Nucleophilic Reactions: Nucleophilic Substitution
As the term suggests, a substitution reaction is one in which one group is substituted for another. For nucleophilic substitution, the reagent is a suitable nucleophile and it displaces a leaving group. As we study the reactions further, we shall see that mechanistically related competing reactions, eliminations, and rearrangements, also need to be considered.
The SN2 reaction: bimolecular nucleophilic substitution The abbreviation SN2 conveys the information ‘substitution–nucleophilic–bimolecular’.
The reaction is essentially the displacement of one group, a leaving group, by another group, a nucleophile. It is a bimolecular reaction since kinetic data indicate that two species are involved in the rate-determining step
Rate = k[RL][Nu]
Nu is the nucleophile, RL is the substrate containing the leaving group L, and k is the rate constant. In general terms, the reaction can be represented as below.
Differences in electronegativities between carbon and the leaving group atom lead to bond polarity. This confers a partial positive charge on the carbon and facilitates the attack of the nucleophile.
As the nucleophile electrons are used to make a new bond to the carbon, electrons must be transferred away to a suitable acceptor to maintain the carbon’s octet.
The suitable acceptor is the electronegative leaving group. The nucleophile attacks from the side opposite the leaving group – electrostatic repulsion prevents attack in the region of the leaving group.
This results in an inversion process for the other groups on the carbon center under attack, rather like an umbrella turning inside out in a violent gust of wind.
The process is concerted, i.e. the bond to the incoming nucleophile is made at the same time as the bond to the leaving group is being broken.
As a consequence, the mechanism involves a high-energy transition state in which both the nucleophile and leaving group are partially bonded, the Nu–C–L bonding is linear, and the three groups X, Y, and Z around carbon are in a planar array.
This is the natural arrangement to minimize steric interactions if we wish to position five groups around an atom, and will involve three sp2 orbitals and a p orbital as shown.
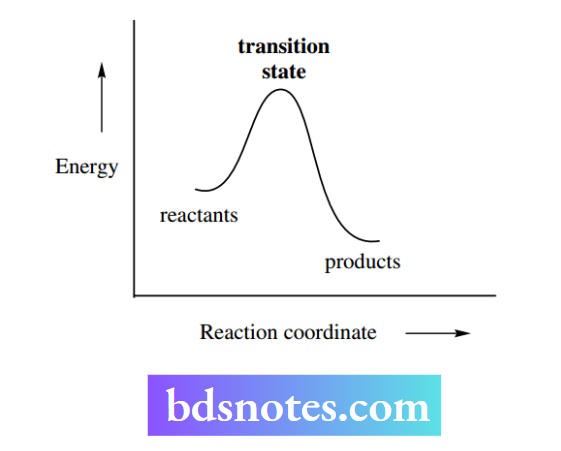
The p orbital is used for the partial bonding; note that we cannot have five full bonds to a carbon atom. The energy profile for the reaction proceeds from reactants to products via a single high-energy transition state.
The rate of an SN2 reaction depends upon several variables. These are:
- The nature of the substituents bonded to the atom attacked by the nucleophile;
- The nature of the nucleophile;
- The nature of the leaving group;
- Solvent effects.
- We can consider these in turn.
- The effect of substituents
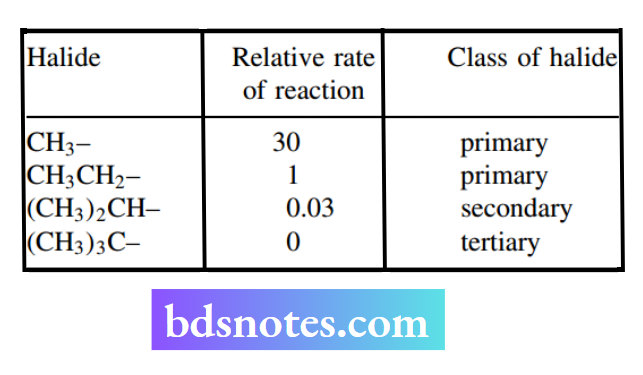
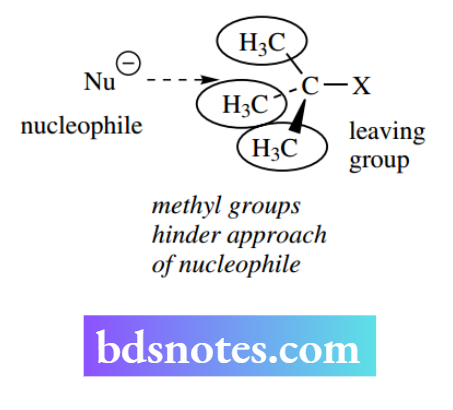
The SN2 mechanism requires the attack of a nucleophile at the rear of the leaving group, and consequently, the size of the groups X, Y, and Z will influence the ease of approach of the nucleophile.
Experimental evidence shows the relative rates for SN2 reactions of halides. This is primarily a result of steric hindrance increasing as one goes from primary to secondary to tertiary compounds.
With the tert-butyl group, the approach of the nucleophile is hindered by three methyl groups, so much so that the SN2 reaction is not normally possible.
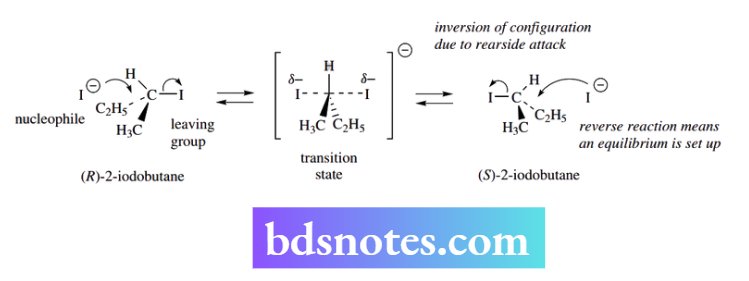
SN2 reactions: The racemization of 2-iodobutane
The inversion in an SN2 reaction can be demonstrated in a rather simple experiment. If (+)-(R)-2-iodobutane is heated in acetone solution, it is recovered unchanged.
However, when sodium iodide is added to the mixture, there is no apparent chemical change, but the optical activity gradually diminishes until it becomes zero, i.e. racemic (±)-(RS )-2-iodobutane has been formed.
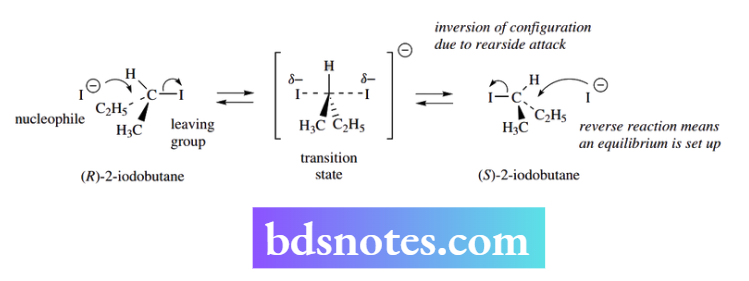
In this reaction, an equilibrium is set up. The nucleophile, iodide, is the same as the leaving group. Therefore, inversion of configuration merely converts the (+)-isomer into the (−)-isomer.
As a result, the optical activity gradually disappears and ultimately becomes zero as the mixture becomes the racemic (±)-form.
We are never going to get a complete conversion of the (+)- into (−)-enantiomer because the reverse reaction will also occur.
This is mechanistically identical to the forward reaction, so either (+)- or (−)-2-iodobutane as starting material would give a racemic product, i.e. it is a racemization reaction.
This is an unusual reaction, in that the energy of the products will be identical to the energy of the reactants, though the interconversion of isomers involves activation energy that must be overcome by the application of heat.
Should a reaction be attempted with tertiary substrates, one does not usually get a substitution, but alternative side reactions occur.
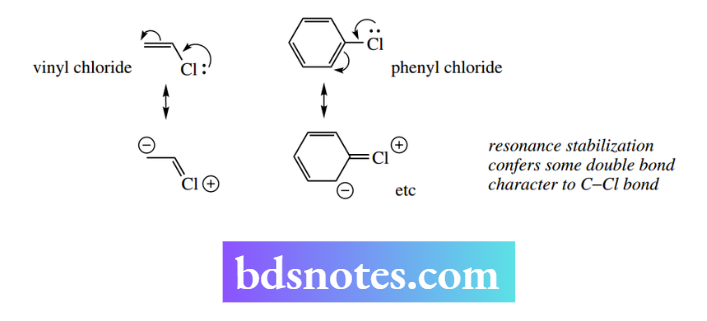
If the potential leaving group is attached to unsaturated carbon, as in vinyl chloride or phenyl chloride, attack by nucleophiles is also extremely difficult, and these compounds are very unreactive in SN² reactions compared with simple alkyl halides.
In these cases, the reason is not so much steric but electrostatic, in that the nucleophile is repelled by the electrons of the unsaturated system.
In addition, since the halide is attached to carbon through a sp2-hybridized bond, the electrons in the bond are considerably closer to carbon than in a sp3-hybridized bond of an alkyl halide.
Lastly, resonance stabilization in the halide gives some double bond character to the C–Hal bond. This effectively strengthens the bond and makes it harder to break. This lack of reactivity is also true for SN¹ reactions.
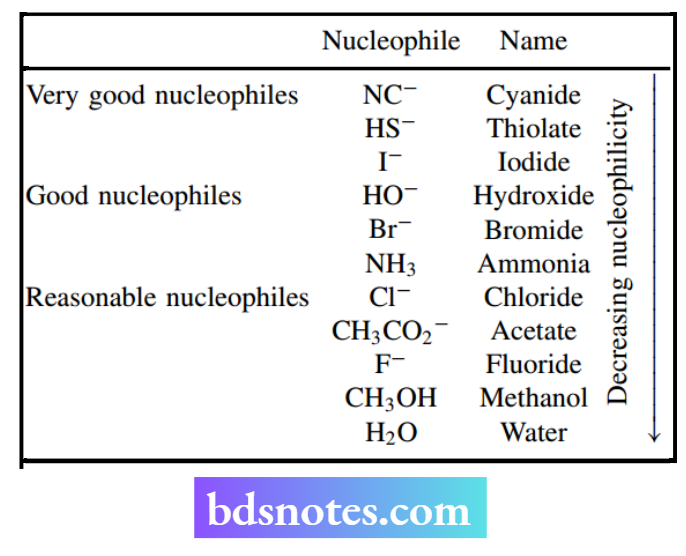
Nucleophiles: nucleophile city and basicity
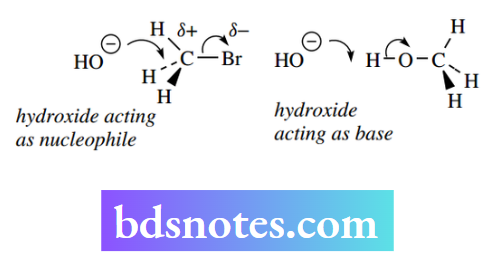
The SN²-type reaction can be considered simply as being initiated by the attack of a nucleophile onto the electron-deficient end of a polarized bond X–Y.

If X = H, then this equates to the removal of a proton and we would consider the nucleophile to be a base. It follows that there is going to be a close relationship between a group’s capacity to act as a nucleophile, i.e. nucleophilicity, and its ability to act as a base, i.e. basicity. Thus, the hydroxide ion can act as a nucleophile or as a base.
In many cases, nucleophilicity can be correlated with basicity, and this forms a helpful way of predicting how good a potential nucleophile may be. The sequences of relative basicity are also reflected in relative nucleophilicities.
The approximation works best for comparisons where the identity of the attacking atom is the same, for Example N, or O, as illustrated. The correlation is useful but not exact.
This is because basicity is a measure of the position of equilibrium between a substrate and its conjugate acid, whereas nucleophilicity relates to the rate of reaction.
The above relationship breaks down when one looks at atoms in the same column of the periodic table.
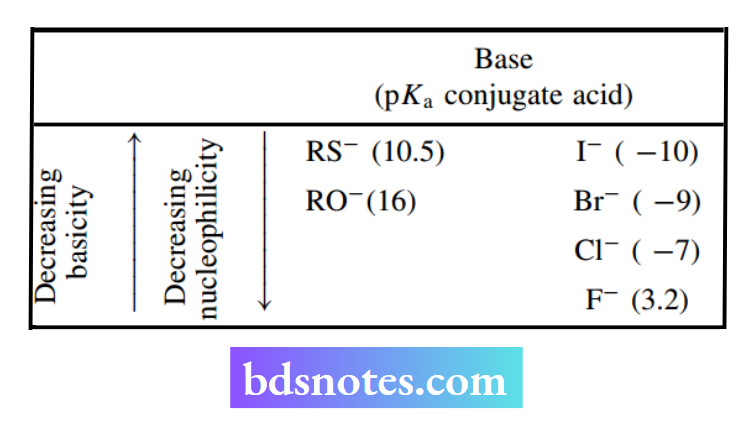
As atomic number increases, basicity decreases, whilst nucleophilicity increases so that electrons associated with larger atoms become less localized, consequently forming weaker bonds with protons the other hand, electrons in the larger atoms are more easily polarizable, and it becomes easier for them to be donated to an electrophile; this leads to greater nucleophilicity. Despite these inconsistencies, there are two important features worth remembering
- An anion is a better nucleophile than an uncharged conjugate acid;
- Strong bases are good nucleophiles.
Selective alkylation of m morphine to codeine and pholcodine Opium is a crude exudate obtained from the opium poppy Papaver somniferum, and it provides several medicinally useful alkaloids. One of these is codeine, which is widely used as a moderate analgesic.
