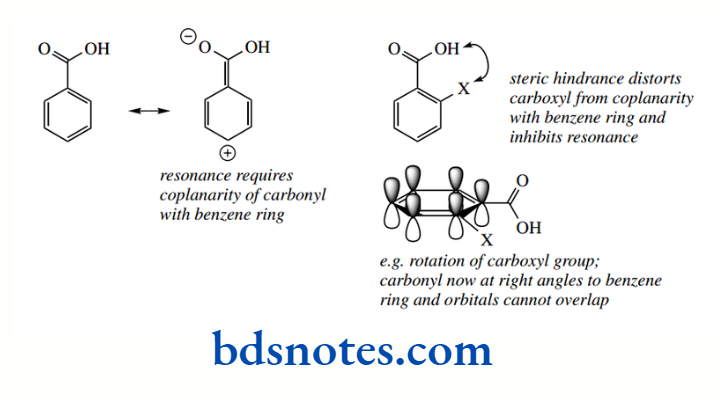
However, we might then expect benzoic acid to be a rather stronger acid than it is, since the phenyl group is closer to the carboxyl group than in phenylacetic acid.
We attribute the lower acid strength to an additional resonance effect in the carboxylic acid that is not favourable in the anion, where it would lead to a carboxylate carrying a double negative charge; therefore, the resonance effect weakens the acid strength.
Further inductive effects from other substituents enhance or counter these effects with predictable results. Thus, a halogen such as chlorine, with a strong inductive effect, produces stronger acids, especially in the case of the ortho derivative. Here, the extra inductive effect is correspondingly closer to the carboxyl group, and it will help to stabilize the conjugate base.
The acid-weakening resonance effects are also diminished by the inductive effects of halogens; it is not favourable to have an electron-withdrawing substituent close to a positive charge.
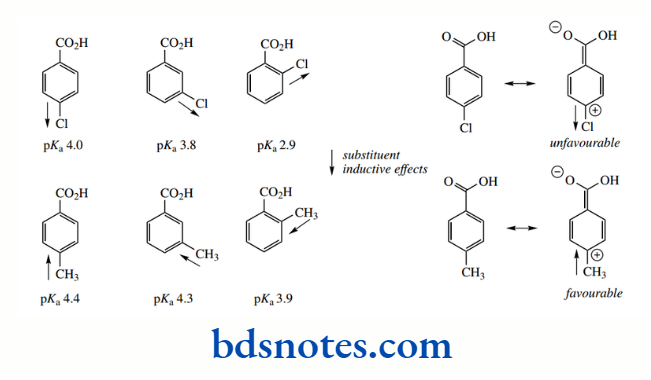
On the other hand, methyl substituents have a weak electron-donating effect opposing that of the aromatic ring. This also favours resonance in the nonionized acid. There is only a modest effect on acidity, except when the methyl is in the ortho position, where the effect is closer to the carboxyl group.
However, ortho substituents add a further dimension that is predominantly steric. Large groups in the ortho position can influence the carboxyl group, forcing it out of the plane of the ring.
The result is that resonance is now inhibited because the orbitals of the carbonyl group are no longer coplanar with the benzene ring. In almost all cases, the ortho-substituted benzoic acid tends to be the strongest acid of the three isomers.
When substituents can also be involved in the resonance effects, changes in acidity become more marked. Consider hydroxy- and methoxy-benzoic acid derivatives. The pKa values are found to be 3.0, 4.1, and 4.6 for the ortho, meta, and para hydroxy derivatives respectively, and 4.1, 4.1, and 4.5 respectively for the corresponding methoxy derivatives.
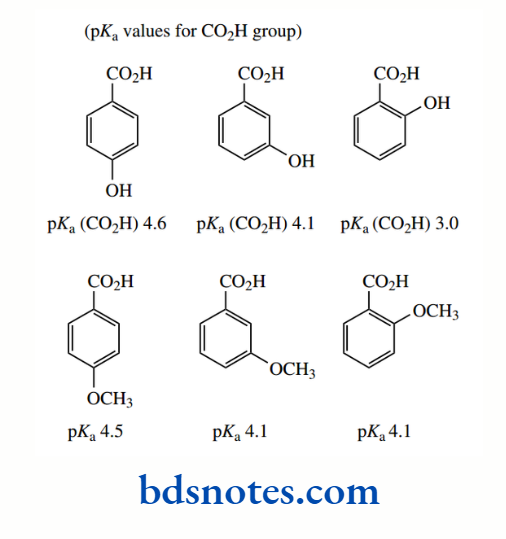
Let us ignore the figure for ortho-hydroxybenzoic acid for the moment since there is yet another feature affecting acidity. We then see that the para derivatives are rather less acidic than we might predict merely from the inductive effect of the OH or OMe groups.
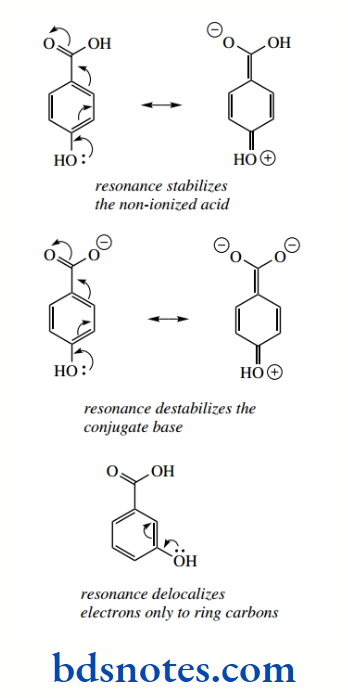
PKa values show that these compounds are less acidic than benzoic acid, whereas the inductive effect would suggest they should be more acidic. This is because of a large resonance effect emanating from the substituent in which electronic charge is transmitted through the conjugated system of the aromatic ring into the carboxyl group.
The electron-donating effect originates from the lone pair electrons on oxygen, with overlap into the π electron system. This electron donation will stabilize the non-ionized acid via electron delocalization but would destabilize the conjugate base by creating a double charge in the carboxylate system.
The net result is lower acidity. This electron-donating effect from lone pair electrons is simply a resonance effect but is often termed a mesomeric effect. A mesomer is another term for a resonance structure. We shall use the ‘resonance effect’ rather than the ‘mesomeric effect’ to avoid having alternative terminologies.
We can write a similar delocalization picture for the ortho-substituted compounds, but this is countered by the opposing inductive effect close to the carboxyl.
However, the steric effect, as described above, means large groups in the ortho position can force the carboxyl group out of the plane of the ring. This weakens the resonance effect since delocalization is dependent upon coplanarity in the conjugate system.
Resonance stabilization is not as important for the meta derivatives, where it is only possible to donate electrons towards the ring carbons, which are, of course, not as electronegative as oxygen.
Meta-substitution is the least complicated, in that groups placed there exert their influence almost entirely through inductive effects. It should be noted that, where we have opposing resonance and inductive effects, the resonance effect is normally of much greater magnitude than the inductive effect and its contribution predominates (but see below for chlorine).
The relatively high acidity of ortho-hydroxybenzoic acid (salicylic acid), compared with the other derivatives just considered, is ascribed to intramolecular hydrogen bonding, which is not possible in the other compounds, even with orthomethoxybenzoic acid.
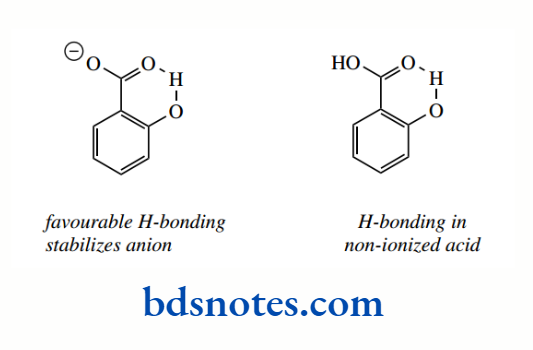
Hydrogen bonding involves a favourable six-membered ring and helps to stabilize the conjugate base. Although some hydrogen bonding occurs in the non-ionized acid, the effect is much stronger in the carboxylate anion.
It should be noted that the electron-donating resonance effects just considered are the result of lone pair electrons feeding into the π electron system.
Potentially, any substituent with a lone pair might do the same, yet we did not invoke such a mechanism with chlorine substituents above. As the size of the atom increases, lone pair electrons will be located in orbitals of higher level, e.g. 3p rather than 2p as in carbon.
Consequently, the ability to overlap the lone pair orbital with the π electron system of the aromatic ring will diminish, a simple consequence of how far from the atom the electrons are mostly located. Chlorine thus produces a low resonance effect but a high inductive effect, and the latter predominates.
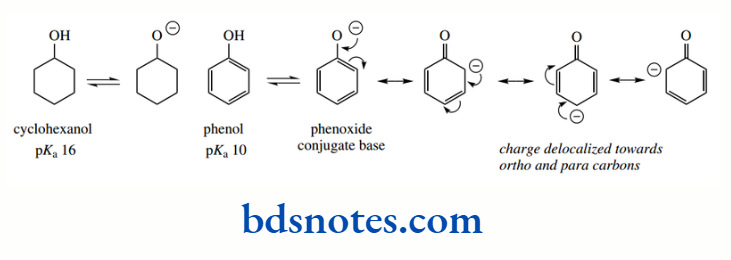
Resonance can also influence the acidity of hydroxyl groups, as seen in phenols. Cyclohexanol has pKa 16, comparable to that of ethanol. On the other hand, phenol has pKa 10, making it considerably more acidic than a simple alcohol, though less so than a carboxylic acid.
This increased acidity is explained in terms of delocalization of the negative charge into the aromatic ring system, with resonance structures allowing ring carbons ortho and para to the original phenol group to become electron rich.
Although the aromatic ring acts as an acceptor of electrons and may be termed an electron sink, the charge is dispersed towards carbon atoms, which is going to be less favourable than if it can be dispersed towards more electronegative atoms such as oxygen.
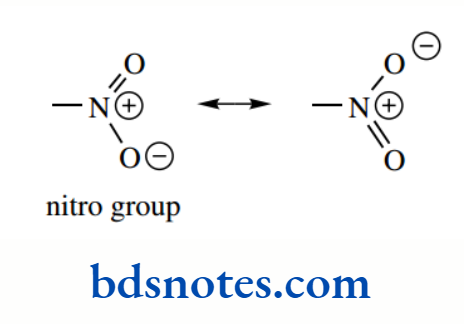
A good illustration of this concept is seen in a series of nitrophenols. The nitro group itself has to be drawn with charge separation to accommodate the electrons and our rules of bonding. However, resonance structures suggest that there is electron delocalization within the nitro group.
With substituted phenols, there can be similar delocalization of charge into the aromatic ring as with phenol, but substituents will introduce their effects, be they inductive or resonance-related.
It can be seen that the nitro group allows further delocalization of the negative charge of the phenoxide conjugate base if it is situated in the ortho or para positions. This increases acidity relative to phenol, and both compounds have essentially the same pKa of 7.2
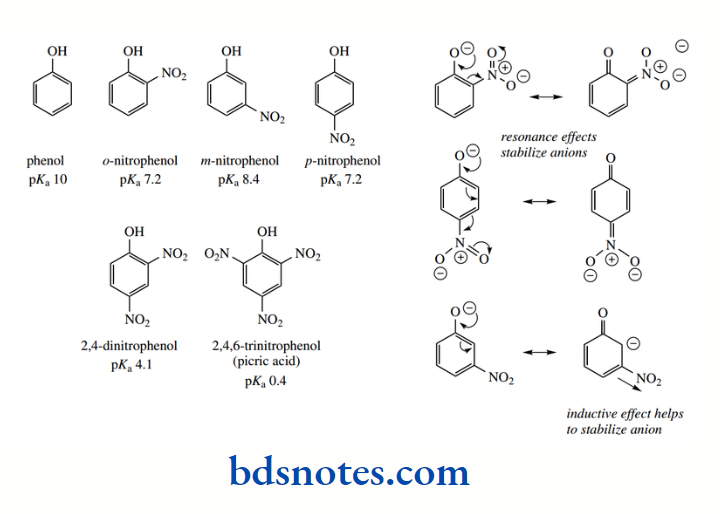
The effect is magnified considerably if there are nitro groups both ortho and para, so that the pKa for 2,4-dinitrophenol is 4.1. A third nitro group, as in 2,4,6-trinitrophenol, confers even more acidity, and this compound has pKa 0.4, making it a strong acid.
This is reflected in its common name, picric acid. Note that m-nitrophenol has pKa 8.4, and is a lot less acidic than o-nitrophenol or p-nitrophenol. We can draw no additional resonance structures here, and the nitro group cannot participate in further electron delocalization.
The increased acidity compared with phenol can be ascribed to the stabilization of resonance structures with the charge on a ring carbon through the nitro group’s inductive effect.
From the above, it should not be difficult to rationalize the effects of other types of substituents on the acidity of phenols. Thus electron-donating groups, e.g. alkyl, reduce acidity, and electron-withdrawing groups, for example, halogens, increase acidity. With strongly electron-withdrawing groups, such as cyano and nitro, the acid-strengthening properties can be quite pronounced.
A summary list of resonance effects emanating from various groups We should also point out that these very same principles will be used to rationalize aromatic substitution. and this is why we have purposely discussed the acidity of aromatic derivatives in some detail.
Basicity
We have already defined a base as a substance that will accept a proton by donating a pair of electrons. Just as we have used pKa to measure the strength of an acid, we need a system to measure the strength of a base. Accordingly, a basicity scale based on pKb was developed in a similar way to pKa. For the ionization of the base B in water.
⇒ \(\mathrm{B}+\mathrm{H}_2 \mathrm{O} \stackrel{K}{\rightleftharpoons} \mathrm{BH}^{\oplus}+\mathrm{HO}^{\ominus}\) the equilibrium constant K is given by the formula.
⇒ \(K=\frac{\left[\mathrm{HO}^{-}\right]\left[\mathrm{BH}^{+}\right]}{[\mathrm{B}]\left[\mathrm{H}_2 \mathrm{O}\right]}\) and since the concentration of water will be essentially constant, the equilibrium constant Kb and the logarithmic pKb may be defined as \(K_{\mathrm{b}}=\frac{\left[\mathrm{HO}^{-}\right]\left[\mathrm{BH}^{+}\right]}{[\mathrm{B}]}\) with \(\mathrm{p} K_{\mathrm{b}}=-\log _{10} K_{\mathrm{b}}\)
This system has been almost completely dropped in favour of using pKa throughout the acidity–basicity scale. To measure the strength of a base, we use the pKa of its conjugate acid, i.e. we consider the equilibrium.
For Which \(K_{\mathrm{a}}=\frac{[\mathrm{B}]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{\left[\mathrm{BH}^{+}\right]}\)
It follows that A strong base has a small Ka and thus a large pKa, i.e. BH+ is favoured over B; A weak base has a large Ka and thus a small pKa, i.e. B is favoured over BH+.
Or, put another way:
The larger the value of p Ka, the stronger the base;
The smaller the value of p Ka, the weaker the base. The relationship between pKa and pKb can be deduced as follows:
⇒ \(\begin{aligned}
K_{\mathrm{b}} & =\frac{\left[\mathrm{HO}^{-}\right]\left[\mathrm{BH}^{+}\right]}{[\mathrm{B}]} \\
K_{\mathrm{a}} & =\frac{[\mathrm{B}]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{\left[\mathrm{BH}^{+}\right]} \\
K_{\mathrm{a}} \times K_{\mathrm{b}} & =\frac{[\mathrm{B}]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{\left[\mathrm{BH}^{+}\right]} \times \frac{\left[\mathrm{HO}^{-}\right]\left[\mathrm{BH}^{+}\right]}{[\mathrm{B}]} \\
& =\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{HO}^{-}\right]
\end{aligned}\)
Thus, Ka × Kb reduces to the ionization constant for water Kw.
In this reaction, one molecule of water acts as a base and accepts a proton from a second water molecule. This second water molecule, therefore, acts as an acid and donates a proton.
The equilibrium constant K for this reaction is given by the formula and because the concentration of water is essentially constant in an aqueous solution, the new equilibrium constant K w is defined as
⇒ \(K=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{HO}^{-}\right]}{\left[\mathrm{H}_2 \mathrm{O}\right]\left[\mathrm{H}_2 \mathrm{O}\right]}\)
For every hydronium ion produced, a hydroxide anion must also be formed, so that the concentrations of hydronium and hydroxide ions must be equal. In pure water at 25 ◦C, this value is found to be 10−7 M.
⇒ \(K_{\mathrm{w}}=\left[\mathrm{HO}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=10^{-7} \times 10^{-7}=10^{-14}\)
We now have the relationship that Ka × Kb = 10−14, or \(\mathrm{p} K_{\mathrm{a}}+\mathrm{p} K_{\mathrm{b}}=14\)
Electronic And Structural Features That Influence Basicity
Basicity relates to the ability of a compound to use its nonbonding electrons to combine with a proton.
We have already seen that features such as inductive or delocalization effects can make an acid stronger. They increase the stability of the conjugate base and consequently favour the loss of a proton from an acid.
It follows that features that stabilize a conjugate base are going to discourage its protonation, i.e. they are going to make it a weaker base. Thus, a compound in which the electrons are delocalized will be less basic than one in which the electrons are localized.
For example, carboxylate anions (delocalized charge) are going to be weaker bases than alkoxide ions (localized charge).
Anionic (charged) bases are naturally going to be more ready to donate electrons to a positively charged proton than a neutral base (uncharged) that uses lone pair electrons.
Most of our organic bases are not anionic, so we need to look at features that affect basicity, just as we have done for acids. Nitrogen compounds are good examples of organic bases and the ones we shall meet most frequently, though oxygen systems will feature prominently in our mechanistic rationalizations.
Electronegativity
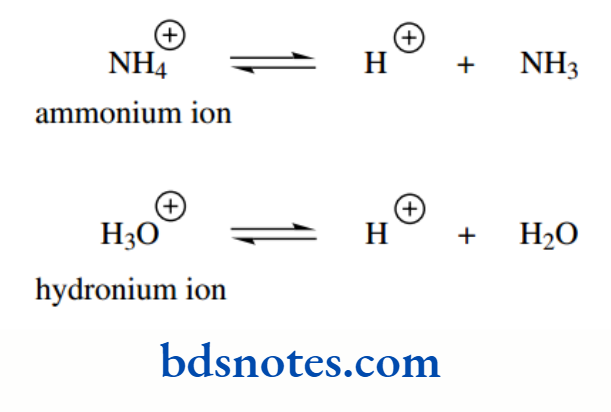
The acidity of an acid HX increases as X becomes more electronegative. Conversely, basicity will decrease as an atom becomes more electronegative. Ammonia is a stronger base than water.
These figures relate to the release of a proton from the conjugate acid, namely ammonium ion and hydronium ion respectively.
This is sometimes confusing; we talk about the pKa of a base when we mean the pKa of its conjugate acid. We cannot avoid this, because it becomes too complicated to use the name of the conjugate acid, but we shall endeavour to show the conjugate acid in structures.
Oxygen is more electronegative than nitrogen, so its electrons are less likely to be donated to a proton. Neutral oxygen bases are generally very much weaker than nitrogen bases, but as we shall see later, the protonation of an oxygen atom is important and the first step in many acid-catalysed reactions, especially carbonyl compounds.
Inductive effects
Electron-donating groups on nitrogen are going to increase the likelihood of protonation and help to stabilize the conjugate acid. They thus increase the basic strength.
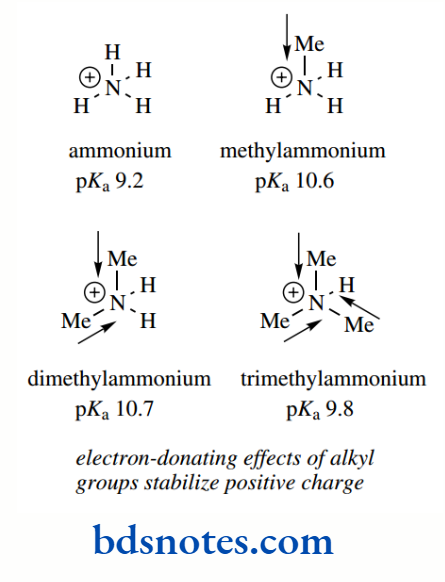
The pKa values for the amines ammonia, methylamine, dimethylamine, and trimethylamine are 9.2, 10.6, 10.7, and 9.8 respectively.
The electron-donating effect of the methyl substituents increases the basic strength of methylamine over ammonia by about 1.4 pKa units, i.e. by a factor of over 25 (101.4 = 25.1).
However, the introduction of a second methyl substituent has a relatively small effect, and the introduction of a third methyl group, as in trimethylamine, actually reduces the basic strength to nearer that of methylamine.
This apparent anomaly is a consequence of measuring pKa values in an aqueous solution, where there is more than ample opportunity for hydrogen bonding with water molecules.
Hydrogen bonding helps to stabilize a positive charge on nitrogen, and this effect will decrease as the number of alkyl groups increases.
Therefore, the observed pKa values are a combination of increased basicity with increasing alkyl groups (as predicted via electron-donating effects) countered by a stabilization of the cation through hydrogen bonding, which decreases with increasing alkyl groups. Note that we saw solvent molecules influencing the acidity of alcohols by stabilizing the conjugate base.
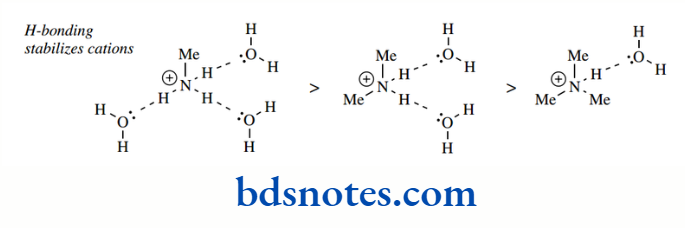
When pKa values are measured in the gas phase, where there are no hydrogen bonding effects, they are found to follow the predictions based solely on electron-donating effects.
In water, mono-, di-, and tri-alkylated amines all tend to have rather similar pKa values, typically in the range 10–11.
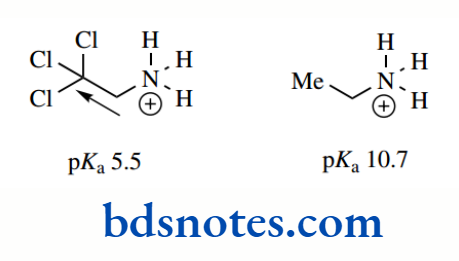
Electron-withdrawing groups will have the opposite effect. They will decrease electron density on the nitrogen, destabilize the conjugate acid, and thus make it less likely to pick up a proton, so producing a weaker base.
Inductive effects from various groups. For example, groups with a strong electron-withdrawing inductive effect, such as trichloromethyl, decrease basicity significantly.
We have already seen that water is a much weaker base than ammonia because oxygen is more electronegative than nitrogen and its electrons are thus less likely to be donated to a proton. Neutral oxygen bases are also generally very much weaker than nitrogen bases.
Nevertheless, the protonation of an oxygen atom is a critical first step in many acid-catalysed reactions.
Oxygen is so electronegative that inductive effects from substituents have rather less influence on basicity than they would in similar nitrogen compounds. Alcohols are somewhat less basic than water, with ethers weaker still.
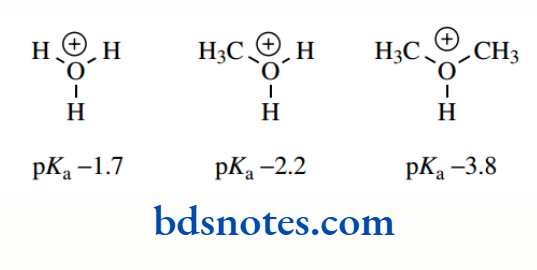
This is precisely opposite to what would be expected from the inductive effects of alkyl groups, and the observations are likely to be the result primarily of solvation (hydrogen bonding) effects. Note, the cations shown all have negative pKa values. In other words, they are very strong acids and will lose a proton readily. Conversely, the non-protonated compounds are weak bases.
Hybridization effects
We have seen above that acidity is influenced by the hybridization of the atom to which the acidic hydrogen is attached. The acidity of a C–H bond was found to increase as the s character of the bond increased. The more characters in the orbital, the closer the electrons are held to the nucleus.
Similar reasoning may be applied to basicity. If the lone pair is in a sp² or sp orbital, it is held closer to the nucleus and is more difficult to protonate than if it is in a sp³ orbital.
Accordingly, we find that nitrile nitrogen (lone pair in an sp orbital) is not at all basic (pKa about −10), though ethylamine (lone pair in an sp3 orbital) has pKa 10.7. Imines (lone pair in an sp2 orbital) are less basic than amines. Cyclohexanimine (the imine of cyclohexanone) has pKa 9.2 and is less basic than cyclohexylamine (pKa 10.6)
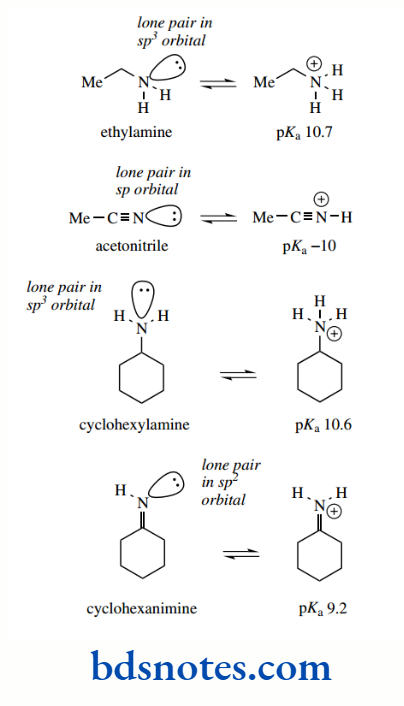
Similarly, alcohols (sp³ hybridization), although they are themselves rather weak bases, are going to be more basic than aldehydes and ketones (sp² hybridization)
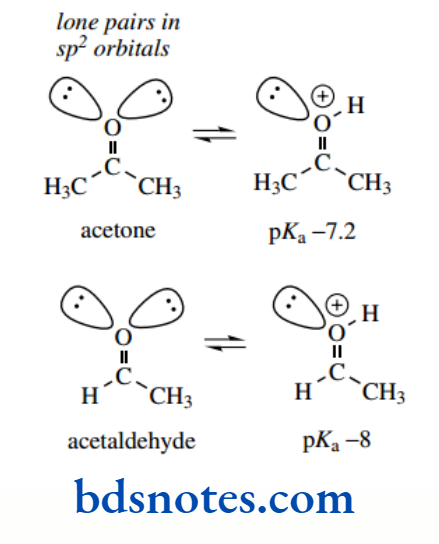
Resonance/delocalize zati on effects
Delocalization of charge in the conjugate base anion contributes to the stabilization of the anion, and thus ionization of the acid is enhanced. Delocalization effects in bases are more likely to stabilize the base rather than the conjugate acid and thus tend to reduce the basicity.
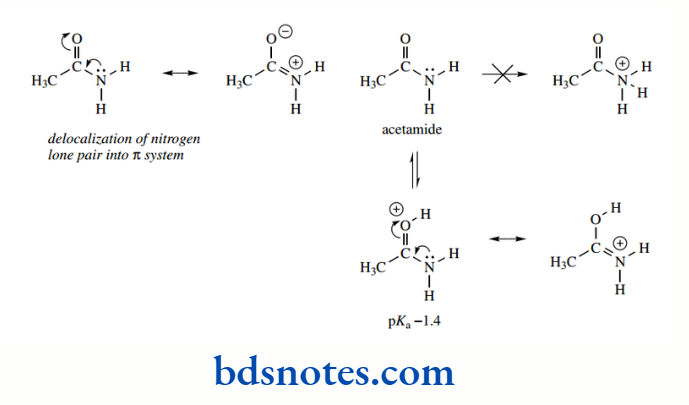
For a summary of various groups that may contribute resonance effects. Pre-eminent among examples is the case of amides, which do not show the typical basicity of amines.
Acetamide, for example, has pKa − 1.4, compared with pKa 10.7 in the case of ethylamine. This reluctance to protonate on nitrogen is caused by delocalization in the neutral amide, in which the nitrogen lone pair can overlap into the π system.
This type of resonance stabilization would not be possible with nitrogen protonated since the lone pair is already involved in the protonation process. Indeed, if amides do act as bases, then protonation occurs on oxygen, not on nitrogen.
Resonance stabilization is still possible in the O-protonated amide, whereas it is not possible in the N-protonated amide. Note that resonance stabilization makes the O-protonated amide somewhat less acidic than the hydronium ion (pKa − 1.7); the amide oxygen is more basic than water.