Opium contains only relatively small amounts of codeine (1–2%), however, most of the codeine for drug use is obtained by semi-synthesis from morphine, which is the major component (12–20%) in opium. Conversion of morphine into codeine requires selective methylation of the phenolic hydroxyl. This can be achieved by an SN2 reaction under basic conditions.
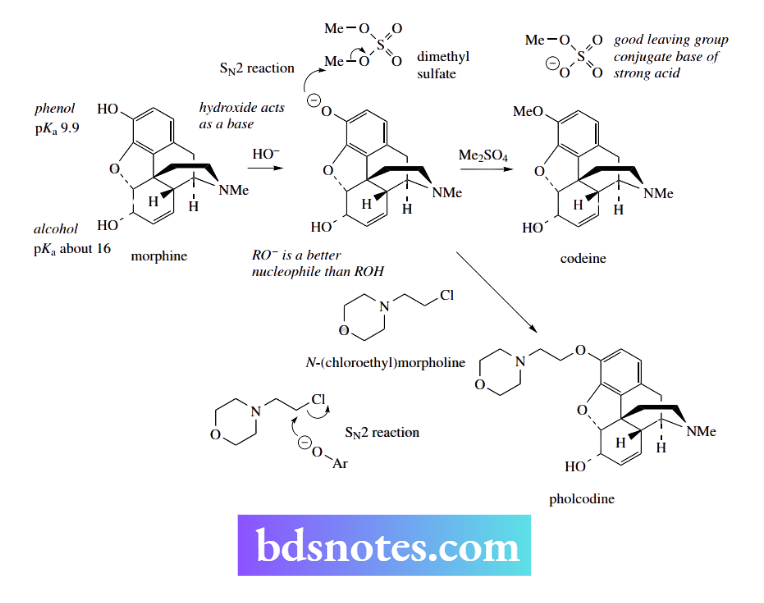
Morphine has two hydroxyls, but one is a phenol and the other is an alcohol. Because phenols (pKa about 10) are considerably more acidic than alcohols (pKa about 16), only the phenol will become ionized under mild basic conditions.
Since the phenolate anion (charged) will then be a much better nucleophile than the alcohol hydroxyl (uncharged), the SN2 reaction will selectively involve the phenolate group. The alcohol group does not react under these conditions.
The methylating agent (electrophile) used in this reaction is dimethyl sulfate; the leaving group is the anion of a sulfuric acid ester and is the conjugate base of a strong acid.
The same type of reasoning allows the production of pholcodine, an effective cough suppressant, from morphine. In this semi-synthesis, the electrophile is N-(chloroethyl)morpholine, and the leaving group is chloride.
Solvent effects
Nucleophilicities are affected by solvents, and any correlations with basicity can break down in protic solvents like methanol or ethanol.
This is because anions are stabilized by hydrogen bonding, and become solvated. These solvation molecules must be lost before the anion can attack as a nucleophile.
Accordingly, better solvents for nucleophilic substitution reactions are the so-called aprotic polar solvents, which contain no protons that allow hydrogen bonding to occur. Anions, consequently, become more nucleophilic in aprotic polar solvents than they are in protic solvents.
As an example, the SN² reaction of chloride with methyl iodide leading to methyl chloride is some 106 times faster in dimethylformamide (DMF) than in methanol. This is because there is no hydrogen bonding possible in DMF.
In sharp contrast, the reaction in the structurally similar solvent N-methyl formamide (HCONHMe), which still contains an N–H that can participate in hydrogen bonding, is only 45 times as fast as in methanol.
Chloride ions actually form stronger hydrogen bonds with methanol than with Nmethylformamide, so there is an increase in reactivity, but hardly as dramatic as with the aprotic solvent DMF.

Leaving groups
The nature of the leaving group is a further important feature of nucleophilic substitution reactions. For the SN² reaction to proceed smoothly,
We need to generate strong bonding between the nucleophile and the electrophilic carbon, at the same time as the bonding between this carbon and the leaving group is weakened.
The high-energy transition state may thus be considered to require the general characteristics shown in the scheme below.
Good leaving groups are those that form stable ions or neutral molecules after they leave the substrate. Consequently, the capacity of a substituent to act as a leaving group can also be related to basicity.
Strong bases (the conjugate bases of weak acids) are poor leaving groups; but, as we have seen above, they are good nucleophiles. On the other hand, weak bases (the conjugate bases of strong acids) are good leaving groups, but they make poor nucleophiles.
We can now understand and predict why some nucleophilic substitution reactions are favored and others are not. Thus, it is easy to convert methyl bromide into methanol by the use of hydroxide as a nucleophile.
On the other hand, it is not feasible to convert methanol into methyl bromide merely by using bromide as the nucleophile.
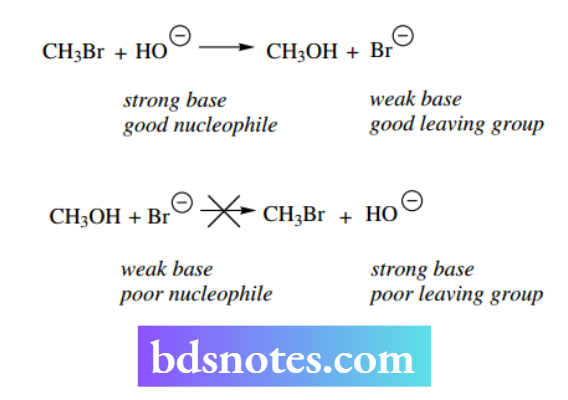
The difference here is primarily due to the nature of the leaving groups. Bromide is a weak base and a good leaving group, whereas hydroxide is a strong base and, therefore, a poor leaving group.
Nevertheless, the latter transformation can be achieved by improving the ability of the leaving group to depart by carrying out the reaction under acidic conditions.
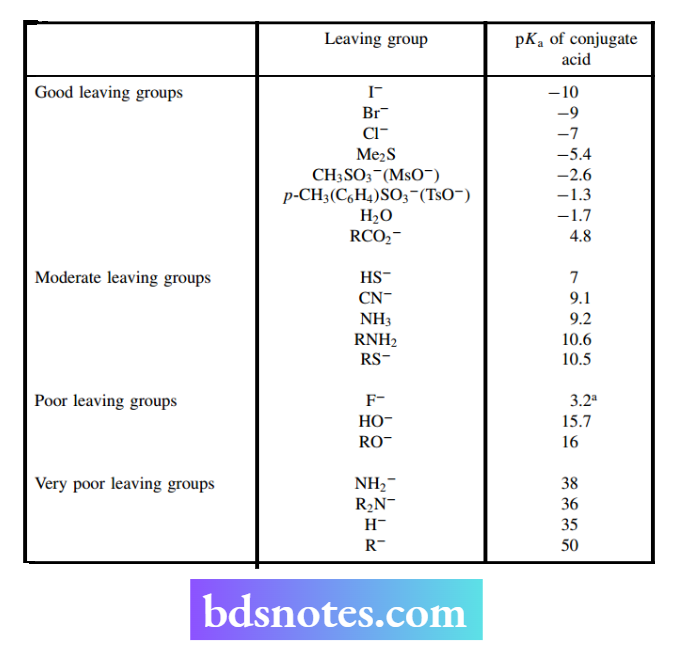
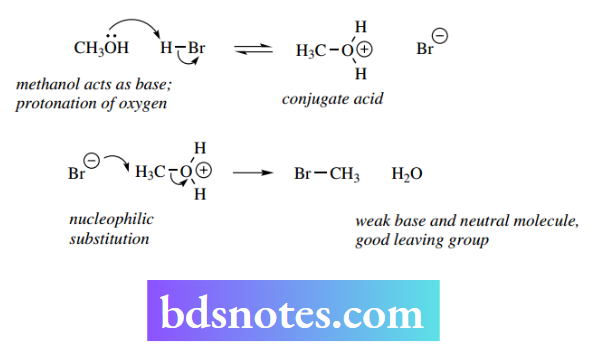
Thus, protonation of the substrate via the oxygen lone pair produces the conjugate acid. This now has greater polarization favoring nucleophilic attack and, most importantly, changes the leaving group from hydroxide (a strong base) to water (a weak base).
The reaction is now facilitated and proceeds readily. Chemical modification of poor leaving groups into good leaving groups may also be considered as a way of enhancing the ease of substitution reactions.
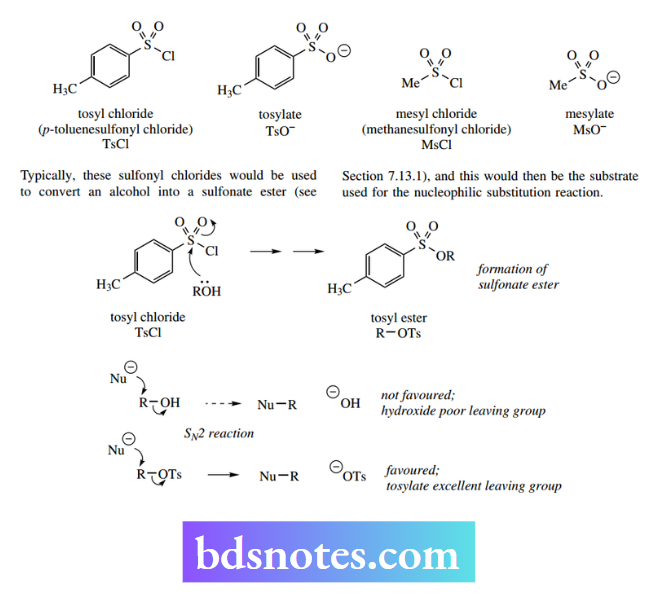
Two important reagents that may be used with alcohols are p-toluenesulfonyl chloride (tosyl chloride) and methanesulfonyl chloride (mesyl chloride). Both anions p-toluenesulfonate (tosylate) and methanesulfonate (mesylate) are excellent leaving groups, being the conjugate bases of strong acids (pKa < 0).
SN2 reactions in cyclic systems
The inversion process accompanying SN2 reactions may have particular significance in cyclic compounds. Thus, if we consider the disubstituted cyclopentane derivative shown undergoing an SN2
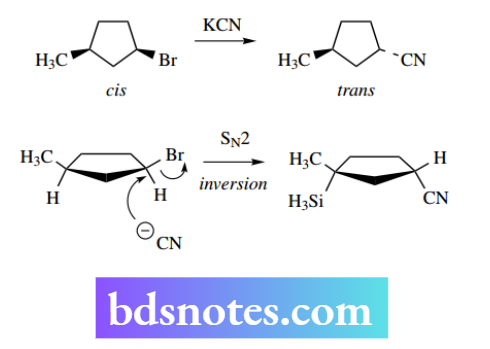
Reaction, we observe that the substituents were arranged in a cis relationship in the original compound and the consequence of inversion is the formation of a trans product. However, it is found that cyclic substrates tend to react much more slowly than similar acyclic compounds.
In small rings this is a consequence of ring strain; the SN2 transition state requires the three groups other than the nucleophile and the leaving group to be spaced 120◦ apart. This would be a severe problem for three- and four-membered rings (angles 60◦ and 90◦ respectively).
It is not a problem for five-membered rings, where this is the normal bond angle in the ring, and such compounds react just as readily as acyclic compounds.
Cyclohexyl derivatives react some 100-fold less readily than acyclic compounds, however, ring strain cannot be an important factor: the 109◦ tetrahedral angles are the same as in an acyclic compound. In cyclohexyl compounds, the rate of reaction is slowed by steric interactions with axial hydrogens.
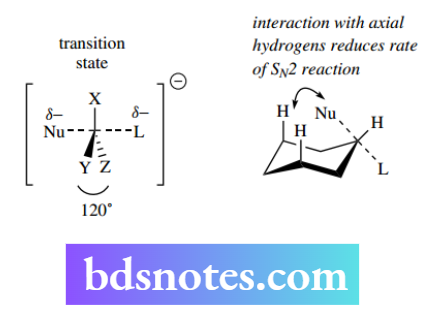
A consequence of the low rate of reaction in SN2 reactions is that side reactions in cyclohexane derivatives, especially elimination reactions (see Section 6.4.1), may often dominate over substitution.
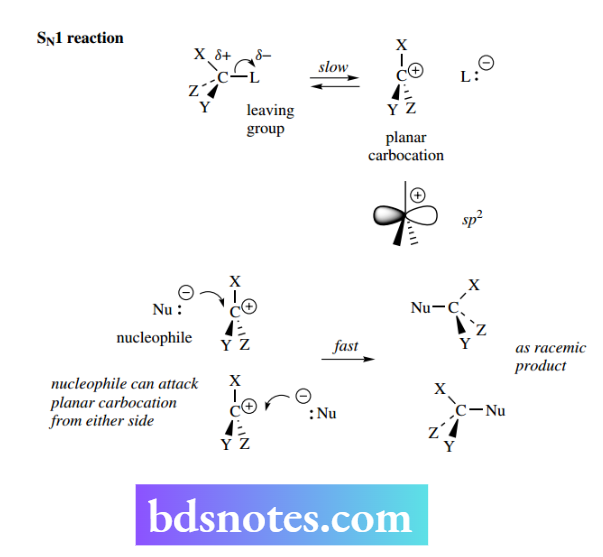
The SN1 reaction: unimolecular nucleophilic substitution
The abbreviation SN1 conveys the information ‘substitution–nucleophilic–unimolecular’. The reaction achieves much the same result as the SN2 reaction, i.e. the replacement of a leaving group by a nucleophile, but is mechanistically different.
It is unimolecular, since kinetic data indicate that only one species is involved in the rate-determining step: Rate = k[RL] where RL is the substrate containing the leaving group L and k is the rate constant. Note that the nucleophile Nu does not figure in the rate equation. In general terms, the reaction can be represented as below.
The first step of the reaction is the loss of the leaving group, transforming the initial polarization ( δ + / δ−) in the molecule into complete charge separation.