The carbonyl oxygen of aldehydes and ketones is less basic than that of an alcohol by several powers of 10. We have just seen above that this arises because the lone pair electrons of the carbonyl oxygen are in orbitals that are approximately sp² in character and are more tightly held than the alcohol lone pairs in sp³ orbitals.
The neutral carbonyl group is thus favoured, and the conjugate acid is correspondingly more acidic. On the other hand, protonated carboxylic acids and esters are shown with the proton on the carbonyl oxygen, despite this oxygen having sp² hybridization, whereas the alternative oxygen has sp³ hybridization.
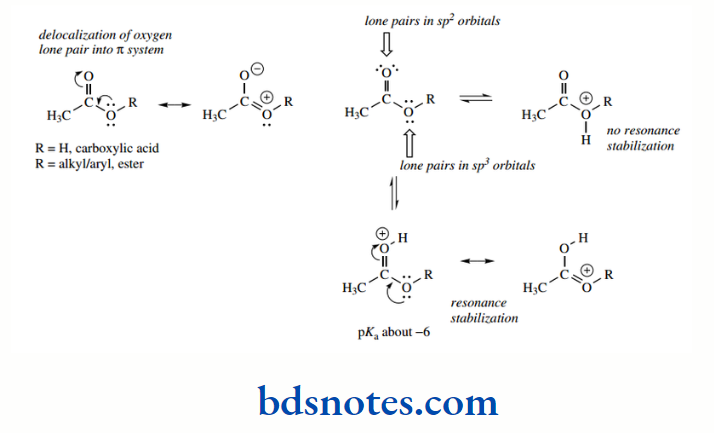
This is a consequence of delocalization, with resonance stabilization being possible when the carbonyl oxygen is protonated, but not possible should the OR oxygen become protonated.
This additional resonance stabilization is not pertinent to aldehydes and ketones, which are thus less basic than the carboxylic acid derivatives. However, these oxygen derivatives are still very weak bases and are only protonated in the presence of strong acids.
In the case of the sulfur analogues thioesters and thioacids, this delocalization is much less favourable.
In the oxygen series, delocalization involves an overlap between the oxygen sp3 orbital and the π system of the carbonyl, which is composed of 2p orbitals.
Delocalization in the sulfur series would require overlap between a sulfur 3p orbital and a carbon 2p orbital, which is much less likely because of the size difference between these orbitals resonance of this type is less favourable in the sulfur esters and acids due to the larger S atom, and less orbital overlap Amidines are stronger bases than amines. The pKa for acetamidine is 12.4.
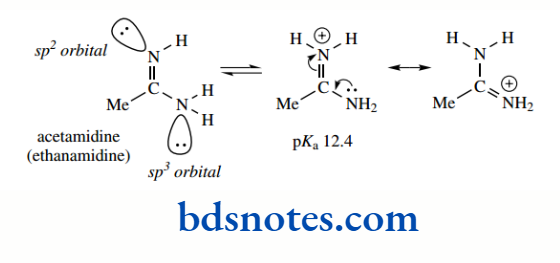
Amidines are essentially amides where the carbonyl oxygen has been replaced with nitrogen, i.e. they are nitrogen analogues of amides. It is the nitrogen replacing the oxygen that becomes protonated. This is easily rationalized, even though the hybridization here is sp², which in theory should be less basic than the sp³-hybridized nitrogen.
Protonation of the imine nitrogen allows resonance stabilization in the cation, which could not happen if the amide nitrogen were protonated.
In addition, the two resonance structures both have a charge on nitrogen and in fact, are identical. We have a similar situation in the carboxylate anion. Amidines, therefore, are quite strong bases, with the potential for electron delocalization being a greater consideration than the hybridization state of the orbital housing the lone pair.
Now let us look at guanidines, which are even stronger bases. Guanidine itself has pKa 13.6. It can be seen that there is delocalization of charge in the conjugate acid, such that in each resonance structure the charge is favourably associated with one of the three nitrogen atoms. No such favourable delocalization is possible in the neutral molecule, so guanidines are readily protonated and, therefore, are strong bases
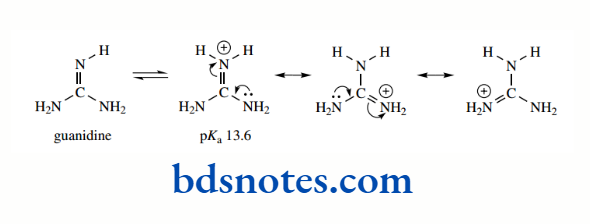
Should you need further convincing that resonance stabilization is an important criterion in acidity and basicity, it is instructive to consider the bond lengths in the carboxylate anion and the guanidinium and guanidinium cations. Now we would expect double bonds to be shorter than single bonds, and this is true in the corresponding non-ionized systems.
However, bond length measurements for the carboxylate anion tell us that the bonds in question (C–O/C=O) are the same length, being somewhere between the expected single and double bond lengths.
The same is true of the C–N/C=N bonds in guanidinium and guanidinium cations. This fits in nicely with the concept that the actual ion is not a mixture of the various resonance forms that we can draw, but something in between.
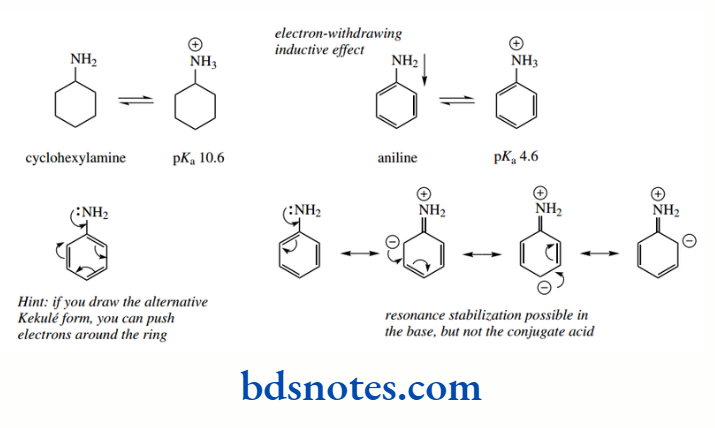
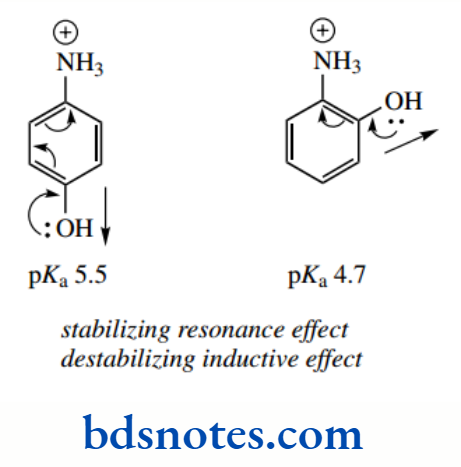
Compare this with the fact that the C–C bond lengths in benzene are somewhere between single and double bonds, and thus do not correspond to either of the Kekule resonance structures.´ If we look at the pKa values for the conjugate acids of cyclohexylamine and the aromatic amine aniline, we see that aniline is the weaker base.
Cyclohexylamine has pKa 10.6, whereas aniline has pKa There is an inductive effect in aniline because the phenyl ring is electron withdrawing.
The carbon atoms of the aromatic ring are sp² hybridized, and more electronegative than sp3-hybridized carbons of alkyl groups. We might, therefore, expect some reduction in basicity.
However, a more prominent effect arises from resonance, which can occur in the uncharged amine, but not in the protonated conjugate acid. This makes the unprotonated amine favourable, and aniline is consequently a very weak base.
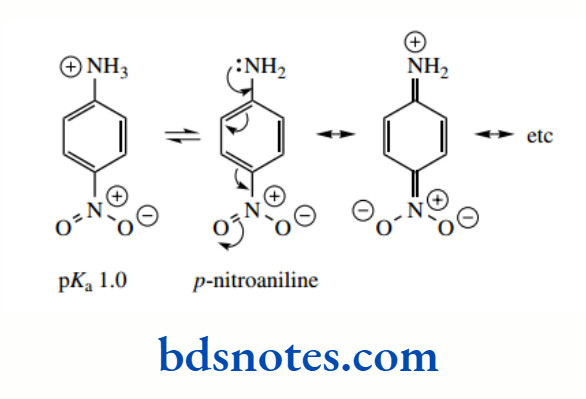
This effect is increased if there is a suitable electron-withdrawing group in the ortho or para position on the aromatic ring. Thus, p-nitroaniline and o-nitroaniline have pKa 1.0 and −0.3 respectively.
These aromatic amines are thus even weaker bases than aniline, a result of improved delocalization in the free base. The increased basicity of the ortho isomer is a result of the very close inductive effect of the nitro group; the meta isomer has only the inductive effect, and its pKa is about 2.5.
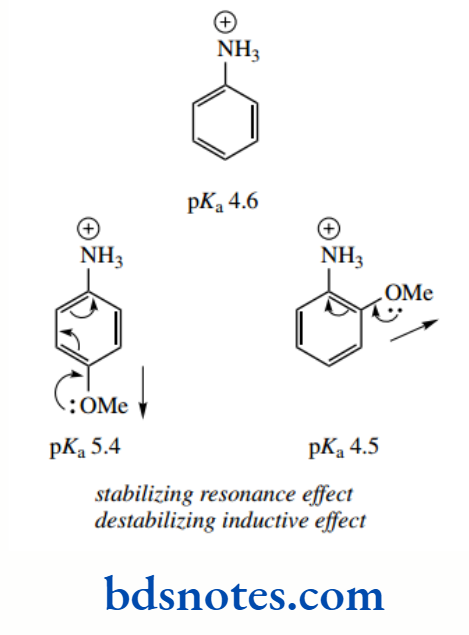
Of course, those groups that can act as electron-donating groups through resonance will produce the opposite effect, and increase the basicity. Through resonance, groups such as hydroxyl and methoxyl can distribute a negative charge towards the amino substituent, facilitating its protonation.
The pKa values for o-methoxyaniline and p-methoxyaniline are about 4.5 and 5.4 respectively, and that for mmethoxyaniline is about 4.2.
The electron-donating resonance effect is countered by the electron-withdrawing inductive effects of these electronegative substituents so that predictions about basicity become a little more complex.
As we pointed out after our considerations of acidity in aromatic derivatives, we wish to emphasize that the very same principles will be used when we consider aromatic substitution reactions. The methods used to understand the basicity of aromatic derivatives will be applied again in a different format.
A word of warning is now needed! Some compounds may have pKa values according to whether they are acting as acids or as bases.
For example, CH3OH has pKa 15.5 and −2.2; the first figure refers to methanol acting as an acid via the loss of a proton and giving CH3O−, and the second value refers to methanol acting as a base, i.e. the conjugate acid losing a proton.
Similarly, CH3NH2 has pKa values of 35 and 10.6, again referring to acid and base behaviour. It is important to avoid confusion in such cases, and this requires an appreciation of typical pKa values for simple acids and bases.
There is no way we would encourage memorizing pKa values, but two easily remembered figures can be valuable for comparisons. These are pKa around 5 for a typical aliphatic carboxylic acid, and pKa around 10 for a typical aliphatic amine.
These then allow us to consider whether the compound in question is more acidic, more basic, etc. It then becomes fairly easy to decide that methanol is not a strong acid, like nitric acid say, so that the pKa − 2.2 is unlikely to refer to its acid properties.
Methylamine ought to be basic rather like ammonia, so the pKa value of 35 would appear well out of the normal range for bases and must refer to its acidic properties. In such cases, there appear to be very good reasons for continuing to use pKb values for bases; unfortunately, however, this is not now the convention.
Basicity Of Nitrogen Heterocycles
Our discussions of the basicity of organic nitrogen compounds have concentrated predominantly on simple amines in which the nitrogen atom under consideration is part of an acyclic molecule.
Many biologically important compounds, and especially drug molecules, are based upon systems in which nitrogen is part of a heterocycle. We shall consider the properties of heterocyclic compounds; here, we mainly want to show how our rationalizations of basicity can be extended to a few commonly encountered nitrogen heterocycles.
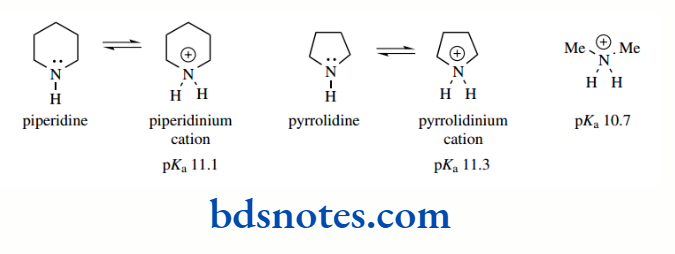
The basicities of the simple heterocycles piperidine and pyrrolidine vary little from that of a secondary amine such as dimethylamine. pKa values for the conjugate bases of these three compounds are 11.1, 11.3, and 10.7 respectively.
However, pyridine and pyrrole are significantly less basic than either of their saturated analogues. The pyridinium cation has pKa 5.2, making pyridine a much weaker base than piperidine, whereas the pyrrolium cation (pKa − 3.8) can be considered a very strong acid, and thus pyrrole is not at all basic.
Although the nitrogen atom in these systems carries a lone pair of electrons, these electrons are not able to accept a proton in the same way as a simple amine. The dramatic differences in basicity are a consequence of the π electron systems, to which the nitrogen contributes.
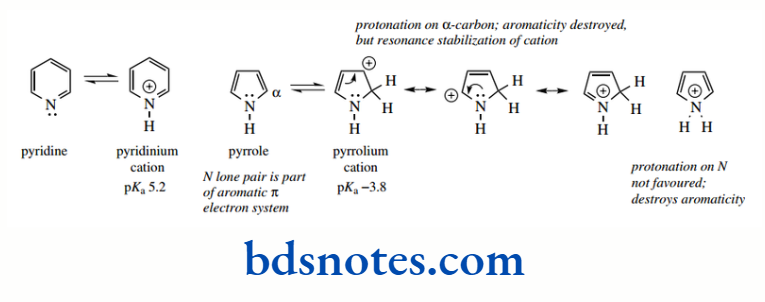
Pyridine, like benzene, is an aromatic system with six π electrons. The ring is planar, and the lone pair is held in an sp² orbital.
The increased s character of this orbital, compared with the sp³ orbital in piperidine, means that the lone pair electrons are held closer to the nitrogen and, consequently, are less available for protonation.
This hybridization effect explains the lower basicity of pyridine compared with piperidine. Pyrrole is also aromatic, but there is a significant difference, in that both of the lone pair electrons are contributing to the six- π-electron system.
As part of the delocalized π electron system, the lone pairs are consequently not available for bonding to a proton.
Protonation of the nitrogen in pyrrole is very unfavourable: it would destroy the aromaticity.
It is possible to protonate pyrrole using a strong acid; but, interestingly, protonation occurs on the α-carbon and not on the nitrogen.
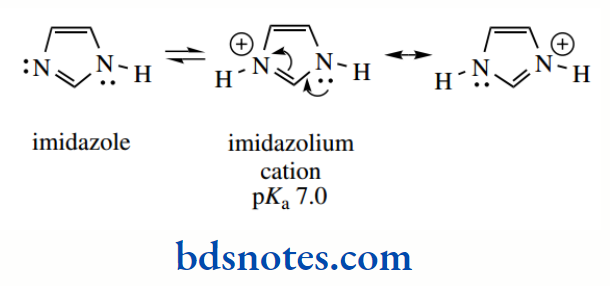
Although this still destroys aromaticity, there is some favourable resonance stabilization in the conjugate acid. Let us consider just one more nitrogen heterocycle here, and that is imidazole, a component of the amino acid histidine.
The imidazolium cation has pKa 7.0, making imidazole less basic than a simple amine, but more basic than pyridine. Imidazole has two nitrogen atoms in its aromatic ring system.
One of these nitrogens contributes its lone pair to make up the aromatic sextet, but the other has a free lone pair that is available for protonation. As with pyridine, this lone pair is in an sp² orbital, but the increased basicity of imidazole compared with pyridine is a result of additional resonance in the conjugate acid.
The basicity of some other heterocyclic systems
Polyfunctional Acids And Bases
We have so far considered acids and bases with a single ionizable group, and have rationalized the measured pKa values concerning structural features in the molecule.
This additional structural feature could well have its own acidic or basic properties, and we thus expect that such a compound will be characterized by more than one pKa value.
Before we consider polyfunctional organic compounds, we should consider the inorganic acids sulfuric acid and phosphoric acid.
Sulfuric acid is termed a dibasic acid, in that it has two ionizable groups, and phosphoric acid is a tribasic acid with three ionizable hydrogens. Thus, sulfuric acid has two pKa values and phosphoric acid has three.
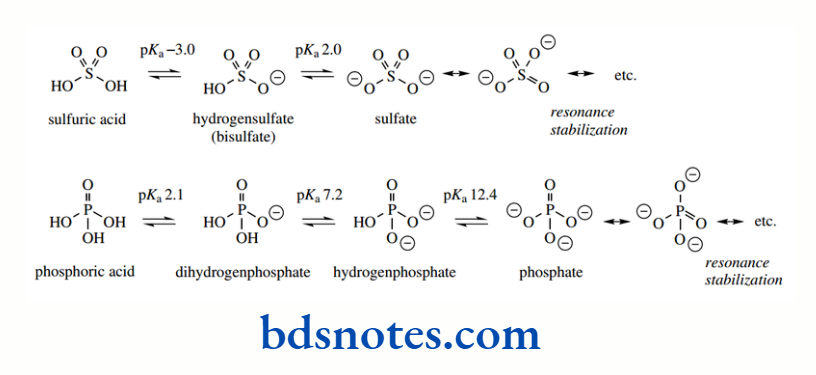
Both acids give rise to resonance-stabilized conjugate bases (compared to carboxylate) and are strong acids. Sulfuric acid is stronger, owing to improved resonance possibilities provided by the two S=O functions, as against just one P=O in phosphoric acid. Note particularly, though, that the pKa values for the second and third ionizations are higher than the first.
This indicates that the loss of a further proton from an ion is much less favourable than the loss of the first proton from the non-ionized acid.
Nevertheless, the sulfate dianion is sufficiently well resonance stabilized via the two S=O functions that hydrogensulfate (bisulfate) is still a fairly strong acid. We can generalize that it is going to be more difficult to lose a proton from an anion than from an uncharged molecule.
This is also true of polyfunctional acids, such as dicarboxylic acids. However, it is found that this effect diminishes as the negative centres become more separated.
Thus, pKa values for some simple aliphatic dicarboxylic acids are as shown, the loss of the first proton being represented by pKa1 and the loss of the second by pKa2.
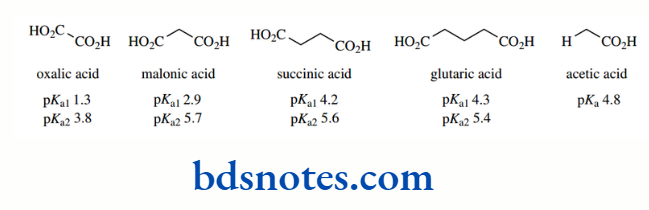
It can be seen that the difference between the first and second pKa values diminishes as the number of methylene groups separating the carboxyls increases, i.e. it becomes easier to lose the second proton as the other functional group is located further away.
It can also be seen that since malonic acid is a stronger acid than acetic acid, then the extra carboxyl is an electron-withdrawing substituent that is stabilizing.
The conjugate base. Again, this effect diminishes rapidly as the chain length increases, as anticipated for an inductive effect. Of course, ionization of a carboxylic acid group to a carboxylate anion reverses the inductive effect, in that the carboxylate will be electron donating, and will destabilize the dianion.
This is reflected in the pKa2 values for malonic, succinic and glutaric acids all being larger than the pKa for acetic acid.
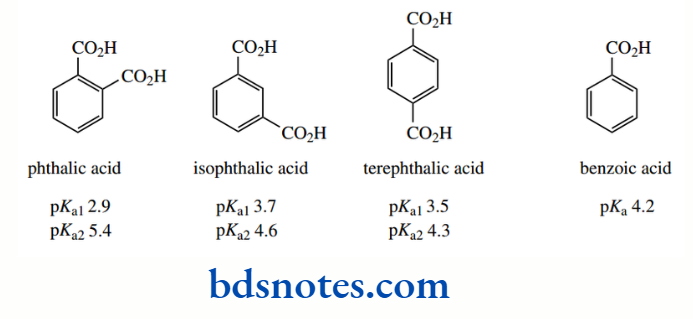
Oxalic acid appears anomalous in this respect, and this appears to be a result of the high charge density associated with the dianion and subsequent solvation effects. In the aromatic benzene dicarboxylic acid derivatives, the pattern is not dissimilar, especially since we have no oxalic acid-like anomaly.
Carboxylic acid groups are electron-withdrawing, and all three diacids are stronger acids than benzoic acid. On the other hand, the carboxylate group is electron donating, and this weakens the second ionization. This makes the second acid a weaker acid than benzoic acid. The effects are greatest in the ortho derivative, where there are also going to be steric factors.
Compounds with two basic groups, for example, diamines, can be rationalized similarly. Here, we must appreciate that both amino groups and ammonium cations are electron withdrawing, the positively charged entity having the greater effect. pKa values for a series of aliphatic diamines.
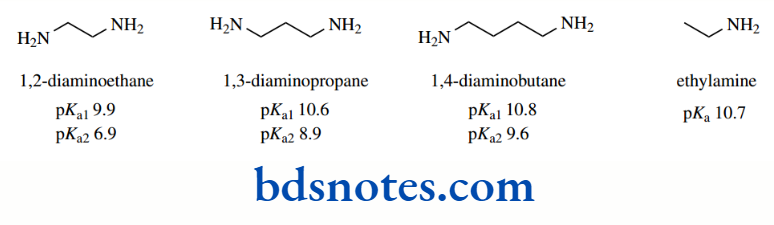
As the distance between the amino groups increases, the effect of the NH2 on the first protonation diminishes, so that pKa1 values for the 1,3- and 1,4-diamino compounds are very similar to that of ethylamine. Only in 1,2-diaminoethane do we see the electron-withdrawing effects of the second amino group decreasing basicity.
However, for the second protonation, it is clear that an ionized amino group has a much larger effect than a nonionized one.
The effects fall off as the separation increases but persist further. Thus, pKa2 values for the 1,3- and 1,4-diamino compounds are now rather different. The aromatic diamines present a much more complex picture, and we do not intend to justify the observed pKa values in detail.
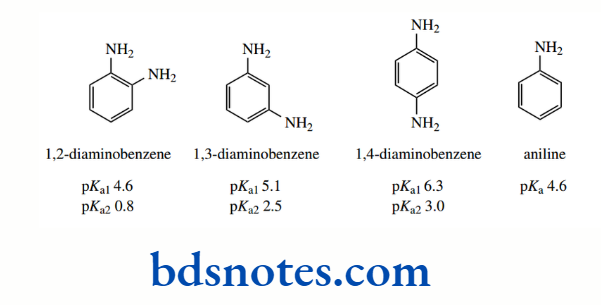
There are going to be several effects here, with some that provide opposing influences. An amino group has an electron-withdrawing inductive effect but has an electron-donating resonance effect that tends to be greater in magnitude than the inductive effect.
A protonated amino group also has an electron-withdrawing inductive effect that is greater than that of an uncharged amino group. On the other hand, it no longer supplies the electron-donating resonance effect.
As with other disubstituted benzenes, the ortho compound also experiences steric effects that may reduce the benefits of resonance. Both the meta and para diamines are stronger bases than aniline, and the protonation of the first amine in all three compounds considerably inhibits the second protonation.
pH
The acidity of an aqueous solution is normally measured in terms of pH. pH is defined as pH = − log10[H3O+] The lower the pH, the more acidic the solution; the higher the pH, the more basic the solution. The pH scale only applies to aqueous solutions and is only a measure of the acidity of the solution.
It does not indicate how strong the acid is (that is a function of pKa) and the pH of an acid will change as we alter its concentration. For instance, dilution will decrease the H3O+ concentration, and thus the pH will increase. In water, the hydronium ion concentration arises by the self-dissociation equilibrium.
⇒ \(\mathrm{H}_2 \mathrm{O}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{\oplus}+\mathrm{HO}^{\ominus}\)
In this reaction, one molecule of water acts as a base, accepting a proton from a second water molecule. The second molecule is acting as an acid and donating a proton.
For every hydronium ion produced, a hydroxide anion must also be formed, so that the concentrations of hydronium and hydroxide ions must be equal. In pure water at 25 ◦C, this value is found to be 10−7 M.
The equilibrium constant K is given by the formula and because the concentration of water is essentially constant in an aqueous solution, the new equilibrium constant K w (the ionization constant for water) is defined as
⇒ \(K=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]\left[\mathrm{HO}^{-}\right]}{\left[\mathrm{H}_2 \mathrm{O}\right]\left[\mathrm{H}_2 \mathrm{O}\right]}\)
K w = [HO−][H3O+] = 10−7 × 10−7 = 10−14
This means that the pH of pure water at 25 ◦C is therefore
pH = − log 10−7 = 7
pH 7 is regarded as neither acidic, nor basic, but neutral. It follows that acids have a pH less than 7 and bases have a pH greater than 7.
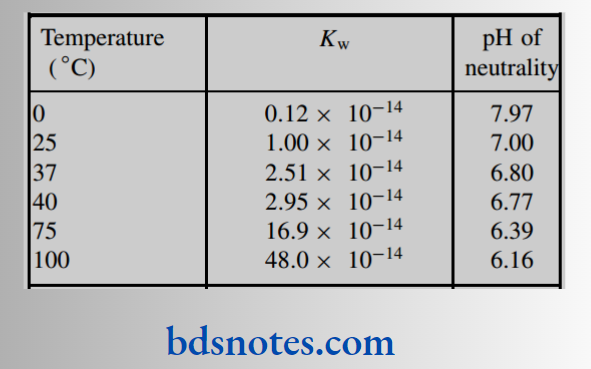
Kw and pH of neutrality at different temperatures We rapidly become accustomed to the idea that the pH of water is 7.0 and that this represents the pH of neutrality.
Unfortunately, this is only true at 25 C; at other temperatures, the amount of ionization varies, so that Kw will consequently be different. We find that the amount of ionization increases with temperature and the pH of neutrality decreases accordingly.
Kw and pH of neutrality at different temperatures
Calculation of pH: strong acids and bases A strong acid is considered to be completely ionized in water so that the hydronium ion concentration is the same as its molarity.
Thus, a 0.1 M solution of HCl in water has [H3O+] = 0.1, and pH = − log 0.1 = 1. Similarly, a 0.01 M solution has [H3O+] = 0.01 and pH = − log 0.01 = 2, and a 0.001 M solution has [H3O+] = 0.001 and pH = − log 0.001 = 3. It follows from this that, because we are using a logarithmic scale, a pH difference of 1 corresponds to a factor of 10 in hydronium ion concentration.
If the pH is known, then we can calculate the hydronium ion concentration. Since pH = − log[H3O+] the hydronium ion concentration is given by [H3O+] = 10−pH For example, if the pH = 4, [H3O+] = 10−4 = 0.0001 M.
When we have a strong base, our calculations need to invoke the ionization constant for water K w = [H3O+][HO−] = 10−14 Thus, the pH of a 0.1 M solution of NaOH in water is calculated from [HO−] = 0.1, and since [H3O+][HO−] = 10−14, [H3O+] must be 10−13.
Hence, the pH of a 0.1 M solution of NaOH in water will be − log 10−13 = 13. A 0.01 M solution of NaOH will have [HO−] = 10−2 and pH = − log 10−12 = 12, and a 0.001 M solution has [HO−] = 10−3 and pH = − log 10−11 = 11.
Weak acids are not completely ionized in aqueous solutions, and the amount of ionization, and thus hydronium ion concentration, is governed by the equilibrium.