To achieve this, we need a good leaving group as with SN² reactions, but also a structure in which the positively charged carbon, a carbocation, is suitably stabilized.
This ionization step constitutes the slow part of the sequence, the rate-determining step, and, since only one molecular species is involved, it is responsible for the observed kinetic data.
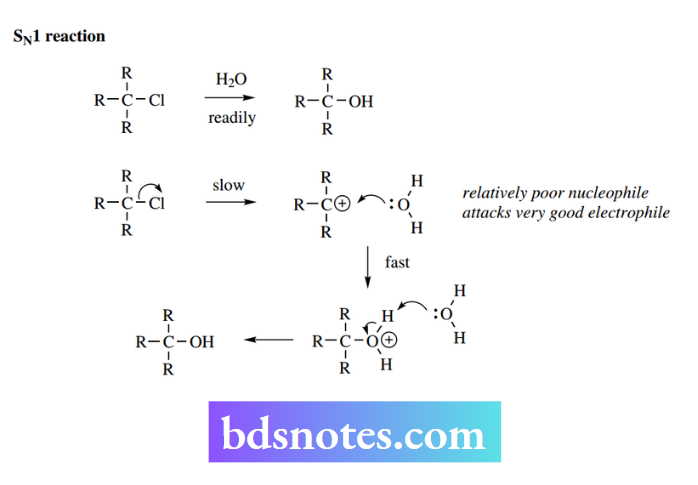
Once the reactive carbocation is formed, it is rapidly attacked by a suitable nucleophilic species, thus generating the final product. In SN1 reactions, the nucleophilicity of the nucleophile is relatively unimportant.
Because of the high reactivity of the carbocation, any nucleophile, charged or uncharged, will rapidly react. Therefore, as the rate equation shows, the nucleophile plays no part in controlling the overall reaction rate.
We have shown carbocation formation as reversible; it would be if the leaving group recombined with the carbocation.
If there is an excess of an alternative nucleophile, however, we shall get the required product. The carbon atom of the carbocation has only six bonding electrons and is a planar entity. The bonding electrons are in sp² orbitals, and there is also an unoccupied p orbital.
The attacking nucleophile can attack from either face of this planar species; so, when X, Y, and Z are different, the product will turn out to be a mixture of two possible stereoisomers.
As there is usually an equal probability of attack at each face, the product will be a racemic mixture. This is in marked contrast to the product from an SN² reaction, where there would be an inversion of configuration and formation of a single enantiomer.
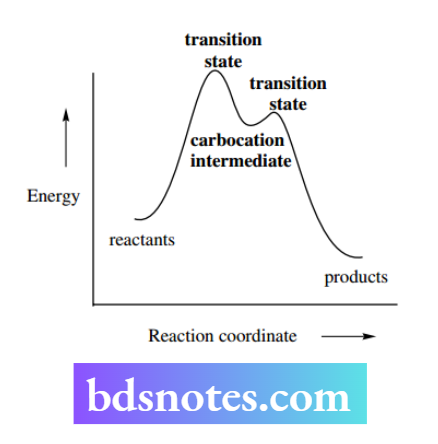
The carbocation is an intermediate in the reaction sequence and corresponds to a minimum in the energy profile.
Its formation depends upon overcoming an activation energy, corresponding to that required for the fission of the bond to the leaving group. Since the carbocation is very reactive, there will be a much smaller activation energy for reaction with the nucleophile.
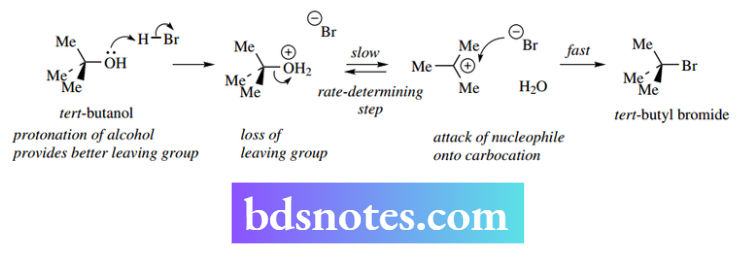
Thus, tert-butanol reacts readily with HBr to give the corresponding bromide. This reaction could not proceed via the SN² mechanism because steric crowding prevents access to the nucleophile. Instead, an SN¹ mechanism can be formulated.
The initial step would be protonation of the alcohol group to improve the nature of the leaving group, i.e. water rather than hydroxide, and allow the formation of the carbocation.
Loss of the leaving group would be the slow, rate-determining step, but the following step, the attack of the nucleophile onto the carbocation, would then be rapid.
Why some SN1 reactions do not lead to racemic products
Notwithstanding the remarks above concerning the equal probability of a nucleophile attacking either face of the planar carbocation and, therefore, producing a racemic product, many SN1 reactions result in varying degrees of inversion and racemization.
This can be rationalized in terms of preferential attack of the nucleophile from the face opposite the leaving group simply because, as the leaving group departs, it hinders attack from that side.
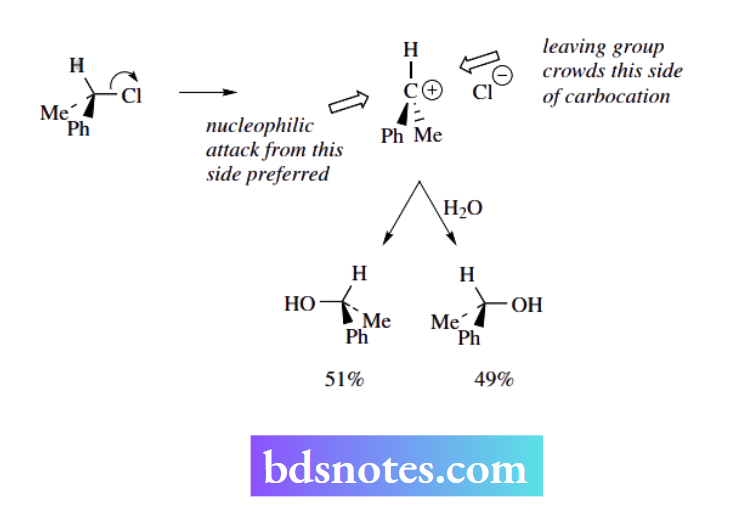
In the example shown, there is slightly more of the ‘inverted’ product in the reaction mixture, though the effect is not especially large. In other recorded examples, up to about 80% of the product might be the inverted form.
It follows that the SN² process is accompanied by complete inversion, whereas an SN¹ process will involve racemization or partial inversion.
The effect of substituents
The SN1 mechanism requires the initial loss of the leaving group to generate a reactive carbocation.

Experimental evidence concerning the relative rates for SN1 reactions of halides. The differences in reactivity reflect structural features that stabilize the intermediate carbocation. Carbocations are stabilized by the electron-donating effect of alkyl groups, which help to disperse the positive charge.
We have noted that alkyl groups have a modest electron-donating effect (see Section 4.3.3). In carbocations, this is not a simple inductive effect, but results from overlap of the σ C–H (or C–C) bond into the vacant p orbital of the carbocation. This leads to a favorable delocalization of the positive charge.
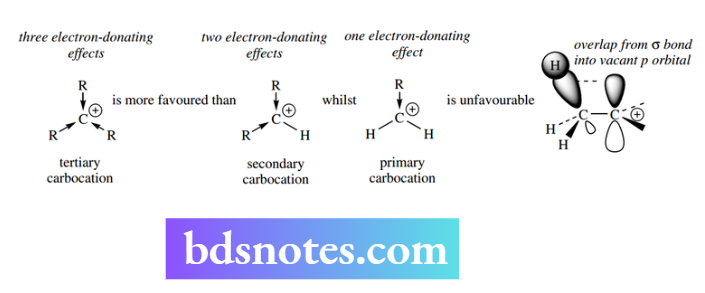
Accordingly, tertiary carbocations benefit from three such effects and are favored over secondary carbocations with two effects, whilst the single effect in primary carbocations is insufficient to provide significant stabilization.
Thus, SN1 reactions are highly favored at tertiary carbon and very much disfavoured at primary carbon. However, in addition, carbocations may be stabilized by resonance.
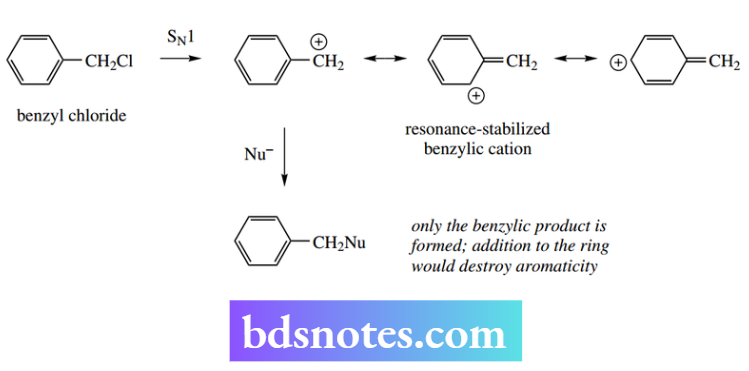
Simple examples of this are met with the allyl and benzyl cations, so that allyl chloride and benzyl chloride react via SN1 reactions, although superficially these appear to involve primary carbocations.
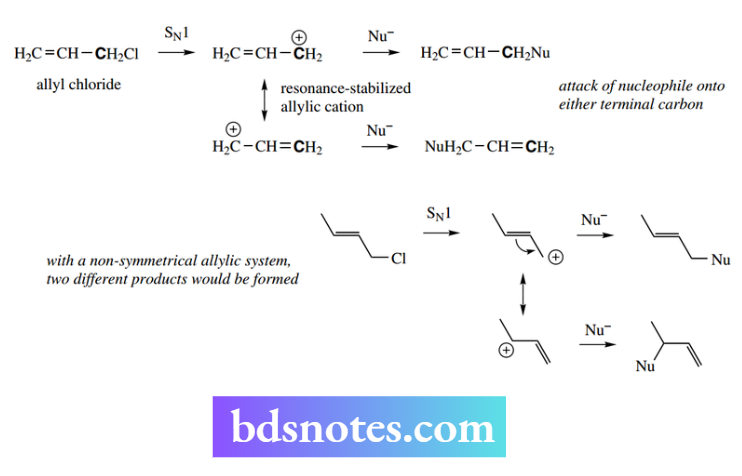
Note that with allyl derivatives there is potential for the nucleophile to react with the different resonance forms, perhaps leading to a mixture of products.
This is not the case with the benzylic substrates, since only the benzylic product is formed; addition to the ring would destroy the stability conferred by aromaticity.
One of the most stable carbocation structures is the triphenylmethyl cation (trityl cation). In this structure, the positive charge is stabilized by resonance employing all three rings. Trityl chloride ionizes readily and can capture an available nucleophile.
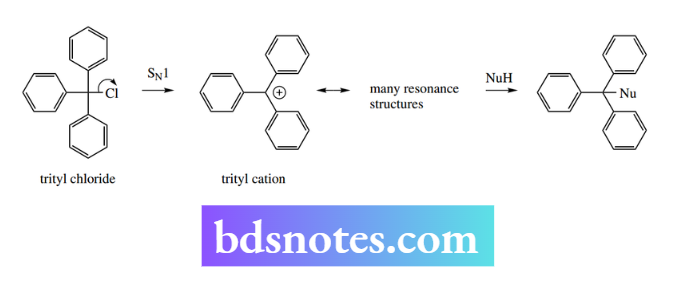
SN1 reactions in cyclic systems
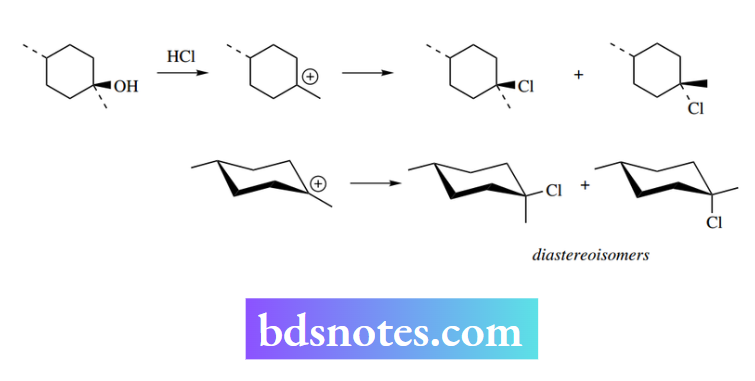
We noted above that the inversion of configuration that accompanied SN2 reactions was particularly apparent in cyclic systems, and that cis derivatives would be converted into trans products in disubstituted rings, and vice versa. Should The net result is that the product mixture consists of two diastereoisomers?
Sn1 Or Sn²?
As we have just seen, SN1 reactions are highly favored at tertiary carbon, and very much disfavoured at primary carbon. This is in marked contrast to SN2 reactions, which are highly favored at primary carbon and not at tertiary carbon.
With SN² reactions, consideration of steric hindrance rationalized the results observed. This leads to the generalizations for nucleophilic substitutions with secondary substrates being able to participate in either type of process.
The most distinguishing feature of the SN1 mechanism is the intermediate carbocation. Formation of the carbocation is the rate-determining step, and this is more favorable in polar solvents that can assist in facilitating the charge separation/ionization.
A useful, though not always exact, SN1 reaction occurs in a similar sort of cyclic system, then there may be stereochemical consequences, though these are easily predicted.
Thus, should the dimethylcyclohexanol shown below participate in an SN1 reaction, then we can deduce that the carbocation will be attacked from either face by the nucleophile, but not necessarily to the same extent.

The guide is that SN1 reactions are going to be favored by an acidic/positive environment, and are less likely to occur under basic/negative conditions. Since good nucleophiles are often also strong bases, this does tend to limit the applicability of SN¹ reactions.