⇒ \(\mathrm{HA}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{H}_3 \mathrm{O}^{\oplus}+\mathrm{A}^{\ominus}\) and the equilibrium constant Ka we defined above
⇒ \(K_{\mathrm{a}}=\frac{\left[\mathrm{A}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{[\mathrm{HA}]}\)
However, since [H3O+] must be the same as [A−], we can write
⇒ \(K_{\mathrm{a}}=\frac{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]^2}{[\mathrm{HA}]}\) and therefore
⇒ \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\sqrt{K_{\mathrm{a}}[\mathrm{HA}]}\)
If we take negative logarithms of both sides, we get \(-\log \left[\mathrm{H}_3 \mathrm{O}^{+}\right]=-\frac{1}{2} \log K_{\mathrm{a}}-\frac{1}{2} \log [\mathrm{HA}]\) which becomes
⇒ \(\mathrm{pH}=\frac{1}{2} \mathrm{p} K_{\mathrm{a}}-\frac{1}{2} \log [\mathrm{HA}]\)
Note: This is simply a variant of the Henderson–Hasselbalch equation below, when [A−] = [H3O+]. The calculation of the pH of a weak base may be approached in the same way. The equilibrium we need to consider is \(\mathrm{B}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{BH}^{\oplus}+\mathrm{HO}^{\ominus}\) and the equilibrium constant Kb will be defined as
⇒ \(K_{\mathrm{b}}=\frac{\left[\mathrm{HO}^{-}\right]\left[\mathrm{BH}^{+}\right]}{[\mathrm{B}]}\)
However, since [HO−] must be the same as [BH+], we can write
⇒ \(K_{\mathrm{b}}=\frac{\left[\mathrm{HO}^{-}\right]^2}{[\mathrm{~B}]}\) and therefore \(\left[\mathrm{HO}^{-}\right]=\sqrt{K_{\mathrm{b}}[\mathrm{B}]}\)
Now we need to remember that K w = [HO−][H3O+] so that we can replace [HO−] with Kw/[H3O+]; this leads to
⇒ \(\frac{K_{\mathrm{w}}}{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}=\sqrt{K_{\mathrm{b}}[\mathrm{B}]}\) so that we can replace [HO−] with Kw/[H3O+]; this leads to \(\frac{K_{\mathrm{w}}}{\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}=\sqrt{K_{\mathrm{b}}[\mathrm{B}]}\) and hence \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=\frac{K_{\mathrm{w}}}{\sqrt{K_{\mathrm{b}}[\mathrm{B}]}}\)
If we now take negative logarithms of both sides, we get \(-\log \left[\mathrm{H}_3 \mathrm{O}^{+}\right]=-\log K_{\mathrm{w}}+\frac{1}{2} \log K_{\mathrm{b}}+\frac{1}{2} \log [\mathrm{B}]\) Which becomes \(\mathrm{pH}=\mathrm{p} K_{\mathrm{w}}-\frac{1}{2} \mathrm{p} K_{\mathrm{b}}+\frac{1}{2} \log [\mathrm{B}]\)
Calculation of pH: weak acids and bases Consider a 0.1 M solution of the weak acid acetic acid (Ka = 1.76 × 10−5; pKa = 4.75). Since the degree of ionization is small, the concentration of undissociated acid may be considered to be approximately the same as the original concentration, i.e. 0.1. The pH can be calculated using the equation \(\mathrm{pH}=\frac{1}{2} \mathrm{p} K_{\mathrm{a}}-\frac{1}{2} \log [\mathrm{HA}]\)
Thus
pH = 2.38 − 0.5 × log 0.1
= 2.38 − (−0.5)
= 2.88
The calculation of the pH of a weak base can be achieved similarly; but again, since we have a base, our calculations need to invoke the ionization constant for water hus
pH = 2.38 − 0.5 × log 0.1
= 2.38 − (−0.5)
= 2.88
The calculation of the pH of a weak base can be achieved similarly; but again, since we have a base, our calculations need to invoke the ionization constant for water Kw = [H3O+][HO−] = 10−14 and pKa + pKb = 14. Thus, for a 0.1 M solution of ammonia (conjugate acid pKa = 9.24
⇒ \(\mathrm{pH}=\mathrm{p} K_{\mathrm{w}}-\frac{1}{2} \mathrm{p} K_{\mathrm{b}}+\frac{1}{2} \log [\mathrm{B}]\)
and pKb is thus 14 − 9.24 = 4.76. This leads to
pH = 14 − 2.38 + 0.5 × log 0.1
= 14 − 2.38 + 0.5(−1)
= 11.12
Alternatively, we could use
⇒ \(\mathrm{pH}=\frac{1}{2} \mathrm{p} K_{\mathrm{w}}+\frac{1}{2} \mathrm{p} K_{\mathrm{a}}+\frac{1}{2} \log [\mathrm{B}]\) To get the same result:
pH = 7 + 4.62 + 0.5 × log 0.1 = 11.12 These calculations are for the pH of weak acids and weak bases. It is well worth comparing the figures we calculated above for strong acids and bases. Thus, a 0.1 M solution of the strong acid HCl had pH 1, and a 0.1 M solution of the strong base NaOH had pH 13.
Although this produces a similar type of expression to that for the pH of a weak acid above, it does employ pKb rather than pKa. To keep to a ‘pKa only’ concept, we need to incorporate the pKa + pKb = pKw expression. Then we get the alternative formula.
⇒ \(\begin{gathered}
\mathrm{pH}=\mathrm{p} K_{\mathrm{w}}-\frac{1}{2}\left(\mathrm{p} K_{\mathrm{w}}-\mathrm{p} K_{\mathrm{a}}\right)+\frac{1}{2} \log [\mathrm{B}] \\
\mathrm{pH}=\frac{1}{2} \mathrm{p} K_{\mathrm{w}}+\frac{1}{2} \mathrm{p} K_{\mathrm{a}}+\frac{1}{2} \log [\mathrm{B}]
\end{gathered}\)
The pH of salt solutions It should be self-evident that solutions comprised of equimolar amounts of a strong acid, for example, HCl, and a strong base, for example, NaOH, will be neutral, i.e. pH 7.0 at 25 ◦C.
We can thus deduce that a solution of the salt NaCl in water will also have a pH of 7.0. However, salts of a weak acid and strong base or a strong acid and weak base dissolved in water will be alkaline or acidic respectively.
Thus, aqueous sodium acetate is basic, whereas aqueous ammonium chloride is acidic. pH values may be calculated from pKa as follows. Consider the ionization of sodium acetate in water; this leads to an equilibrium in which AcO− acts as base \(\Theta_{\mathrm{OAc}}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{HOAc}+\mathrm{HO}^{\ominus}\)
⇒ \(\begin{aligned}
\mathrm{pH} & =\frac{1}{2} \mathrm{p} K_{\mathrm{w}}+\frac{1}{2} \mathrm{p} K_{\mathrm{a}}+\frac{1}{2} \log [\mathrm{B}] \\
& =7+2.38+0.5 \times \log 0.1 \\
& =7+2.38+0.5 \times(-1) \\
& =8.88
\end{aligned}\)
If we now consider a 0.1 M solution of ammonium chloride in water, where pKa for the conjugate acid NH4+ is 9.24, we have the equilibrium.
⇒ \(\mathrm{NH}_4^{\oplus}+\mathrm{H}_2 \mathrm{O} \rightleftharpoons \mathrm{NH}_3+\mathrm{H}_3 \mathrm{O}^{\oplus}\) in which the ammonium ion is acting as an acid. For the ionization of a weak acid, we calculated above that the pH is given by the equation. \(\mathrm{pH}=\frac{1}{2} \mathrm{p} K_{\mathrm{a}}-\frac{1}{2} \log [\mathrm{HA}]\)
Thus \(\begin{aligned}
\mathrm{pH} & =4.62-0.5 \times \log 0.1 \\
& =4.62-0.5(-1) \\
& =5.12
\end{aligned}\).
In both cases, we are assuming that the concentration of the ion (either AcO− or NH4+) is not significantly altered by the equilibrium and can, therefore, be considered to be equivalent to the molar concentration.
The Henderson–Hasselbalch equation
Ka for the ionization of an acid HA has been defined as
⇒ \(K_{\mathrm{a}}=\frac{\left[\mathrm{A}^{-}\right]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{[\mathrm{HA}]}\) and this can be rearranged to give \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=K_{\mathrm{a}} \times \frac{[\mathrm{HA}]}{\left[\mathrm{A}^{-}\right]}\)
Taking negative logarithms of each side, this becomes \(-\log \left[\mathrm{H}_3 \mathrm{O}^{+}\right]=-\log K_{\mathrm{a}}+\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}\)
or \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}\)
This is referred to as the Henderson–Hasselbalch equation, and it is sometimes written as
⇒ \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{[\text { base }]}{\text { [acid] }}\)
Using this relationship, it is possible to determine the degree of ionization of an acid at a given pH. An immediate outcome from this expression is that the pKa of an acid is the pH at which it is exactly half dissociated. This follows from \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}\)
But when the concentrations of acid HA and conjugate base A− are equal, then
⇒ \(\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=\log 1=0\)
So that pH = pKa
This means we can determine the pKa of an acid by measuring the pH at the point where the acid is half neutralized. As we increase the pH, the acid becomes more ionized; as we lower the pH, the acid becomes less ionized. For a base, Ka is defined as
⇒ \(K_{\mathrm{a}}=\frac{[\mathrm{B}]\left[\mathrm{H}_3 \mathrm{O}^{+}\right]}{\left[\mathrm{BH}^{+}\right]}\)
which can be rearranged to give \(\left[\mathrm{H}_3 \mathrm{O}^{+}\right]=K_{\mathrm{a}} \times \frac{\left[\mathrm{BH}^{+}\right]}{[\mathrm{B}]}\)
so that the Henderson–Hasselbalch equation is written \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{[\mathrm{B}]}{\left[\mathrm{BH}^{+}\right]}\) \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{[\mathrm{B}]}{\left[\mathrm{BH}^{+}\right]}\) or, as previously
⇒ \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\text { [base] }}{\text { [acid] }}\)
Again, we can see that the pKa of a base is the pH at which it is half ionized. As we increase the pH, the base becomes less ionized; as we lower the pH, the base becomes more ionized.
A further useful generalization can be deduced from the Henderson–Hasselbalch equation. This relates to the ratio of ionized to non-ionized forms as the pH varies.
A shift in pH by one unit to either side of the pKa value must change the ratio of ionized to non-ionized forms by a factor of 10. Every further shift of pH by one unit changes the ratio by a further factor of 10.
Thus, for example, if the pKa of a base is 10, at pH 7 the ratio of free base to protonated base is 1:103. An acid with pKa 2 at pH 7 would produce a ratio of acid to anion of 105:1.
Calculation of percentage ionization
Using the Henderson–Hasselbalch equation, we can easily calculate the amount of ionized form of an acid or base present at a given pH, provided we know the pKa. For example, consider aqueous solutions of acetic acid (pKa 4.75) first at pH 4.0 and then at pH 6.0. Since \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}\) at pH 4.0
⇒ \(\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=\mathrm{pH}-\mathrm{p} K_{\mathrm{a}}=4.0-4.75=-0.75\)
Thus
⇒ \(\frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=10^{-0.75}=0.18\)
If we consider [A−] = I, the fraction ionized, then [HA] is the fraction non-ionized, i.e. 1 − I, and I/(1 − I) = 0.18, from which I may be calculated to be about 0.15 or 15%. At pH 6.0, pH – pKa = 1.25, and the calculation yields I/(1 − I) = 101.25 = 17.8, so that I = 0.95, i.e. 95% ionized. With a base such as ammonia (pKa 9.24), the percentages ionized at pH 8.0 and 10.0 are calculated as follows:
⇒ \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{[\mathrm{B}]}{\left[\mathrm{BH}^{+}\right]}\)
At pH 8.0
⇒ \(\log \frac{[\mathrm{B}]}{\left[\mathrm{BH}^{+}\right]}=\mathrm{pH}-\mathrm{p} K_{\mathrm{a}}=8.0-9.24=-1.24\)
Now with bases, [B] is the non-ionized fraction 1 − I and [BH+] is the ionized fraction I, so (1 − I)/I = 0.057, and therefore I = 0.95, i.e. 95% ionized. At pH 10.0, the calculation yields (1 − I)/I = 100.76 = 5.75, so that I = 0.15, i.e. 15% ionized.
The ionization a-aminoino acids a t p H 7
Peptides and proteins are composed of α-amino acids linked by amide bonds. Their properties, for example, the ability of enzymes to catalyse biochemical reactions, are dependent upon the degree of ionization of various acidic and basic side chains at the relevant pH.
This aspect will be discussed, but, here, let us consider a simple amino acid dissolved in water at pH 7.0. An α-amino acid has an acidic carboxylic acid group and a basic amine group. Both of these entities need to be treated separately.
The carboxylic acid groups of amino acids have pKa values in a range from about 1.8 to 2.6 (see Section 13.1). Let us consider a typical carboxylic acid group with pKa 2.0. Using the Henderson–Hasselbalch equation
We can deduce that \(\log \frac{\left[\mathrm{RCO}_2^{-}\right]}{\left[\mathrm{RCO}_2 \mathrm{H}\right]}=\mathrm{pH}-\mathrm{p} K_{\mathrm{a}}=7.0-2.0=5.0\)
Thus \(\frac{\left[\mathrm{RCO}_2{ }^{-}\right]}{\left[\mathrm{RCO}_2 \mathrm{H}\right]}=10^5=10000: 1\)
Therefore, the carboxylic acid group of an amino acid can be considered to be completely ionized in solution at pH 7.0. Now let us consider the amino group in α-amino acids. The pKa values of the conjugate acids are found to range from about 8.8 to 10.8. We shall consider a typical group with pKa 10.0. From
⇒ \(\begin{gathered}
\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\left[\mathrm{RNH}_2\right]}{\left[\mathrm{RNH}_3{ }^{+}\right]} \\
\log \frac{\left[\mathrm{RNH}_2\right]}{\left[\mathrm{RNH}_3{ }^{+}\right]}=\mathrm{pH}-\mathrm{p} K_{\mathrm{a}}=7.0-10.0=-3.0 \\
\frac{\left[\mathrm{RNH}_2\right]}{\left[\mathrm{RNH}_3{ }^{+}\right]}=10^{-3}=1: 1000
\end{gathered}\)
Therefore, as with the carboxylic acid group, we find that the amino group of an amino acid is effectively ionized completely, i.e. fully protonated, in solution at pH 7.0. Therefore, we can deduce that α-amino acids in solution at pH 7.0 exist as dipolar ions; these are called zwitterions (German; Twitter = hybrid)
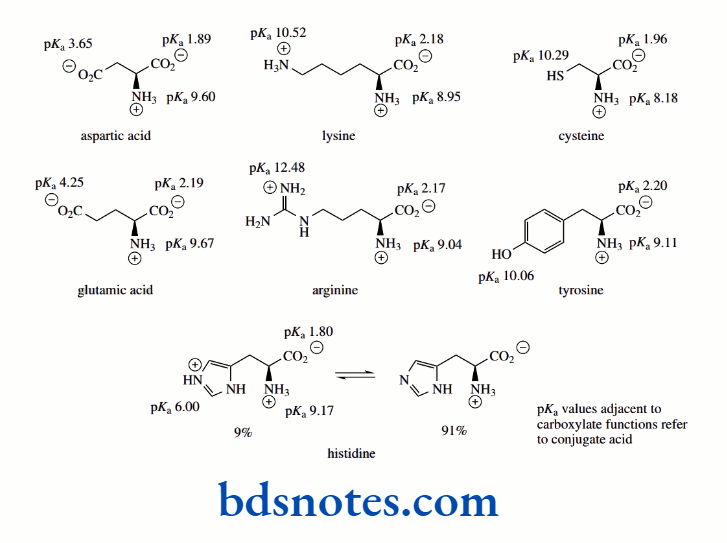
Some amino acids have additional ionizable groups in their side chains. These may be acidic or potentially acidic (aspartic acid, glutamic acid, tyrosine, cysteine), or basic (lysine, arginine, histidine).