Indeed, under strongly basic conditions, side reactions such as elimination are more likely to occur than nucleophilic substitution reactions.
However, all is not lost, because the carbocation is a particularly good electrophile and can be used with relatively poor nucleophiles. This is illustrated in the following examples.
Finally, do appreciate that, depending upon conditions, both SN1 and SN2 mechanisms might be operating at the same time, with each contributing its stereochemical characteristics to the product.
Biological SN1 reactions involving allylic cations
The living groups most commonly employed in nature are phosphates and diphosphates. These good leaving groups are anions of the strong acids phosphoric (pKa 2.1) and diphosphoric (pKa 1.5) acids respectively. The pKa values given refer to the first ionization of these polyfunctional acids.
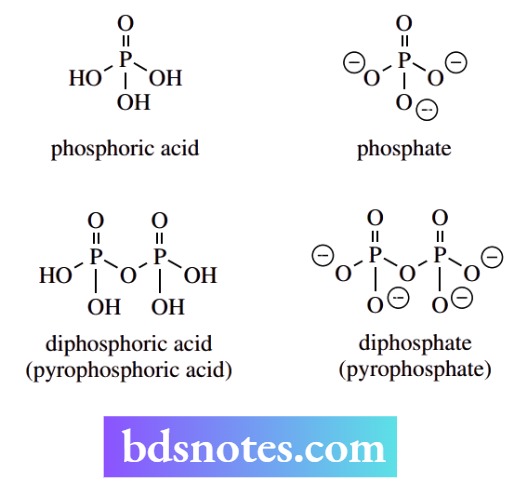
The compound dimethylallyl diphosphate provides an excellent example of a natural product with a diphosphate leaving group that can be displaced in a nucleophilic substitution reaction.
Suitable nucleophiles are hydroxyl groups, Example a phenol, though frequently an electron-rich nucleophilic carbon is employed.
Dimethylallyl diphosphate is a precursor of many natural products that contain in their structures branched-chain C5 subunits termed isoprene units.
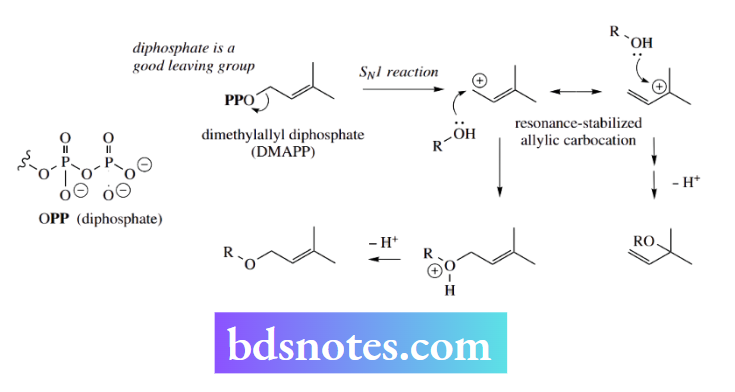
Although both SN2 and SN1 mechanisms might be formulated for such reactions, all the available evidence favors an SN1 process. This is rationalized in terms of the formation of a favorable resonance-stabilized allylic cation by loss of the leaving group.
In the majority of natural product structures, the nucleophile has attacked the allylic system on the same carbon that loses the diphosphate, but there are certainly examples of nucleophilic attack on the alternative tertiary carbon.
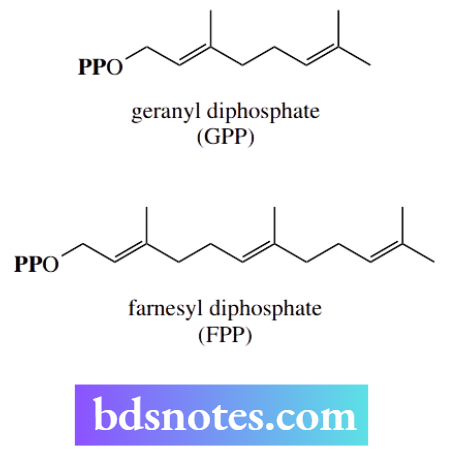
Geranyl diphosphate and farnesyl diphosphate are analogs of dimethylallyl diphosphate that contain two and three C5 subunits respectively; they can undergo the same SN1 reactions as dimethylallyl diphosphate. In all cases, a carbocation mechanism is favored by the resonance stabilization of the allylic carbocation.
Dimethylallyl diphosphate, geranyl diphosphate, and farnesyl diphosphate are precursors for natural terpenoids and steroids. The possibility of nucleophilic attack on different carbons in the resonance-stabilized carbocation facilitates another modification exploited by nature during terpenoid metabolism.
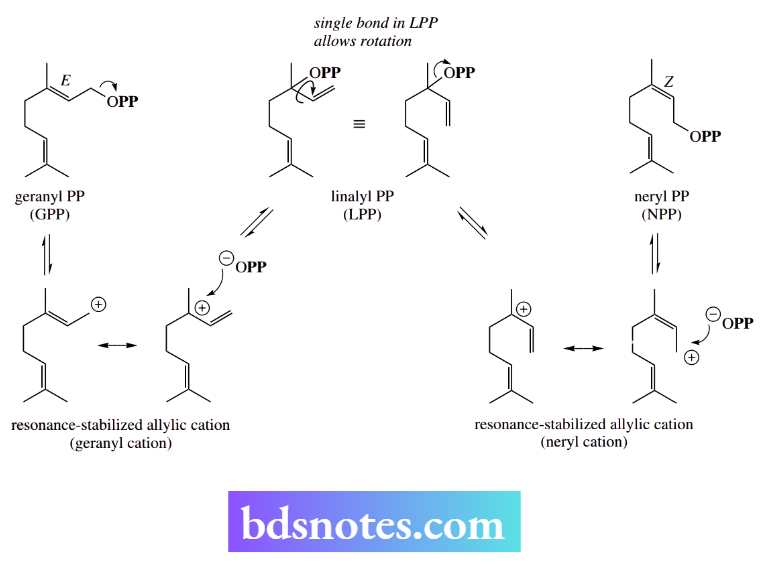
This is a change in double-bond stereochemistry in the allylic system. The interconversions of geranyl diphosphate, linalyl diphosphate, and neryl diphosphate provide neat but satisfying examples of the chemistry of simple allylic carbocations.
Thus, geranyl diphosphate ionizes to the resonance-stabilized geranyl carbocation; in nature, this can recombine with the diphosphate anion in two ways, reverting to geranyl diphosphate or forming linalyl diphosphate.
In linalyl diphosphate, the original double bond from geranyl diphosphate has now become a single bond, and free rotation is possible. Ionization of linalyl diphosphate occurs, giving a resonance-stabilized neryl carbocation, one form of which now has a Z double bond. Recombination of this with diphosphate leads to neryl diphosphate, a geometric configurational isomer of geranyl diphosphate.
It is normally very difficult to change the configuration of a double bond. Nature achieves it easily in this allylic system via carbocation chemistry.
Nucleophilic Substitution Reactions
Halide as a nucleophile: alkyl hali des Halide can be employed as a nucleophile in either SN2 or SN1 reactions to generate an alkyl halide. However, note that, in the general example shown, protonation by the acidic reagent HBr is required to improve the leaving group.
The utility of this simple transformation is often to increase the reactivity of the substrate, in that halide is a good leaving group and so can participate in other nucleophilic substitution reactions.
Oxygen and sulfur as nucleophiles: Ethers, Esters, Thioethers, Epoxides
Alkyl halides can react with water or alcohols by either SN2 or SN1 mechanisms to give alcohols or ethers respectively.
It is often preferable to use basic conditions with hydroxide or alkoxide as a better nucleophile, though this may lead to elimination and alkene formation as a competing reaction.
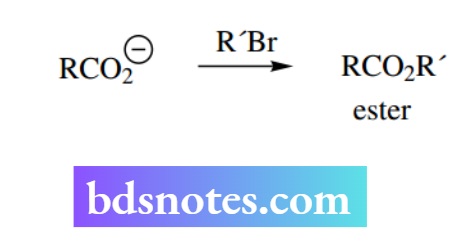
Although a carboxylate anion is only a relatively modest nucleophile, it is possible to exploit an SN² reaction to prepare esters from carboxylic acids as an alternative to the usual esterification methods. Such methods might be useful, depending upon the nature and availability of starting materials.
Sulfur nucleophiles behave similarly to oxygen compounds. Again, the anion will be a better nucleophile than the thiol; and since thiols are more acidic than alcohols, the conjugate bases are more easily generated.
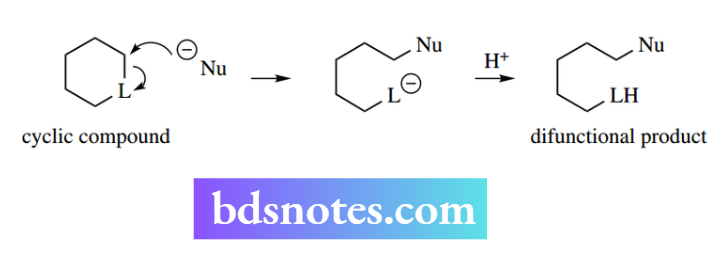
Note also that ring-opening nucleophilic substitution reactions may be possible, and that these will give a product with two functional groups since the leaving group is still attached to the original molecule through another bond.
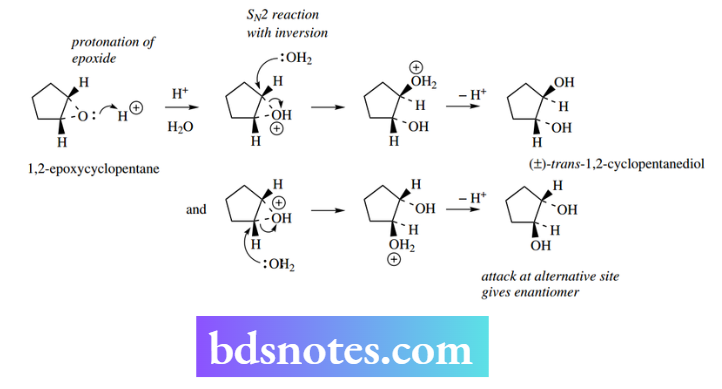
A simple example of a ring-opening substitution reaction is the acid-catalyzed hydrolysis of epoxides. In the example shown, protonation of the epoxide oxygen improves the leaving group, and an SN2 reaction may then proceed using water as the nucleophile.
Three-membered rings must of necessity be cis-fused and the inversion process, therefore, generates a trans-1,2-diol.
This is true even if the other end of the epoxide ring system is attacked, though it will produce the enantiomeric product. Since both reactions can occur with equal probability, the product here is racemic.
S -Adenosylmethionine in biological methylation reactions
In biological methylation, the S-methyl group of the amino acid L-methionine is used to methylate suitable O, N, S, and C nucleophiles. First, methionine is converted into the methylating agent S-adenosylmethionine (SAM).
SAM is a nucleoside derivative. Both the formation of SAM and the subsequent methylation reactions are nice examples of biological SN2 reactions.
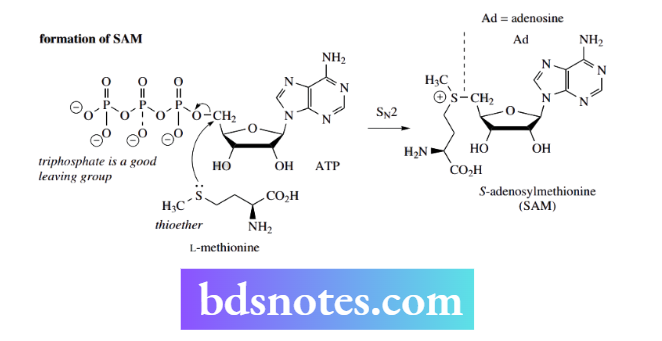
L-methionine is a thioether that acts as a sulfur nucleophile to react with adenosine triphosphate (ATP);. Sulfur is a good nucleophile, and ATP contains a good leaving group, the triphosphate moiety.
The leaving group is in a primary position, favoring an SN² reaction, the product of which is SAM. This can be regarded as similar to a protonated alcohol in nucleophilic substitution, in that it now contains a good leaving group that is a neutral molecule, in this case, the thioether S-adenosylhomocysteine.
Subsequent SN² reactions with appropriate nucleophiles (alcohols, phenols, amines, etc.) produce the methylated compounds.
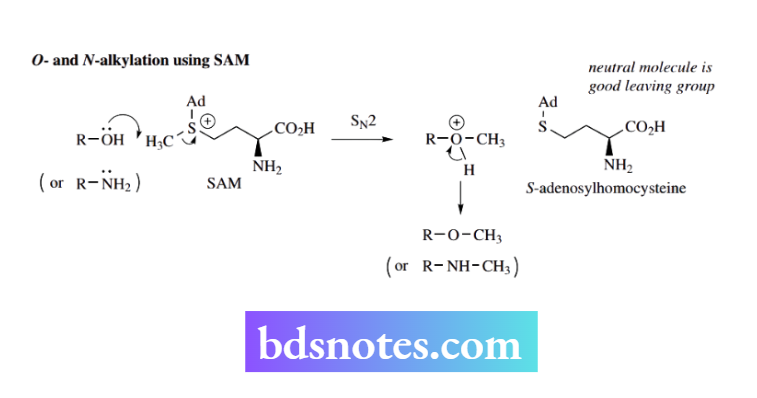
Note that in nature, these are all enzyme-catalyzed reactions. This makes the reactions specific.
It means possible competing SN2 reactions involving attack at either of the two methylene carbons in SAM are not encountered. It also means that where the substrate contains two or more potential nucleophiles, a reaction occurs at only one site, dictated by the enzyme.