We use the term ‘potentially acidic’ to describe the phenol and thiol groups of tyrosine and cysteine respectively; under physiological conditions, these groups are unlikely to be ionized.
It is relatively easy to calculate the amount of ionization at a particular pH and to justify that latter statement.
Similar calculations as above for the basic side-chain groups of arginine (pKa 12.48) and lysine (pKa 10.52), and the acidic side-chains of aspartic acid (pKa 3.65) and glutamic acid (pKa 4.25) show essentially complete ionization at pH 7.0.
However, for cysteine (pKa of the thiol group 10.29) and for tyrosine (pKa of the phenol group 10.06) there will be negligible ionization at pH 7.0. For cysteine at pH 7.0, the Henderson–Hasselbalch equation leads to
⇒ \(\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=\mathrm{pH}-\mathrm{p} K_{\mathrm{u}}=7.0-10.29=-3.29\)
And \(\frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=10^{-3.29}=5.1 \times 10^{-4}\)
Interestingly, the heterocyclic side-chain of histidine is partially ionized at pH 7.0. This follows from
⇒ \(\begin{gathered}
\log \frac{[\mathrm{B}]}{\left[\mathrm{BH}^{+}\right]}=\mathrm{pH}-\mathrm{p} K_{\mathrm{z}}=7.0-6.00=-1.0 \\
\frac{[\mathrm{B}]}{\left[\mathrm{BH}^{+}\right]}=10^{-1.0}=10
\end{gathered}\)
Which translates to approximately 9.1% ionization. We shall see that this modest level of ionization is particularly relevant in some enzymic reactions where histidine residues play an important role.
Note, however, that when histidine is bound in a protein structure, pKa values for the imidazole ring vary somewhat in the range 6–7 depending upon the protein, thus affecting the level of ionization. The ionic states at pH 7.0 of these amino acids with ionizable side chains are shown below.
Buffers
A buffer is a solution that helps to maintain a reasonably constant pH environment by countering the effects of added acids or bases.
They are used extensively for the handling of biochemicals, especially enzymes, as well as in chromatography and drug extractions. The simplest type of buffer is composed of a weak acid–strong base combination or a weak base–strong acid combination.
This may be prepared by combining the weak acid (or base) with its salt. For example, the sodium acetate–acetic acid combination is one of the most common buffer systems.
Although tabulated data are available for the preparation of buffer solutions, a sodium acetate–acetic acid buffer could be prepared simply by adding sodium hydroxide to an acetic acid solution until the required pH is obtained.
For maximum efficiency, this pH needs to be within about 1 pH unit on either side of the pKa of the weak acid or base used.
Since acetic acid is only weakly dissociated, the concentration of acetic acid will be almost the same as the amount put in the mixture.
On the other hand, the sodium acetate component will be almost completely dissociated, so the acetate ion concentration can be considered the same as that of the sodium acetate used for the solution.
The addition of an acid such as HCl to the buffer solution provides H+, which combines with the acetate ion to give acetic acid.
This has a twofold effect: it reduces the amount of acetate ion present and, by so doing, also increases the amount of undissociated acetic acid.
Provided the amount of acid added is small relative to the original concentration of base in the buffer, the alteration in base: acid ratio in the Henderson–Hasselbalch equation is relatively small and has little effect on the pH value.
Similar considerations apply if a base such as NaOH is added to the buffer solution. This will decrease the amount of undissociated acid, and increase the amount of acetate ion present. The Henderson–Hasselbalch equation may be employed in calculations relating to the properties and effects of buffer solutions.
Preparation of a buffer
One litre of 0.1 M sodium acetate buffer with a pH of 4.9 is required. The pKa of acetic acid is 4.75. From the Henderson–Hasselbalch equation \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}\)
Therefore \(4.9=4.75+\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}\)
So that \(\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=0.15\) and \(\frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}=10^{0.15}=\frac{1.41}{1}\)
This means that the buffer solution requires 1.41 parts sodium acetate to 1 part acetic acid. Therefore, this can be prepared by mixing 1.41/2.41 = 0.585 l of 0.1 M sodium acetate with 1/2.41 = 0.415 l of 0.1 M acetic acid. The amount of sodium acetate in 1 l of solution will thus be 0.0585 M, and the amount of acetic acid will be 0.0415 M.
Buffering effect
If 1 ml of 1 M HCl is added to this sodium acetate buffer solution, the pH change may be calculated as follows. Again, we require the Henderson–Hasselbalch equation:
⇒ \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\left[\mathrm{A}^{-}\right]}{[\mathrm{HA}]}\)
We are adding [H3O+] of 0.001 M, and this reacts
⇒ \(\Theta_{\mathrm{OAc}}+\mathrm{HCl} \longrightarrow \mathrm{HOAc}+\mathrm{Cl}^{\ominus}\)
so effectively reducing the amount of acetate base by 0.001 M and also increasing the amount of acetic acid by 0.001 M. We can ignore the small change in volume arising from the addition of the acid. The Henderson–Hasselbalch equation becomes
⇒ \(\mathrm{pH}=4.75+\log \frac{0.0585-0.001}{0.0415+0.001}\)
So
⇒ \(\begin{aligned}
\mathrm{pH}=4.75+\log \frac{0.0575}{0.0425} & =4.75+\log 1.35 \\
& =4.75+0.13=4.88
\end{aligned}\)
It can be seen, therefore, that the effect of the addition of the acid is to change the pH value from 4.90 to 4.88, i.e. by just 0.02 of a pH unit.
This contrasts with the effect of adding 0.001 M of HCl to 1 litre of water (pH 7). The new [H3O+] of 0.001 M gives pH = − log[H3O+] = − log 0.001 = 3, i.e. a change of four pH units. If 1 ml of 1 M NaOH was added to this buffer solution, the pH change may be calculated similarly.
We are adding [HO−] of 0.001 M, and this reacts effectively increasing the amount of acetate base by 0.001 M and also decreasing the amount of acetic acid by 0.001 M. The Henderson–Hasselbalch equation becomes
⇒ \(\mathrm{HOAc}+\mathrm{HO}^{\ominus} \longrightarrow \mathrm{H}_2 \mathrm{O}+{ }_{\mathrm{OAc}}\)
⇒ \(\mathrm{pH}=4.75+\log \frac{0.0575+0.001}{0.0415-0.001}\)
So
⇒ \(\begin{aligned}
\mathrm{pH}=4.75+\log \frac{0.0585}{0.0405} & =4.75+\log 1.44 \\
& =4.75+0.16=4.91
\end{aligned}\)
Again, the pH change is minimal, which is the whole point of a buffer solution. Note also that the pH of a buffer solution is essentially independent of dilution. An unbuffered solution of an acid or base would suffer a pH change on dilution because pH relates to hydronium ion concentration.
Dilution of a buffered solution does not affect pH because any such changes are accommodated in the log([A−]/[HA]) component and, therefore, cancel out. We have made certain approximations in deriving the equations, and at very high dilutions the pH does begin to deviate.
The sodium acetate–acetic acid combination is one of the most widely used buffers and is usually referred to simply as acetate buffer.
Other buffer combinations commonly employed in chemistry and biochemistry include carbonate–bicarbonate (sodium carbonate–sodium hydrogen carbonate), citrate (citric acid–trisodium citrate), phosphate (sodium dihydrogen phosphate–disodium hydrogen phosphate), and tris [tris(hydroxymethyl)aminomethane–HCl].
The buffering effect of blood plasma
In humans, the pH of blood is held at a remarkably constant value of 7.4 ± 0.05. In severe diabetes, the pH can drop to pH 7.0 or below, leading to death from acidotic coma.
Death may also occur at pH 7.7 or above because the blood is unable to release CO2 into the lungs.
The pH of blood is normally controlled by a buffer system, within rather narrow limits to maintain life and within even narrower limits to maintain health. The buffering system for blood is based on carbonic acid (H2CO3) and its conjugate base bicarbonate (HCO3−)
⇒ \(\mathrm{H}_2 \mathrm{CO}_3 \rightleftharpoons \mathrm{H}^{\oplus}+\mathrm{HCO}_3^{\ominus}\)
From the Henderson–Hasselbalch equation \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\left[\mathrm{HCO}_3^{-}\right]}{\left[\mathrm{H}_2 \mathrm{CO}_3\right]}\)
we can see that maintaining the pH depends upon the ratio of bicarbonate to carbonic acid concentrations.
Large quantities of acid formed during normal metabolic processes react with bicarbonate to form carbonic acid. This, however, dissociates and rapidly loses water to form CO2 that is removed via the lungs.
⇒ \(\mathrm{H}_2 \mathrm{CO}_3 \rightleftharpoons \mathrm{H}_2 \mathrm{O}+\mathrm{CO}_2\)
The pH is maintained, therefore, in that a reduction in [HCO3−] is countered by a corresponding decrease in [H2CO3]. The increase in metabolic acid is compensated by a corresponding increase in CO2. If the pH of blood rises, [HCO3−] temporarily increases.
The pH is rapidly restored when atmospheric CO2 is absorbed and converted into H2CO3. It is a reservoir of CO2 that enables the blood pH to be maintained so rigidly. This reservoir of CO2 is large and can be altered quickly via the breathing rate.
Using pKa values
Predicting acid-base interactions
With knowledge of pKa values, or a rough idea of relative values, one can predict the outcome of acid-base interactions.
This may form an essential preliminary to many reactions, or provide us with an understanding of whether a compound is ionized under particular conditions, and whether or not it is in a soluble form.
As a generalization, acid-base interactions result in the formation of the weaker acid and the weaker base, a consequence of the most stable species being favoured at equilibrium. Thus, a carboxylic acid such as acetic acid will react with aqueous sodium hydroxide to form sodium acetate and water.
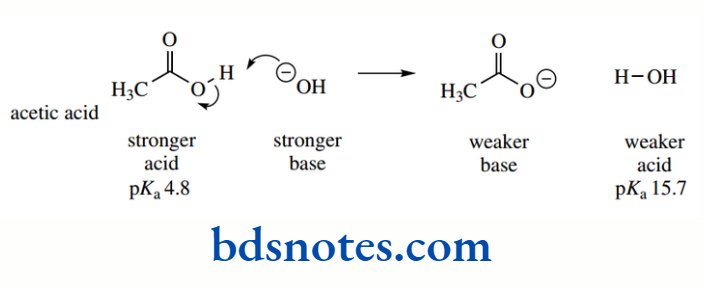
Consider the pKa values. Acetic acid (pKa 4.8) is a stronger acid than water (pKa 15.7), and hydroxide is a stronger base than acetate. Accordingly, hydroxide will remove a proton from acetic acid to produce acetate and water, the weaker base–weaker acid combination.
Because of the large difference in pKa values, the position of equilibrium will greatly favour the products, and we can indicate this by using a single arrow and considering the reaction to be effectively irreversible.
Even acids that are not particularly soluble in water, for example, benzoic acid, will participate in this reaction because the conjugate base benzoate is a water-soluble anion.
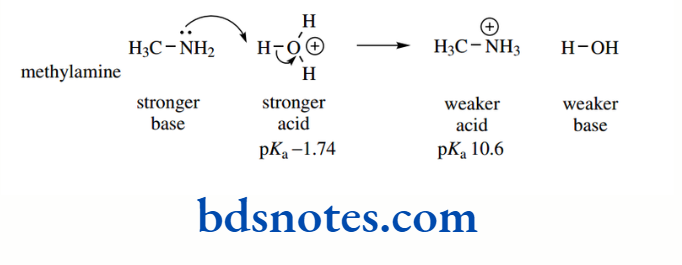
Bases can be considered in the same way. Thus, methylamine will react with aqueous HCl to produce methylammonium chloride and water. Hydronium is a stronger acid than methylammonium, and methylamine is a stronger base than water, so methylamine will become protonated in aqueous acid.
Again, there is a large difference in pKa values, so the position of equilibrium is well over to the righthand side. Bases that are not particularly soluble in water, e.g. aniline, can easily be made soluble by conversion to the salt form.
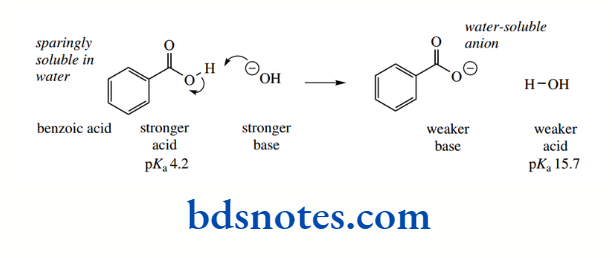
However, attempts to make an aqueous solution of the base sodium amide would result in the formation of sodium hydroxide and ammonia.
The amide ion is a strong base and abstracts a proton from water, a weak acid. The reverse reaction is not favoured, in that hydroxide is a weaker base than the amide ion, and ammonia is a weaker acid than water.
Take care with the terminology ‘amide’: the amide anion H2N− is quite different from the amide molecule RCONH2.
In general, in aqueous solutions, water can donate a proton to any base stronger than the hydroxide ion. If we wish to use bases that are stronger than the hydroxide ion, then we must employ a solvent that is a weaker acid than water.
For example, hydrocarbons (pKa about 50), ethers (pKa about 50), or even liquid ammonia (pKa 38). Since these are all extremely weak acids, they will not donate a proton even to a strong base such as an amide ion. Thus, in liquid ammonia, the amide ion may be used to convert an acetylene to its acetylide ion conjugate base.
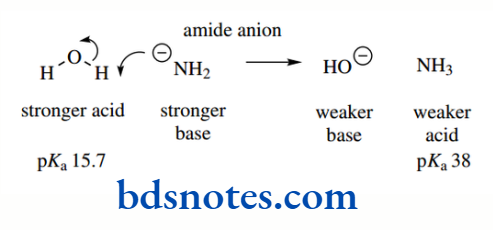
Similarly, the amide ion could be used to abstract a proton from a ketone to produce an enolate anion in an essentially irreversible reaction, since the difference in acidities of the ketone and ammonia is so marked.