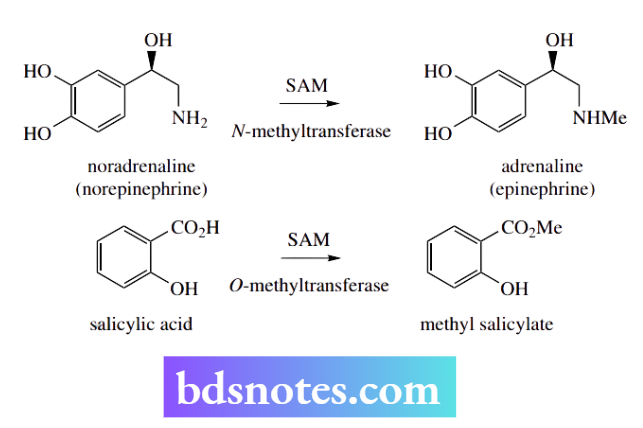
The enzymes are usually termed methyltransferases. Thus, in animals, an N-methyltransferase is responsible for SAM-dependent N-methylation of noradrenaline (norepinephrine) to adrenaline (epinephrine), whereas an O-methyltransferase in plants catalyzes the esterification of salicylic acid to methyl salicylate.
Glutathione a s a sulfur nucleophile in the metabolism of foreign compounds
Glutathione is a tripeptide containing a thiol grouping, which is part of the amino acid cysteine.
This SH group plays an important role as a nucleophile in the metabolism of potentially dangerous foreign compounds taken in by the body.
The potential of SH as a nucleophile is exploited in metabolic reactions catalyzed by enzymes termed glutathione S-transferases, which conjugate the foreign compounds, i.e. bind them to glutathione.
Conjugation markedly reduces the biological activity of the compound, and most conjugates are inactive. In addition, conjugation usually increases the polarity of the substrate, thus increasing its water solubility and its potential to be excreted.
There may be further modification to the glutathione part of the conjugate before the foreign compound is finally excreted.
Care: this is not the structural “conjugation”. Glutathione can react with many potentially toxic electrophiles, including halides and epoxides that react via simple SN2 reactions.
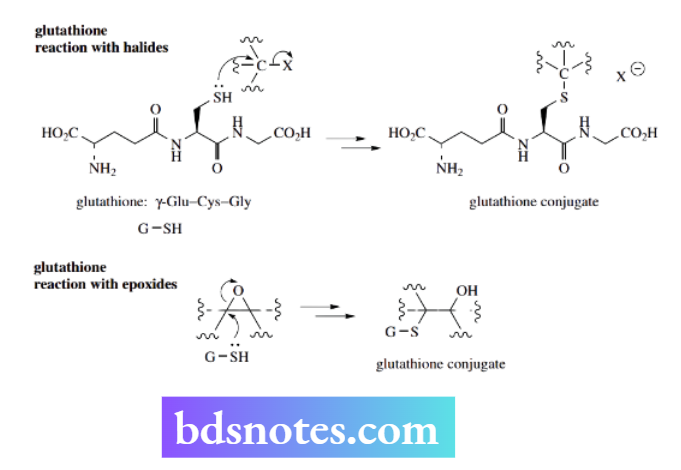
A specific example involving aflatoxins is shown in Box 6.8. We shall see other examples of glutathione reacting as a nucleophile in detoxification reactions, where conjugation is not the result of nucleophilic substitution.
For example, it might be a nucleophilic addition to an electrophile such as an unsaturated carbonyl compound.
Nitrogen as a nucleophile: ammonium salts, amines
Amines react with alkyl halides to give initially ammonium salts, from which an amine product is liberated in the presence of a base, typically an excess of the amine.
However, this is not always a useful reaction, in that the product amine is usually just as nucleophilic as the starting amine, allowing further SN² reactions to occur. Depending upon conditions, mixtures of amines together with the quaternary salt may be produced.
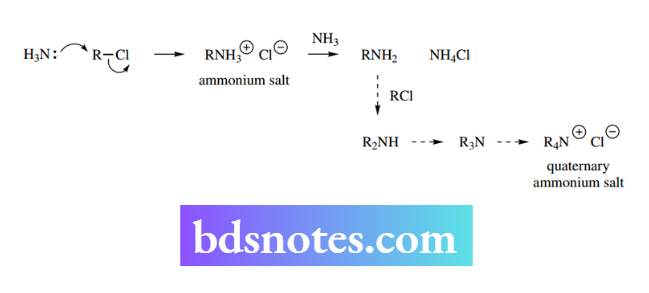
Nevertheless, it offers a convenient route to amino acids, both natural and unnatural, since the amino group in amino acids is less basic (pKa about 9.8) than a simple amine (pKa about 10.6) and is consequently rather less nucleophilic. a about 9.8) than a simple amine (pKa about 10.6) and is consequently rather less nucleophilic.
Curare-like muscle relaxants: quaternary ammonium salts
The production of a quaternary ammonium salt from a tertiary amine and an alkyl halide forms the synthetic route to decamethonium, the first of a range of synthetic muscle relaxants having an action like the natural materials found in the arrow-poison curare.
Decamethonium is a di-quaternary salt, as are more modern analogs, such as suxamethonium.
Suxamethonium superseded decamethonium as a drug because it has a shorter and more desirable duration of action in the body. This arises because it can be metabolized by ester-hydrolysing enzymes (esterases)
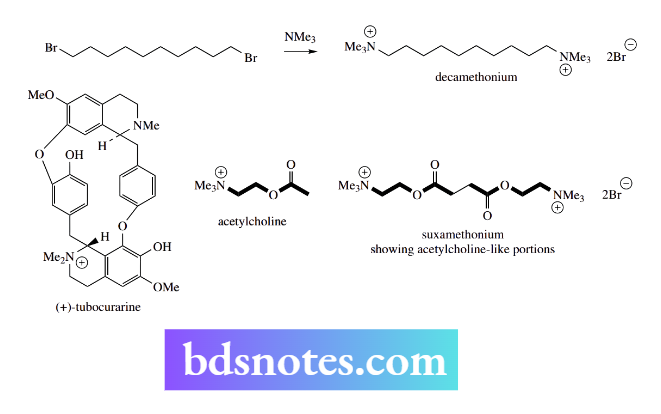
Curare-like muscle relaxants act by blocking acetylcholine receptor sites, thus eliminating the transmission of nerve impulses at the neuromuscular junction.
There are two acetylcholine-like groupings in the molecules, and the drugs, therefore, probably span and block several receptor sites.
The neurotransmitter acetylcholine is also a quaternary ammonium compound. The natural material present in curare is tubocurarine, a complex alkaloid that is a mono-quaternary salt.
Under physiological conditions, the tertiary amine will be almost completely protonated, and the compound will similarly possess two positively charged centers.
A Flatoxins And DNA Damage
The aflatoxins are rather unpleasant fungal toxins. At high levels, they can cause severe liver damage in animals and humans, and at lower levels, they are implicated in liver cancer.
These toxins are produced by the fungus Aspergillus flavus, a common contaminant on nuts and grains. Aflatoxin B1 is the most commonly encountered example, and it is also one of the most toxic.
We now know that the toxicity is initiated by the oxidative metabolism of the toxin in the body, converting aflatoxin B1 into an electrophilic epoxide.
This epoxide is attacked in an SN2 reaction by a nitrogen atom in a guanine residue of DNA. This leads to irreversible binding of the toxin to DNA, inhibition of DNA replication and RNA synthesis, and initiation of mutagenic activity.
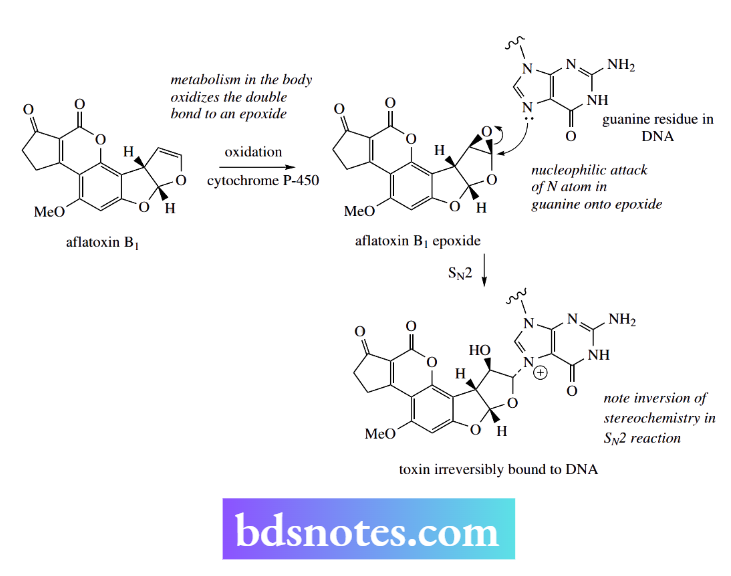
Fortunately, nature provides an alternative nucleophile whose role is to mop up dangerous electrophiles such as aflatoxin B1 epoxide before they can do damage, and to remove them from the body. This compound is glutathione, a tripeptide composed of glutamic acid, cysteine, and glycine.
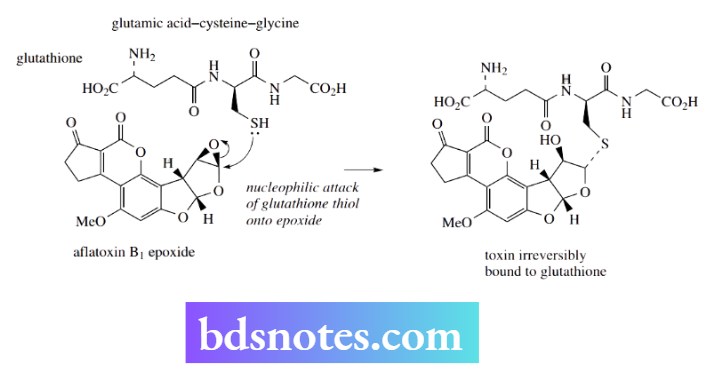
It is the thiol grouping that acts as a nucleophile, attacking the epoxide function of the toxin.
In this way, the toxin becomes irreversibly bound to glutathione, and the additional polar functionalities in the adduct mean that the product becomes water-soluble. The glutathione–toxin adduct can thus be excreted from the body.
Carbon as a nucleophile: nitriles, Gri guard reagents, acetylides
Nucleophilic substitution reactions employing carbon as a nucleophile are important in synthetic chemistry in that they create a new C–C bond.
A carbon nucleophile, of course, must be in the form of anionic carbon, or its equivalent. One of the simplest sources of anionic carbon is the cyanide anion. HCN is a weak acid (pKa 9.1) and forms a series of stable salts.
Sodium and potassium cyanides are convenient sources of cyanide, which in many reactions behaves similarly to a halide nucleophile. Thus, the reaction of an alkyl halide with cyanide creates a nitrile and extends the carbon chain in the substrate by one carbon.
It is easy to rationalize why cyanide can displace a halide such as bromide: HCN is a weak acid (pKa 9.1), so cyanide is a good nucleophile, whereas HBr is a strong acid (pKa − 9), and bromide is a good leaving group.
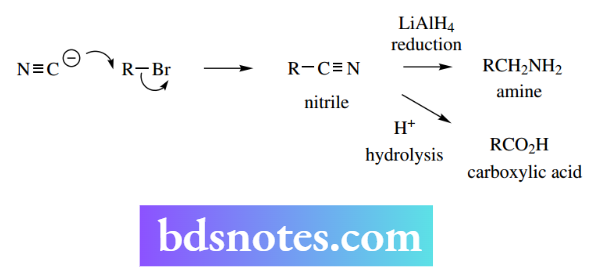
As we shall see later, other reactions of nitriles extend the usefulness of this reaction. Thus, reduction of nitriles gives amines, whereas hydrolysis generates carboxylic acid.
Organometallic reagents also provide carbon nucleophiles that can be considered to behave as carbanions. Although there are a variety of organometallic reagents available, we include here only two types of reagents, namely Grignard reagents and acetylides.
Reacting an alkyl or aryl halide, usually the bromide, with metallic magnesium in ether solution produces Grignard reagents. An exothermic reaction takes place in which the magnesium dissolves, and the product is a solution of the Grignard reagent RMgBr or ArMgBr.

The formation of this product need not concern us, but its nature is important. We can deduce from the ions Mg2+ and Br− that it contain the equivalent of R− or Ar−, i.e. the alkyl or aryl group has been transformed into its carbanion equivalent. This carbanion equivalent can behave as a nucleophile in typical nucleophilic substitution reactions.
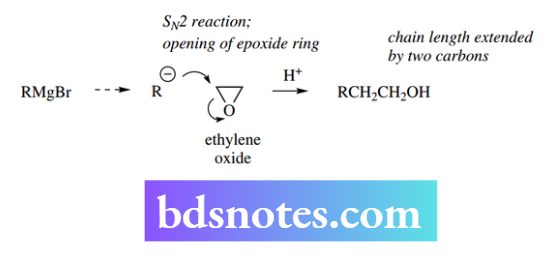
In the example shown, the reaction of a Grignard reagent with the epoxide electrophile ethylene oxide proceeds as expected, and after acidification results in the formation of an alcohol that is two carbons longer than the original nucleophile The carbanion equivalent from a Grignard reagent is also a strong base.
pKa values for alkanes are typically about 50 and for aromatics about 44. Not surprisingly, a Grignard reagent reacts readily with Acetylides are formed by treating terminal acetylenes with a strong base, sodium amide in liquid ammonia being the one most commonly employed.