However, if the base chosen were ethoxide, then enolate anion formation would be dependent on an equilibration reaction since the two acids are more comparable in acidity. As we shall see, this latter system may be used, provided the equilibrium can be disturbed in favour of the products.
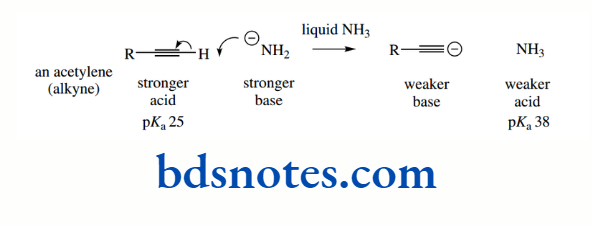
Sodium ethoxide may be produced by treating ethanol with sodium hydride. Again, hydride is the strong base, the conjugate base of the weak acid hydrogen, so the reaction proceeds readily.
Sodium tert-butoxide (from sodium hydride treatment of tertbutanol) is a stronger base than sodium ethoxide since tert-butanol (pKa 19) is less acidic than ethanol (pKa 16).
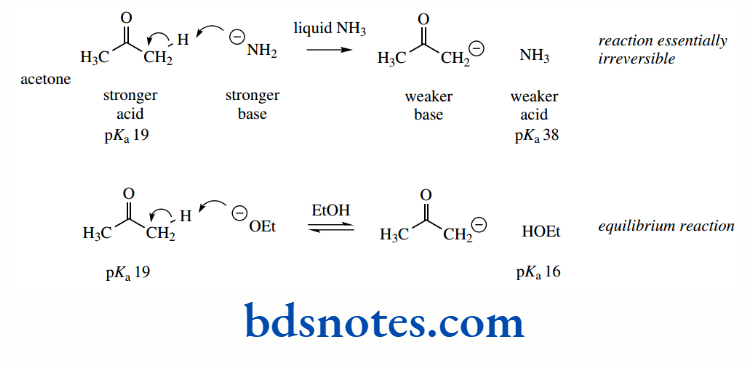
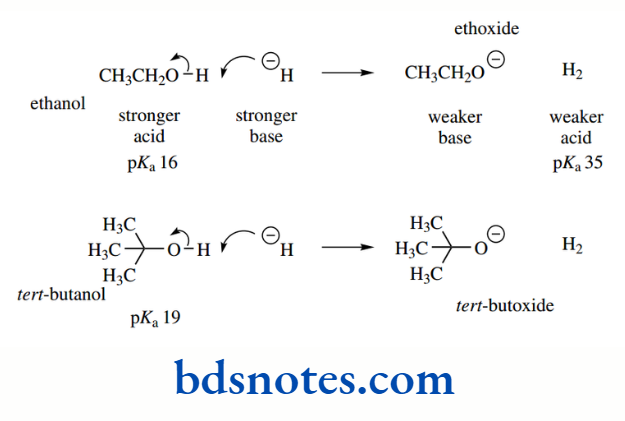
Some bases and acids that are commonly used as reagents to initiate reactions are listed in Table 4.10, in decreasing order of basicity or acidity.
Isotopic labelling using basic reagents By definition, an acid will donate a proton to a base, and it is converted into its conjugate base. Conversely, a base will accept a proton from a suitable donor, generating its conjugate acid.
We can utilize these properties to label certain compounds with isotopes of hydrogen, namely deuterium (2H or D) and tritium (3H or T). To differentiate normal hydrogen (1 H) from deuterium and tritium, it is sometimes referred to as protium.
Because these isotopes are easily detectable and measurable by spectroscopic methods, labelling allows us to follow the fate of particular atoms during chemical reactions, or metabolic studies.
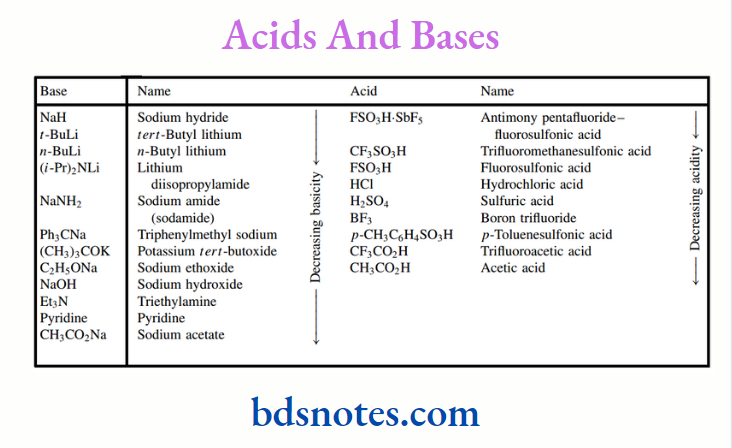
In general, the labelling process is one of exchange labelling, removing protium from an acid, and allowing the conjugate base to accept isotopic hydrogen from a suitable donor, most conveniently and cheaply supplied as labelled water. If the labelled compound is going to be of use, say in metabolic studies, the labelling must be achieved at a position that does not easily exchange again in an aqueous environment. This rules out hydrogens attached to oxygen or nitrogen that can be exchanged through simple acid-base equilibria.
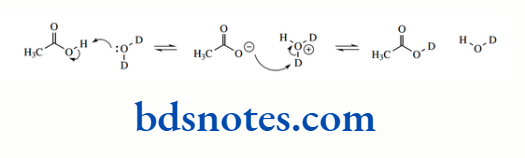
Thus, dissolving acetic acid in deuterated water will rapidly give deuterated acetic acid by acid-base equilibria. However, if the deuterated acetic acid were then dissolved in normal water, the reverse process would wash out the label equally rapidly.
Useful labelled compounds containing deuterium or tritium normally require the isotopic hydrogen to be attached to carbon, so the acid-base equilibrium will require cleavage of a C–H bond, where acid strength is usually very weak.
This will necessitate the use of a very strong base to achieve the formation of the conjugate base. A simple example follows from the reactions considered in Section 4.11.1. We saw that we needed to use a strong base such as the amide ion to form the conjugate base of an acetylene. This reaction was favoured, in that the products were the weaker base acetylide and the weaker acid ammonia.
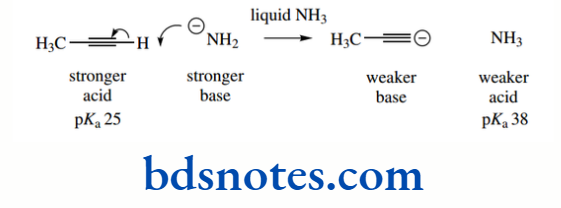
Upon completion of this ionization, we can then add labelled water D2O. Under these conditions, labelling occurs through the abstraction of a deuteron 2H+ from D2O. This is feasible because acetylide is a stronger base than hydroxide and water is a stronger acid than acetylene.
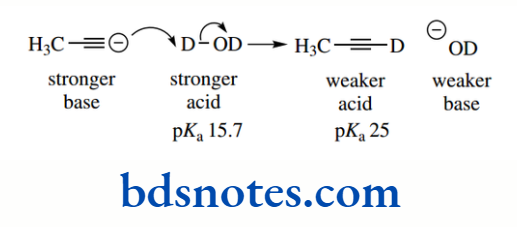
It is also possible to produce deuterium-labelled acetaldehyde by an exchange reaction with D2O and NaOD. This results in an exchange of all three α-hydrogens for deuterium and depends upon the generation of the conjugate base of acetaldehyde under basic conditions.
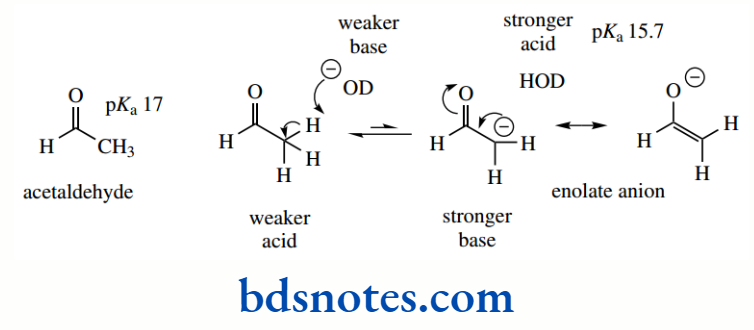
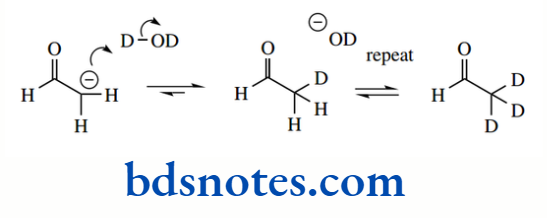
Unlike the example of the acetylene above, the feature of this process is that it is an equilibrium. This is because acetaldehyde is a weak acid (pKa 17), a weaker acid in fact than water (pKa 15.7).
In other words, the conjugate base of acetaldehyde (the enolate anion) is a stronger base than hydroxide.
Treatment of acetaldehyde with hydroxide thus generates an equilibrium mixture, with only a small amount of enolate anion present. Nevertheless, since we get a small amount of conjugate base, this can abstract a deuteron from the solvent D2O in the reverse reaction.
Provided excess D2O is available, equilibration allows the exchange of all three hydrogens in the methyl group of acetaldehyde. The label introduced is unfortunately not stable enough, in that similar treatment with a strong base and H2O will reverse the process and incorporate 1 H.
Note that the aldehydic hydrogen is not acidic and, therefore, not removed by base. This follows from consideration of the conjugate base, which has no stabilizing features.
The amount of enolate anion present at equilibrium may be calculated from the pKa values:
⇒ \(K=\frac{[\text { enolate }]\left[\mathrm{H}_2 \mathrm{O}\right]}{[\text { acetaldehyde }]\left[\mathrm{HO}^{-}\right]}\)
⇒ \(\begin{aligned}
& =\frac{[\text { enolate }]\left[\mathrm{H}^{+}\right]}{[\text {acetaldehyde }]} \times \frac{\left[\mathrm{H}_2 \mathrm{O}\right]}{\left[\mathrm{HO}^{-}\right]\left[\mathrm{H}^{+}\right]}=10^{-17} \frac{1}{10^{-15.7}} \\
& =10^{-1.3}=0.05
\end{aligned}\)
i.e. the proportion of enolate is about 5%. Despite the unfavourable equilibrium, this type of reaction works surprisingly well under relatively mild conditions.
By using a much stronger base, example sodium hydride or lithium diisopropylamide, the generation of the conjugate base would be essentially complete.
Treating the enolate anion with D²O would give a deuterium-labelled acetaldehyde, but only one atom of deuterium would be introduced. It is the equilibration process that allows the exchange of all three hydrogens.
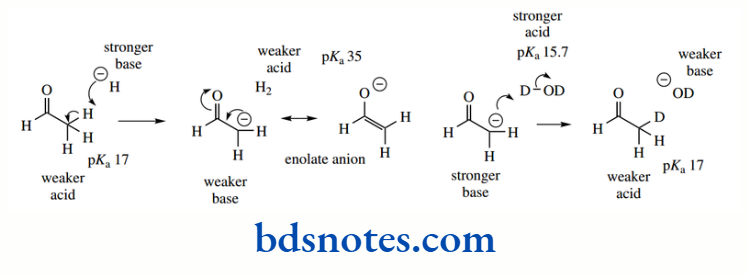
Amphoteric compounds: ami no acid ds Amphoteric compounds are compounds that may function as either acid or base, depending upon conditions. We have already met this concept in Section 4.5.4, where simple alcohols and amines have two pKa values according to whether the compound loses or gains a proton.
Of course, with alcohols and amines, acidity and basicity involve the same functional group. Other amphoteric compounds may contain separate acidic and basic groups. Particularly important examples of this type are the amino acids that makeup proteins.
The carboxylic acid groups of protein amino acids have pKa values of about 2, ranging from about 1.8 to 2.6, making them significantly more acidic than simple alkanoic acids (pKa about 5).
For the amino groups, the pKa values of the conjugate acids are found to range from about 8.8 to 10.8, with most of them in the region 9–10. These values are thus much closer to those of simple amines (pKa about 10).
Protein amino acids are α-amino acids, the amino and carboxylic acid groups being attached to the same carbon. Thus, the groups are close and will exert maximum inductive effects.
The increased acidity of the carboxylic group, therefore, reflects the electron-withdrawing inductive effect of the amino group, or, more correctly, the ammonium ion. This is because we should not consider the amino acid as the nonionized structure but as the doubly charged form termed a zwitterion (German: Twitter = hybrid)
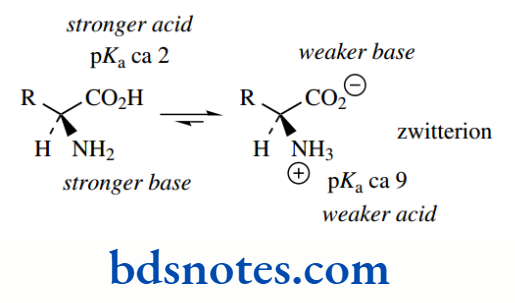
Consider the two pKa values. The carboxylic acid group (pKa²) is a stronger acid than the protonated NH² group (pKa 9). Thus, the carboxylic acid will protonate the amino group, and, in pure water (pH 7), amino acids having neutral side chains will exist predominantly as the doubly charged zwitterion.
This may be considered an internal salt, and could be compared to ammonium acetate, the salt formed when ammonia (pKa 9.2) reacts with acetic acid (pKa 4.8)
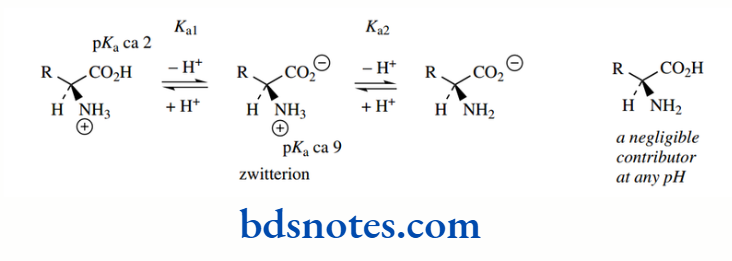
At low pH (acidic solution), an amino acid will exist as the protonated ammonium cation, and at high pH (basic solution) as the amino carboxylate anion. The intermediate zwitterion form will predominate at pHs between these extremes.