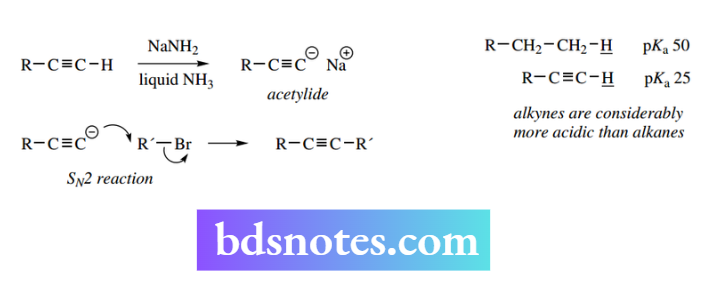
Acetylenes with a hydrogen atom attached to the triple bond are weakly acidic (pKa about 25) due to the stability of the acetylide anion water to form the hydrocarbon, so these reactions must be conducted under anhydrous conditions and this anion can thus act as a nucleophile.
It reacts with appropriate electrophiles, e.g. alkyl halides, in the manner expected. This reaction extends a carbon chain by two or more atoms, depending on the acetylide used.
Probably the most significant examples of carbon nucleophiles are enolate anions. These can participate in a wide variety of important reactions, and simple nucleophilic substitution reactions are included among these.
However, we shall consider these reactions at a later stage, when the nature and formation of enolate anions are discussed we shall consider hydride as a nucleophile that participates in a typical SN2 process. This achieves the replacement of a leaving group by hydrogen and, therefore, is a reduction of the substrate.
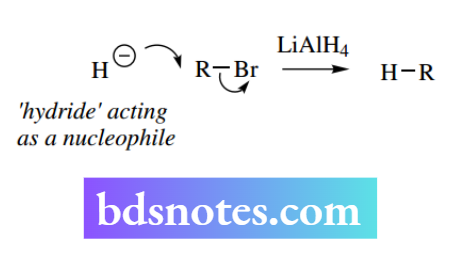
Hydride as nucleophilicle: lithium aluminium hydride and sodium borohydride de reduction ons Many complex metal hydrides such as lithium aluminium hydride (LiAlH4, abbreviated to LAH) and sodium borohydride (NaBH4) can deliver hydride in such a manner that it appears to act as a nucleophile.
We shall look at the nature of these reagents later under the reactions of carbonyl compounds, where we shall see that the complex metal hydride never actually produces hydride as a nucleophile, but the aluminium hydride anion can effect a transfer of hydride.
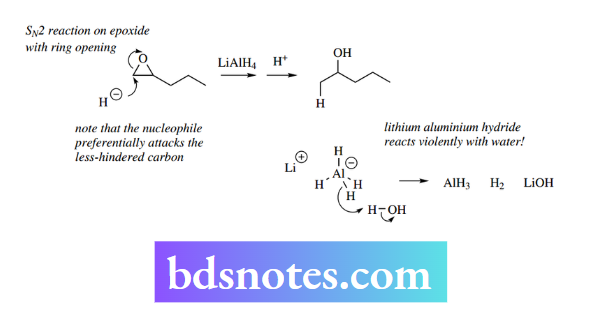
Hydride itself, for Example from sodium hydride, never acts as a nucleophile; owing to its small size and high charge density it always acts as a base. Nevertheless, to understand the transformations In the example shown overleaf where hydride attacks the epoxide function, the product is an alcohol, and the reaction is completed by supplying a proton source, usually water.
Lithium aluminium hydride reacts violently with water, liberating hydrogen, and the heat of the reaction usually ignites the hydrogen.
LAH must, therefore, be used in rigorously anhydrous conditions, usually in ether solution. Any solvent containing OH or NH groups would destroy the reagent by acting as a proton donor for hydride.
The addition of water as a proton source has to be carried out with considerable caution since any unreacted LAH will react violently with this water. In the laboratory, safe removal of excess LAH may be achieved by adding small amounts of an ester such as ethyl acetate.
Note that LAH is a powerful reducing agent and will attack many other functional groups, especially carbonyl groups.
An analogous series of reactions is involved when sodium borohydride is used as the reducing agent.
Sodium borohydride is considerably less reactive than LAH, and reactions proceed much more slowly. This reagent may be used in alcoholic or even aqueous solutions, so there are no particular hazards associated with its use.
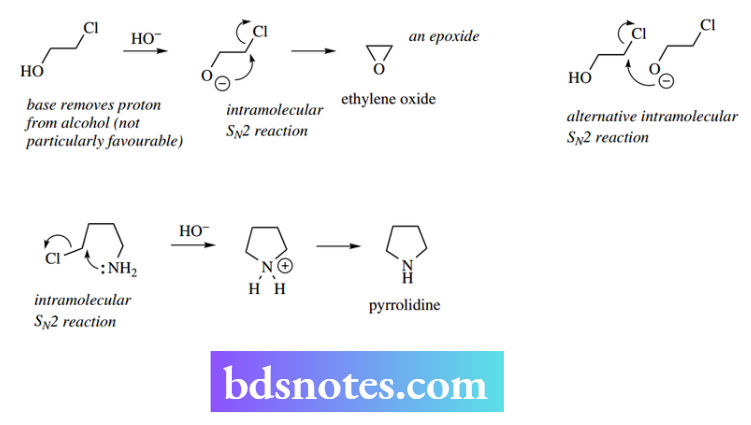
Simple examples shown above are the base-catalysed formation of oxygen- and nitrogen-containing ring systems. We have shown base-initiated ionization of the alcohol to an alkoxide anion in epoxide formation; the anion is a better nucleophile than the alcohol.
For pyrrolidine synthesis, the amino group is sufficiently nucleophilic for a reaction to occur, but a base is required to remove a proton from the first formed intermediate.
Formation of cyclic compounds
In substrates where there is a good leaving group in the same molecule as the nucleophile, one may get an intramolecular process and create a ring system.
It is usually necessary to find conditions that favour an intramolecular process over the alternative intermolecular reaction. This is typically achieved by carrying out the reaction at relatively higher dilutions, thereby minimizing the intermolecular processes.
Competing Reactions Eliminations And Rearrangements
When nucleophilic substitution reactions are attempted, the expected product may often be accompanied by one or more additional products that arise from competing reactions.
Since these competing reactions share features of the nucleophilic substitution mechanism, they are readily rationalized, and it is possible to devise conditions to minimize or maximize the formation of such products.
The most common alternative reactions are eliminations and rearrangements, which we shall consider in turn.
Elimination reactions
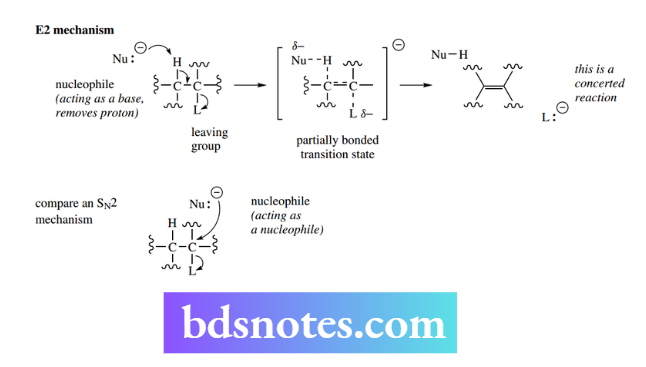
The E2 reaction: bimolecular elimination
The abbreviation E2 conveys the information ‘elimination–bimolecular’. The reaction is a concerted process in which a nucleophile removes an electrophile at the same time as a leaving group departs. It is bimolecular since kinetic data indicate that two species are involved in the rate-determining step:
Rate = k[RL][Nu]
Nu is the nucleophile, RL is the substrate containing the leaving group L, and k is the rate constant. The electrophile removed is usually hydrogen, so we can consider that the nucleophile is acting as a base.
We have seen above the close relationship between basicity and nucleophilicity, so the E2 mechanism provides an example of how the alternative property of nucleophiles may come into play and lead to different products. To achieve an SN2 reaction, the nucleophile must approach to the rear of the leaving group and then displace it.
If a rear-side approach is hindered by adjacent groups, or perhaps because the nucleophile is rather large, it becomes energetically easier for the nucleophile to act as a base and remove a proton from the substrate.
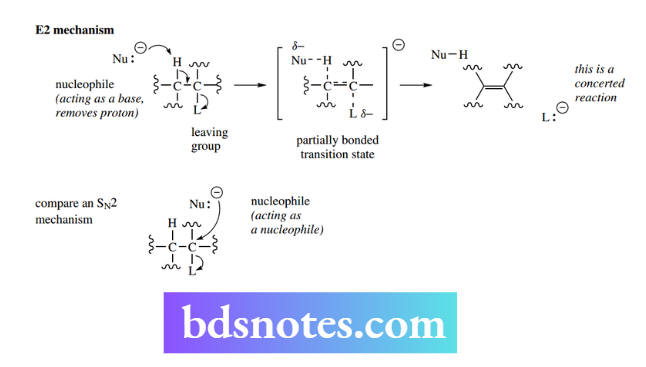
As the proton is removed, electrons that were involved in bonding the proton to the substrate are then used to form the double bond; however, to maintain the octet of electrons on the neighbouring carbon, the electrons will have to be transferred to a suitable acceptor, in this case, the leaving group.
As with the SN2 mechanism, the reaction is concerted and proceeds through a high-energy transition state, in which partial bonds have been established. The energy profile will look the same as that of an SN2 reaction The elimination reaction generates a new π bond in a planar alkene.
Since the π bond is perpendicular to the plane of the alkene, we can predict that the most favourable way to achieve the new π bonding is to start with the H–C–C–L atoms in a planar array. This will line up the orbitals and allow easy development of the π bond.
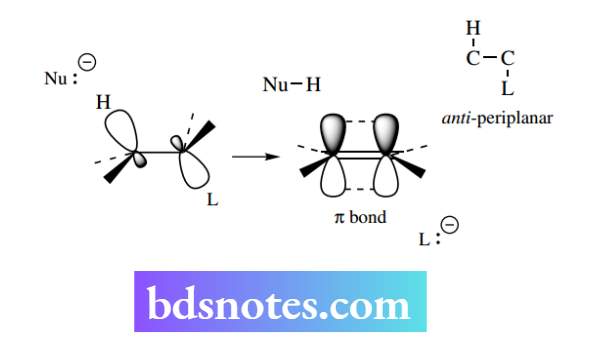
The nucleophile approaches from the side opposite the electronegative leaving group – electrostatic repulsion discourages attack in the region of the leaving group. With the substrate in the favoured staggered conformation, we describe this arrangement of atoms as anti-periplanar.
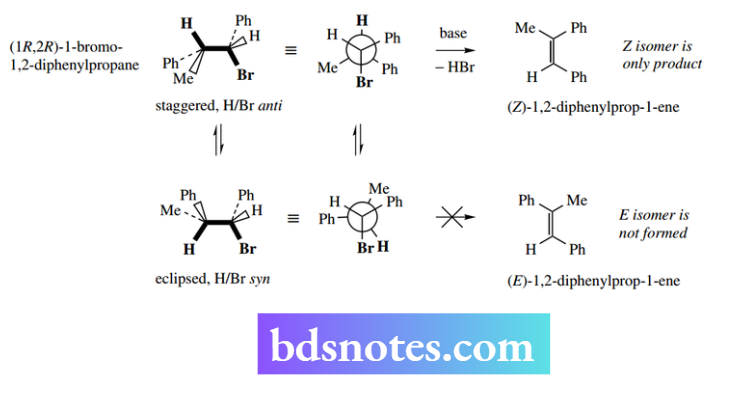
The requirement for the proton electrophile and the leaving group involved in the elimination to be anti to each other is demonstrated by the nature of the product obtained from a suitable substrate, Example (1R,2R)-1-bromo-1,2-diphenylpropane, when treated with base. The only product formed is (Z)-1,2- diphenylprop-1-ene.
This is the product of an anti-elimination of H and Br when the substrate is in a staggered conformer.
If H and Br were positioned on the same side of the conformer, then it would need to be in an unfavourable eclipsed conformer to line up the orbitals.
Elimination of H and Br in this fashion is termed a syn elimination and would lead to the E-product. However, this is not the product formed, and, in general, syn eliminations are very rare.
The anti-stereochemical relationship is obligatory by observing elimination reactions in suitable cyclohexane derivatives.
The only way to achieve a planar arrangement of the H–C–C–L atoms is when H and L are both axial and, consequently, trans to each other (transaxial). Thus, consider menthyl chloride and neomenthyl chloride, which are stereoisomers differing in configuration at just one centre.
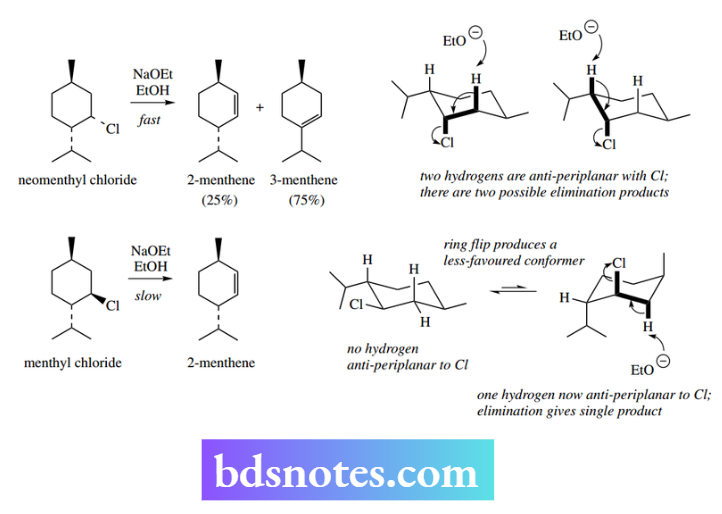
Treatment of neomenthyl chloride with base rapidly produces two different alkenes, i.e. 2- 2-menthene and 3-menthene.