The uncharged amino acid has no real existence at any pH. Ironically, we are so familiar with the terminology amino acid, yet such a structure has no real existence! Amino acids are ionic compounds, solids with a high melting point.
We can appreciate that the ionization of the carboxylic acid is affected by the electron-withdrawing inductive effect of the ammonium residue; hence the increased acidity when compared with an alkanoic acid.
Similarly, the loss of a proton from the ammonium cation of the zwitterion is influenced by the electron-donating inductive effect from the carboxylate anion, which should make the amino group more basic than a typical amine. That this is not the case is thought to be a solvation effect (compare simple amines).
The pH at which the concentration of the zwitterion is a maximum is equal to the isoelectric point pI, strictly that pH at which the concentrations of cationic and anionic forms of the amino acid are equal. With a simple amino acid, this is the mean of the two pKa values:
⇒ \(\mathrm{pH}=\frac{\mathrm{p} K_{\mathrm{al}}+\mathrm{p} K_{\mathrm{a} 2}}{2}\)
This is deduced from \(K_{\mathrm{a} 1}=\frac{\left[\mathrm{H}^{+}\right][\mathrm{zw} \text { zwitterion }]}{[\text { cation }]} \text { and } K_{\mathrm{a} 2}=\frac{\left[\mathrm{H}^{+}\right][\text {anion }]}{[\text { zwitterion }]}\)
It follows, therefore, that pH = pKa1 when [cation] = [zwitterion], and that pH = pKa2 when [zwitterion] = [anion]. At the isoelectric point, [cation] = [anion]; thus
⇒ \(\begin{aligned}
{[\text { cation }] } & =\frac{\left[\mathrm{H}^{+}\right][\mathrm{zwitterion}]}{K_{\mathrm{a} 1}} \\
& =[\text { anion }]=\frac{K_{\mathrm{a} 2}[\mathrm{zwitterion}]}{\left[\mathrm{H}^{+}\right]}
\end{aligned}\)
Therefore \(K_{\mathrm{a} 1} \times K_{\mathrm{a} 2}=\left[\mathrm{H}^{+}\right]^2\) and by taking negative logarithms, it follows that \(\mathrm{pH}=\frac{\mathrm{p} K_{\mathrm{a} 1}+\mathrm{p} K_{\mathrm{a} 2}}{2}\)
For alanine, pKa1 = 2.34 and pKa2 = 9.69, so pI = 6.02. Most amino acids have a pI around them.
Note that a number of the protein amino acids also have ionizable functions in the side-chain R group. These may be acidic or potentially acidic (aspartic acid, glutamic acid, tyrosine, cysteine), or basic (lysine, arginine, histidine). These amino acids are thus characterized by three pKa values.
We have used the term ‘potentially acidic’ to describe the phenol and thiol groups of tyrosine and cysteine respectively; under physiological conditions, these groups are unlikely to be ionized. Let us consider lysine, which has a second amino group in its side chain.
The pKa values for lysine are 2.18 (CO2H), 8.95 ( α-NH2), and 10.52 ( ε-NH2). Note that the ε-amino group has a typical amine pKa value, and is a stronger base than the α-amino group. In a strongly acidic solution, lysine will be present as a di-cation, with both amino groups protonated.
As the pH is raised, the most acidic proton will be lost first, and this is the carboxyl proton (pKa 2.18). As the pH increases further, protons will be lost first from the more acidic α-ammonium cation (pKa 8.95), and lastly from the least acidic ε-ammonium cation (pKa 10.52). Again, there is no intermediate with uncharged functional groups.
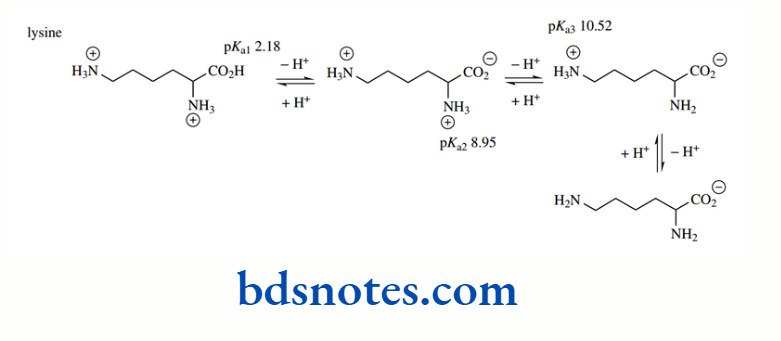
The isoelectric point for lysine is the pH at which the compound is in an electrically neutral form, and this will be the average of pKa2 (the cation) and pKa3 (the dipolar ion). For lysine, pKa² = 8.95 and pKa3 = 10.52, so pI = 9.74.
Glutamic acid is an example of an amino acid with an acidic side chain. The pKa values are 2.19 (CO2H), 4.25 ( γ-CO2H), and 9.67 (NH2). Here, the γ-carboxyl is more typical of a simple carboxylic acid.
In a strongly acidic solution, glutamic acid will be present as a cation, with the amino group protonated. As the pH is raised, a proton will be lost from the most acidic group, namely the 1- 1-carboxyl (pKa 2.19), followed by the γ-carboxyl (pKa 4.25). Lastly, the proton from the α-ammonium cation (pKa 9.67) will be removed. Yet again, there is no intermediate with uncharged functional groups.
The isoelectric point for glutamic acid is the pH at which the compound is in an electrically neutral form, and this will be the average of pKa1 (the cation) and pKa2 (the dipolar ion). For glutamic acid, pKa1 = 2.19 and pKa2 = 4.25, so pI = 3.22.
Isoelectric points are useful concepts for the separation and purification of amino acids and proteins using electrophoresis. Under the influence of an electric field, compounds migrate according to their overall charge.
As we have just seen for amino acids, this very much depends upon the pH of the solution. At the isoelectric point, there will be no net charge, and, therefore, no migration towards either anode or cathode.
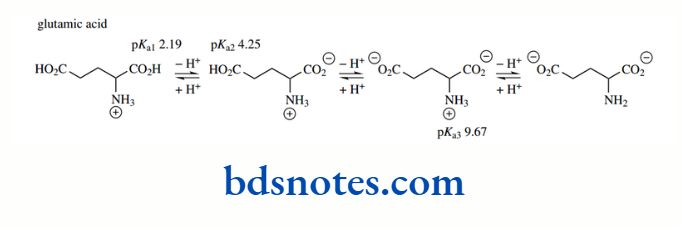
Ionization of morphine: extraction from opium
Morphine is the major alkaloid in opium, the dried latex obtained from the opium poppy, Papaver somniferum. About 25% of the mass of opium is composed of alkaloids, with morphine constituting about 12–15%. Morphine is a powerful analgesic and remains one of the most valuable for relief of severe pain.
However, most of the morphine extracted from opium is processed further to give a range of semi-synthetic drugs, with enhanced or improved properties. A means of extracting morphine from the other alkaloids in opium is thus desirable.
Alkaloids are found mainly in plants and are nitrogenous bases, typically primary, secondary, or tertiary amines. The basic properties facilitate their isolation and purification.
Water-soluble salts are formed in the presence of mineral acids, and this allows the separation of the alkaloids from any other compounds that are neutral or acidic. It is a simple matter to take a plant extract in a water-immiscible organic solvent and extract this solution with aqueous acid.
Salts of the alkaloids are formed, and, being water soluble, these transfer to the aqueous acid phase. On basifying the acid phase, the alkaloids revert to an uncharged form and may be extracted into fresh organic solvent.
Opium contains over 40 different alkaloids, all of which will be extracted from opium by the procedure just described. It then remains to separate morphine from this mixture. Of the main opium alkaloids, only morphine displays some acidic properties as well as basic properties.
Although a tertiary amine, morphine also contains a phenolic group. The acidity of this group can be exploited for the preferential extraction of morphine from an organic solvent by partitioning with an aqueous base.
Thus, if a solution of opium alkaloids in an organic solvent, for example, dichloromethane, is shaken with aqueous NaOH, only morphine will ionize at this pH, and it will form the water-soluble phenolate anion.
The other alkaloids will remain non-ionized and stay in the organic layer, allowing their separation from the aqueous morphine phenolate fraction.
By adding acid to the aqueous fraction, the phenolate will become protonated to give the non-ionized phenol, which may be extracted by shaking with the organic solvent.
Care is needed during the acidification since the addition of too much acid would ionize the amine and create another water-soluble ion, the protonated amine.
This would stay in the aqueous phase and not be extracted by an organic solvent. The optimum pH will be the isoelectric point as described under amino acids.
This is the pH at which the concentrations of cationic and anionic forms of morphine are equal and is the mean of the two pKa values.
Morphine has pKa (phenol) 9.9, and pKa (amine) 8.2, so that pI = 9.05. Note that the protonated amine is a stronger acid than the phenol so the intermediate between the two ionized forms will be the non-ionized alkaloid.
We can then use the Henderson–Hasselbalch equation \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\text { [base] }}{\text { [acid] }}\) to calculate the relative amounts of the ionic forms at this pH.
For the phenol, pKa 9.9 \(\log \frac{[\text { base }]}{\text { [acid] }}=\mathrm{pH}-\mathrm{p} K_{\mathrm{a}}=9.05-9.9=-0.85\)
Thus [base]/[acid] = 10−0.85 = 0.14 or about 1:7.
For the amine, pKa 8.2
⇒ \(\log \frac{[\text { base ] }}{\text { [acid] }}=\mathrm{pH}-\mathrm{p} K_{\mathrm{a}}=9.05-8.2=0.85\)
Thus [base]/[acid] = 100.85 = 7.08 or about 7:1.
At pH 9.05, the phenol–phenolate equilibrium favours the phenol by a factor of 7: 1, and the amine–ammonium ion equilibrium favours the amine by a factor of 7: 1. In other words, the non-ionized morphine predominates, and this can thus be extracted into the organic phase. What about the amounts in ionized form; are these not extractable?
By solvent extraction of the non-ionized morphine, we shall set up a new equilibrium in the aqueous phase, so that more non-ionized morphine is produced at the expense of the two ionized forms.
A second solvent extraction will remove this, and we shall effectively recover almost all the morphine content. A third extraction would make certain that only traces of morphine were left in ionized forms.
pKa and drug absorption
Cell membranes are structures containing lipids and proteins as their main components. Many drug molecules are weak acids or bases and can, therefore, exist as ionized species, depending upon their pKa values and the pH of the environment.
One of the more important concepts relating to drug absorption is that ionized species have very low lipid solubility, and are unable to permeate through membranes. Only the non-ionized drug is usually able to cross membranes. A range of pKa values covered by some common drugs
If we invoke the Henderson–Hasselbalch equation \(\mathrm{pH}=\mathrm{p} K_{\mathrm{a}}+\log \frac{\text { [base] }}{\text { [acid] }}\)
where pH = pKa for the drug, then, for a weak acid
⇒ \(\log \frac{[\text { base }]}{\text { [acid }]}=\log \frac{\text { [ionized }]}{\text { [non-ionized }]}=0\)
In other words, [ionized] = [non-ionized] = 50%. As we saw in Section 4.9, a shift in pH by one unit to either side of the pKa value must change the ratio of ionized to non-ionized forms by a factor of 10.
For pH=pka+1
⇒ \(\begin{aligned}
\log \frac{[\text { ionized] }}{\text { [non-ionized] }} & =1 \text { and therefore } \frac{\text { [ionized] }}{\text { [non-ionized] }} \\
& =10
\end{aligned}\)
for ph=pKa-1
⇒ \(\begin{aligned}
\log \frac{\text { [ionized] }}{[\text { non-ionized] }} & =-1 \text { and therefore } \frac{\text { [ionized] }}{\text { [non-ionized] }} \\
& =0.1
\end{aligned}\)
For a weak base, we have a similar relationship, though \(\log \frac{[\text { base }]}{[\text { acid }]}=\log \frac{\text { non-ionized] }}{\text { [ionized] }}\)
The human body has many different pH environments. For example, blood plasma has a rigorously controlled pH of 7.4 gastric juice is usually strongly acidic (pH from about 1 to 7), and urine can vary from about 4.8 to 7.5.
It is possible to predict the qualitative effect of pH changes on the distribution of weakly acidic and basic drugs, especially concerning gastric absorption and renal excretion:
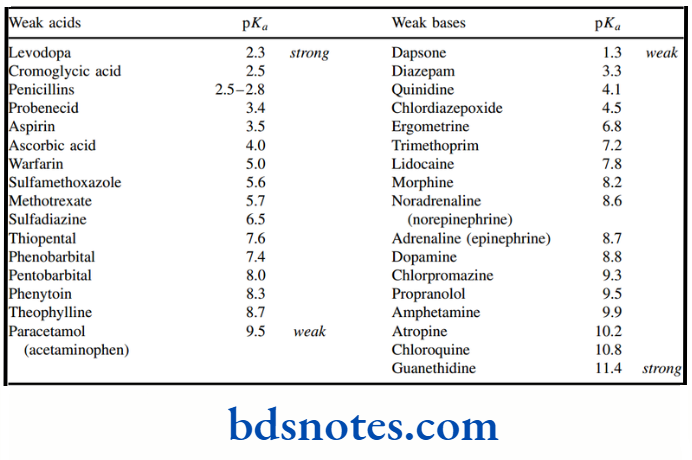
Very weak acids with pKa values greater than 7.5 will be essentially non-ionized at all pH values in the range of 1–8 so that absorption will be largely independent of pH.
Acids with pKa values in the range 2.5–7.5 will be characterized by significant changes in the proportion of non-ionized drugs according to the pH. As the pH rises, the percentage of nonionized drugs decreases, and absorption therefore also decreases.
Absorption of stronger acids (pKa less than about 2.5) should also depend upon pH, but the fraction that is non-ionized is going to be very low except under the most acidic conditions in the stomach. Absorption is typically low, even under acidic conditions.
Basic drugs will not be absorbed from the stomach, where the pH is strongly acidic.
The excretion of drugs will be affected by the pH of the urine. If the urine is acidic, weak bases are ionized and there will be poor re-absorption. With basic urine, weak bases are non-ionized and there is more re-absorption.
The pH of the urine can be artificially changed in the range of 5–8.5: oral administration of sodium bicarbonate (NaHCO3) increases pH values, whereas ammonium chloride (NH4Cl) lowers them.
Thus, urinary acidification will accelerate the excretion of weak bases and retard the excretion of weak acids. Making the urine alkaline will facilitate the excretion of weak acids and retard that of weak bases.
Leave a Reply