If one considers the three-dimensional shape of neomenthyl chloride, it can be seen that, in the preferred conformer with the two alkyl groups equatorial, the chlorine is an axial substituent.
This means there are two different hydrogen atoms adjacent that are also axial and anti-periplanar to the chlorine.
As a consequence, two different E2 eliminations can occur; hence the two observed products. That the two products are not formed in equal amounts will be considered in the next section.
On the other hand, menthyl chloride is only slowly converted by treatment with base, and into a single product, i.e. 2-menthene. In the preferred conformation of menthyl chloride, all three substituents are equatorial, and no adjacent hydrogen is in a planar relationship to the chlorine leaving group.
The fact that slow elimination occurs at all is a result of conformational isomerism into the less-favoured conformer that has all three substituents axial.
In this conformer, there is a single hydrogen anti-periplanar with the chlorine, so elimination occurs giving just one product. The conformational equilibrium is slowly disturbed because the elimination removes the small concentration of unfavoured conformers.
Direction of elimination
The E2 elimination of HCl from neomenthyl chloride described above produced two products, namely 2- 2-menthene and 3-menthene in a ratio of about 1 : 3.
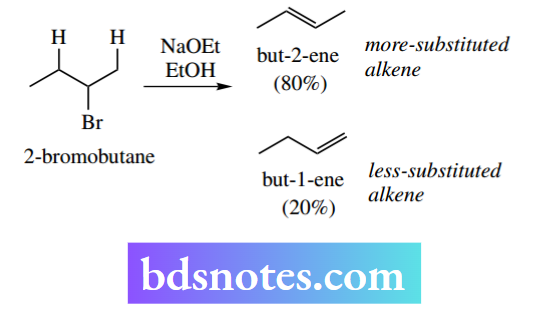
It is a general observation that, where different alkene products can arise through E2 elimination, the more-substituted alkene predominates.
2-Menthene contains a double bond with two alkyl substituents, whereas the double bond in 3-menthene has three substituents.
The more-substituted alkene is termed the Saytzeff product; the less-substituted alkene is termed the Hofmann product. We recommend you disregard the proper names and think of the products in terms of ‘more-substituted alkene’ and ‘less-substituted alkene’.
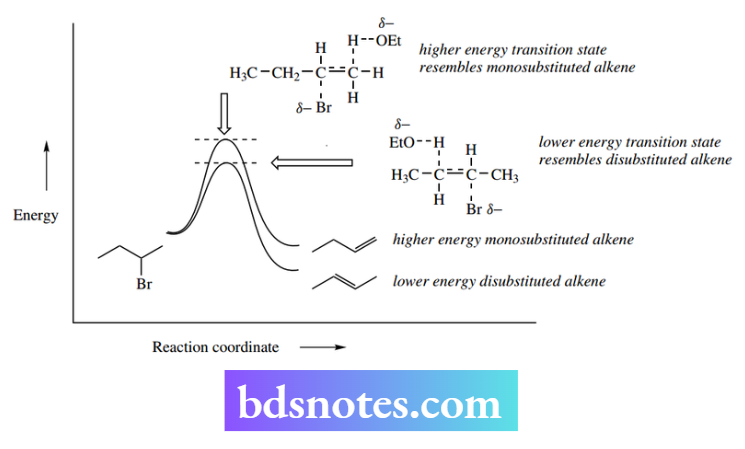
A further example of the more-substituted alkene predominating is found in the elimination of HBr from 2-bromobutane. The major product is the more-substituted alkene but-2-ene, which predominates over the less-substituted alkene but-1-ene by a factor of 4: 1.
The reasoning for this direction of elimination is twofold. The more-substituted alkene is actually of lower energy than the less-substituted alkene because of the stabilizing electron-donating effect of alkyl groups, and a similar effect will occur in the transition state where the double bond is developing. This is seen in the energy profile for the reaction.
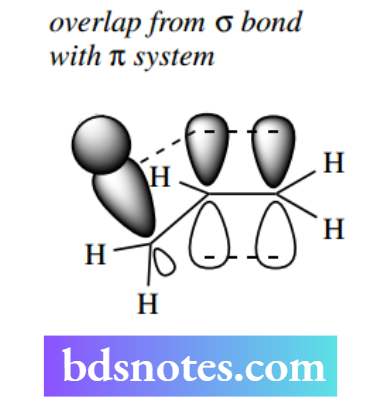
The stabilizing effect of alkyl groups appears to involve the overlap of σ C–H (or C–C) orbitals with the π system of the alkene, rather as we have seen with carbocations (see Section 6.2.1). The more alkyl groups attached, the more stabilization the alkene derives.
This effect is relatively small and both products are formed, usually with one predominating. The more substituted Saytzeff product typically predominates when the leaving group is small, for example, halide.
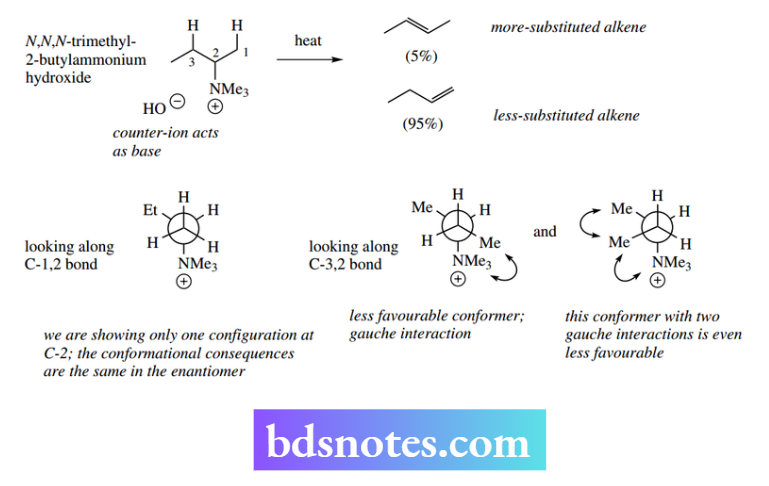
On the other hand, when there is a large leaving group present, Example quaternary ammonium, then steric effects become more important than the stabilizing effects of alkyl groups.
This is exemplified by the heatinitiated decomposition of the quaternary ammonium salt below. The elimination is now governed by which is the more favourable conformer of the substrate where a hydrogen atom is positioned anti to the quaternary ammonium substituent.
Two such possibilities can be considered. The conformer set up for 1,2-elimination is more favourable than the conformer for 2,3-elimination since the latter conformer would necessitate a less favourable gauche interaction.
An alternative conformer for 2,3-elimination has two unfavourable gauche interactions. Thus, it is the large leaving group that now dictates the direction of elimination, and the less-substituted alkene (Hofmann product) predominates.
Again, it should be noted that both products are obtained – the effect is not sufficiently great to produce one product exclusively.
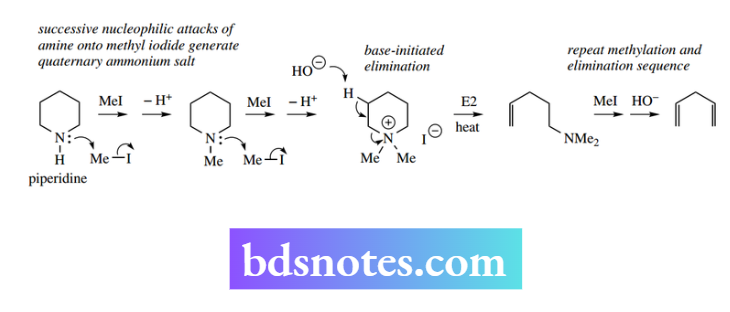
Note that with some cyclic substrates, the leaving group may remain as part of the product alkene. Elimination reactions played an important role in the early structural analysis of alkaloids (typically cyclic amines).
Combination of N-methylation followed by elimination may be used to open up nitrogen heterocycles, as shown with piperidine.
One further consideration relating to the nature of the products in eliminations is the stereochemistry of the double bond. For instance, base-catalysed elimination of HBr from 2-bromopentane gives three products.
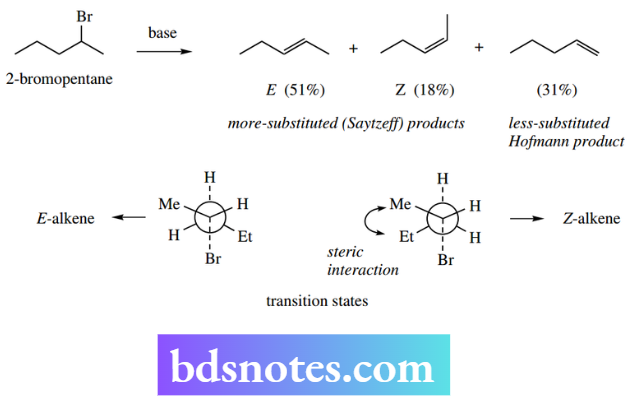
This elimination involves a small leaving group, so the more substituted alkene predominates. However, E and Z isomers of this Saytzeff product are produced, and in unequal amounts.
The major product is the E-alkene can be rationalized in terms of minimizing steric repulsion during the transition state.
Note the terminology that can be used to describe product distribution in this type of reaction. Reactions are termed regiospecific where one product is formed exclusively, or regioselective where one product predominates.
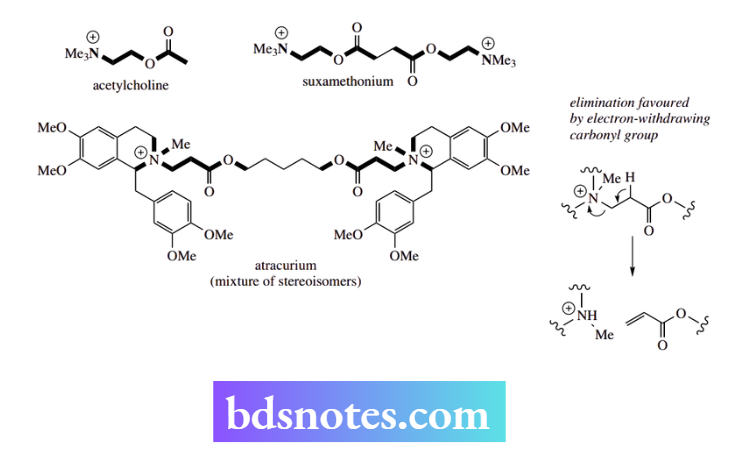
Atracurium is a curare-like muscle relaxant that is metabolized via an elimination reaction We have seen above that the muscle relaxant properties of curare and synthetic analogues result from competing with acetylcholine at receptors, thus blocking nerve impulses at the neuromuscular junction.
As quaternary ammonium salts, there are two well-separated acetylcholine-like groupings in the molecules, and the drugs probably span and block several receptor sites.
These agents work rapidly and are of considerable value in surgery. However, artificial respiration is required until the agent is metabolized, and thus broken down by the patient. Recent developments have led to agents with a built-in functional group that allows more rapid metabolism.
Initially, the presence of ester groupings, as in suxamethonium, allowed fairly rapid metabolism in the body via esterase enzymes that hydrolyse these linkages. The enzyme involved appears to be a non-specific serum acetylcholinesterase. Even better is the inclusion of functionalities that allow additional degradation via an elimination reaction.
Such an agent is atracurium. In addition to enzymic ester hydrolysis, atracurium is also degraded in the body by a non-enzymic elimination reaction that is independent of liver or kidney function. Normally, this elimination would require strongly alkaline conditions and a high temperature, but the presence of the carbonyl group increases the acidity of the proton and thus facilitates its removal.
Elimination can proceed readily under physiological conditions, giving atracurium a half-life of about 20 minutes. This is particularly valuable where patients have low or atypical esterase enzymes.
Atracurium contains four chiral centres (including the quaternary nitrogens) and is supplied as a mixture of stereoisomers; a single isomer cisatracurium has now been introduced. This isomer is more potent than the mixture, has a slightly longer duration of action, and produces fewer cardiovascular side effects.
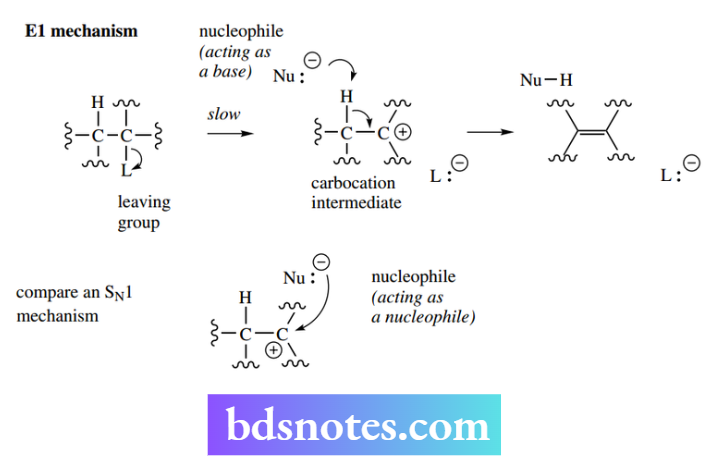
The E1 reaction: unimolecular elimination The abbreviation E1 conveys the information ‘elimination–unimolecular’. The reaction achieves the same result as the E2 reaction but is mechanistically different in that it involves a carbocation intermediate.
It is unimolecular since kinetic data indicate that only one species is involved in the rate-determining step where RL is the substrate containing the leaving group L and k is the rate constant.
The nucleophile Nu does not figure in the rate equation. Just as the E2 mechanism shares features of the SN2 mechanism, the E1 mechanism shares features of the SN1 reaction.
The initial step is the formation of a carbocation intermediate through the loss of the leaving group. This slow step becomes the rate-determining step for the whole reaction, i.e. the E1 mechanism is unimolecular. In general terms, the reaction can be represented as follows.
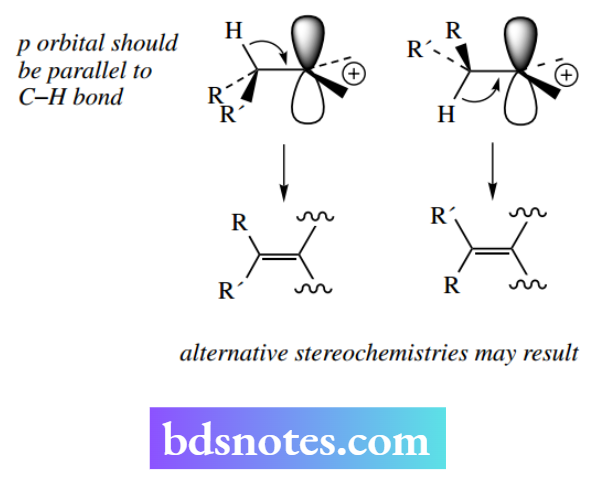
Once formed, the carbocation could be attacked by a nucleophile – the SN1 reaction. However, if the nucleophile acts as a base, then it removes a proton from a position adjacent to the positive centre and the original bonding electrons are used to discharge the positive charge and make a new double bond.
A stereochemical consequence of this is that the proton lost should be perpendicular to the plane of the carbocation to achieve maximum overlap with the unfilled p-orbital during the formation of the π bond.
We do not have the same strict stereochemical requirements as in the E2 mechanism, and isomeric alkenes may well be produced. If several hydrogens are available for elimination, then the preferred product formed is the more substituted Saytzeff alkene.
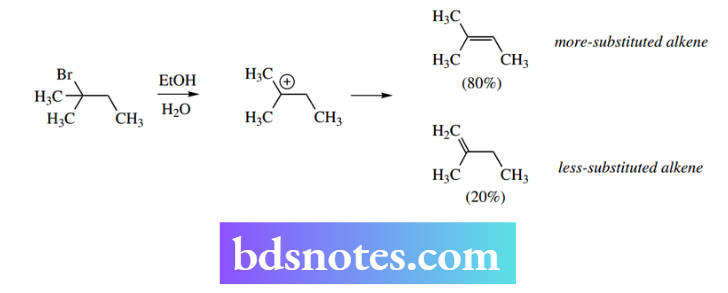
E1 elimination in the synthesis of tamoxifen
We have already employed the tamoxifen structure as an example of defining the configuration of double bonds.
Tamoxifen is a highly successful oestrogen-receptor antagonist used in the treatment of breast cancer. It may be synthesized by the following sequence.
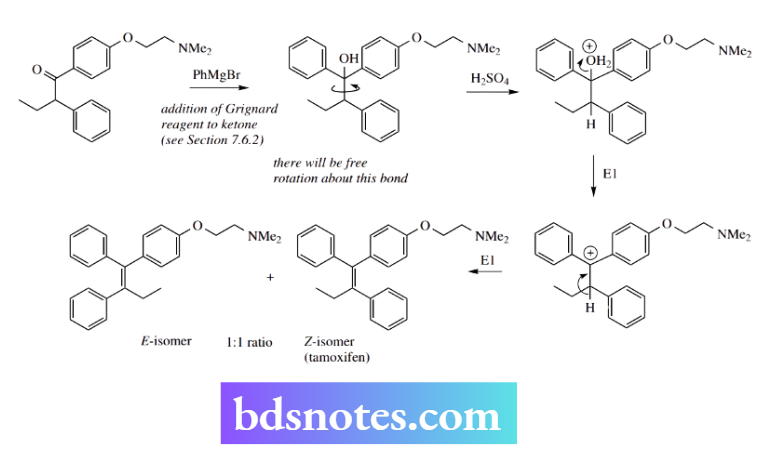
The main skeleton of the drug is constructed by a Grignard addition reaction on the appropriate ketone using phenyl magnesium bromide. This produces a tertiary alcohol.
It now remains to eliminate water from this structure. This is achieved under acid conditions.
An E1 mechanism is involved: protonation of the tertiary alcohol allows loss of water as the leaving group and generation of a carbocation, which is favoured since it is both tertiary and benzylic.
However, completion of the elimination by proton loss gives a 1: 1 mixture of the E- and Z-alkenes, since there is no stereocontrol at this stage – free rotation about the C–C bond in the alcohol and subsequent structures until the double bond is formed means both stereochemistries will be produced. The drug material tamoxifen is the Z-isomer.
E1 or E2 ?
We have seen above that the structure of the substrate is the most important feature that dictates the mechanism of substitution reactions. Thus, the SN2 mechanism is favoured when the reaction takes place at a primary centre, whereas an SN1 mechanism is preferred at tertiary centres, or where stable intermediate carbocations can be produced.
We can use similar reasoning to predict that an E2 mechanism might be preferred when the leaving group departs from a primary centre and that an E1 mechanism is likely when structural features facilitate carbocation formation.
By structural features, we mean tertiary, allylic, or benzylic centres. When a secondary centre is involved, then either E1 or E2 might occur, depending upon reaction conditions.
In general, these predictions are found to be sound. However, there is an apparent anomaly, in that E2 reactions also frequently occur with tertiary substrates. If we think a little deeper, we shall discover that it is not unreasonable for this to be so.
The E2 reaction is initiated by the base removing a proton, and this is still possible even where there is a tertiary centre.
Although, for steric reasons, a nucleophile cannot approach a tertiary centre to displace a leaving group (SN2 reaction), it is still feasible for a base to remove a proton from an adjacent carbon.
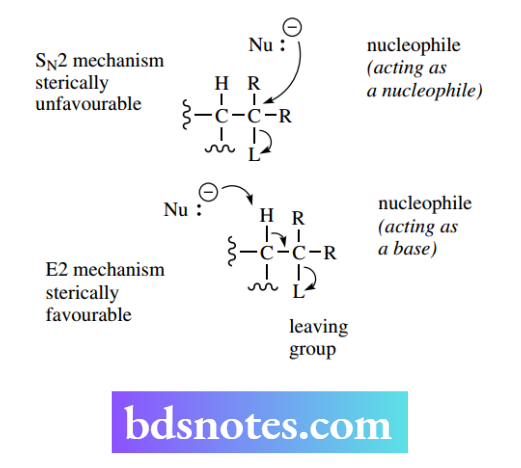
Accordingly, the E2 mechanism becomes relatively favourable, even with tertiary substrates, when we use a strong base or more concentrated base. We are thus more likely to get an E1 mechanism when we have a tertiary centre, and weak bases or bases in low concentration.
Polar solvents are also going to be conducive to carbocation mechanisms. Just as acidic conditions help to favour SN1 reactions, they also going to favour E1 reactions.
Elimination or substitution? Elimination can be a troublesome side reaction during substitution reactions. In general terms:
- Strong Bases Favour Elimination;
- Large Bases Favour Elimination;
- Steric Crowding In The Substrate Favours Elimination;
- High Temperatures And Low Solvent Polarity Favour Elimination.
Carbocati on rearrangement reactions
Most organic reactions involve changes to functional groups whilst the fundamental molecular skeleton remains unchanged.
In molecular rearrangements, groups migrate within the molecule and the molecular skeleton is modified. In most rearrangements, the groups migrate to the next atom, a 1,2-shift, though 1,3-shifts and other migrations are known.
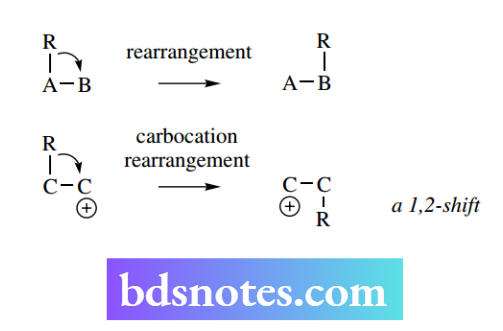
The most common examples of rearrangements involve an electron-deficient atom, and pre-eminent among these are carbocations.
Since carbocations are a feature of the SN1 and E1 mechanisms, it follows that rearrangements can be side-reactions of these types of transformation. The driving force in carbocation rearrangements is to form a more stable carbocation.
Consider a proposed nucleophilic substitution reaction on the secondary alcohol shown using aqueous HBr.
As a secondary alcohol, either SN2 or SN1 mechanisms are possible (see Section 6.2.3), but SN1 is favoured because of the acidic environment and the large tert-butyl group hindering the approach of the nucleophile. The expected SN1 bromide product is formed, together with a smaller amount of the E1-derived alkene in a competing reaction.
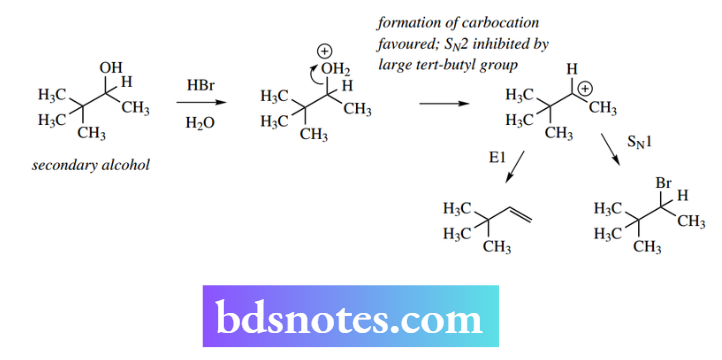
However, other products are also produced. These are isomers of the above products and have a rearranged carbon skeleton. Their formation is rationalized as follows:
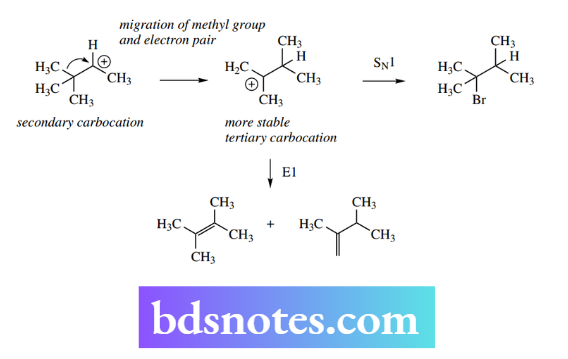
The first-formed carbocation is secondary. This carbocation can become a more stable tertiary carbocation via rearrangement, in which a methyl group with its pair of electrons migrates from one carbon to the adjacent positive centre.
Now the rearranged tertiary carbocation can yield SN1- and E1-type products in much the same manner as the original secondary carbocation.
A rearranged bromide is formed, together with two alkenes from an E1 process, with both more-substituted Saytzeff and less-substituted Hofmann alkenes being produced. The formation of such rearranged products proves that this unexpected transformation must occur.
These carbocation rearrangements are termed Wagner–Meerwein rearrangements. They are most commonly encountered with secondary carbocations where rearrangement produces a more stable tertiary carbocation.
They are less common with tertiary carbocations, which are already stabilized by the maximum number of alkyl groups, and where any rearrangement would tend to produce only a less stable secondary carbocation.
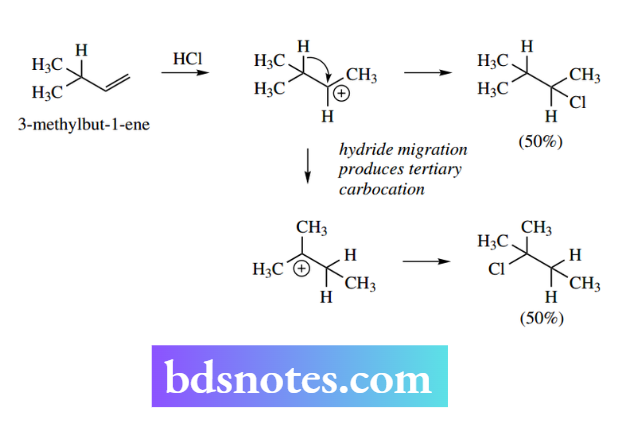
Wagner–Meerwein rearrangements are not restricted to methyl migrations, and we may also see the transfer of hydrogen with an electron pair, i.e. a hydride migration.
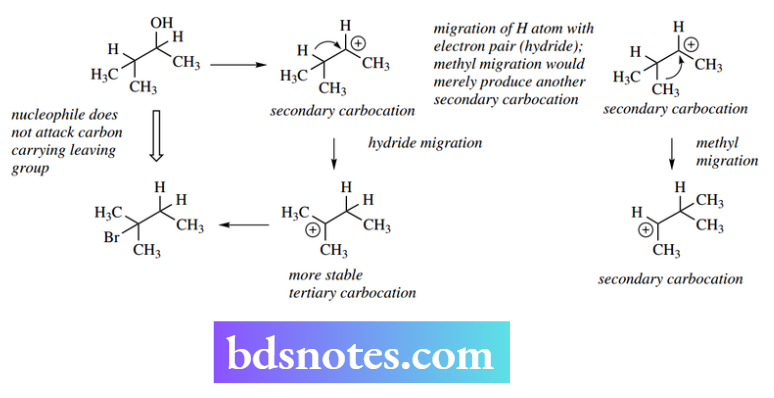
This is observed in the case of the secondary alcohol illustrated, where a secondary carbocation would be generated.
A methyl migration would merely lead to another secondary carbocation, and this serves no stabilizing effect.
However, a hydride migration produces a tertiary carbocation, so this process will stabilize the system. This is what happens, and the major product is a bromide where the halogen appears to have attacked the wrong position, i.e. different from that which originally carried the leaving group. This is the pointer to something unusual occurring.
Again, the driving force is the conversion of a secondary carbocation into a more stable tertiary carbocation.
Hydride migration also accounts for one of the observed products from the treatment of the cyclohexanol tosylate with acetic acid.
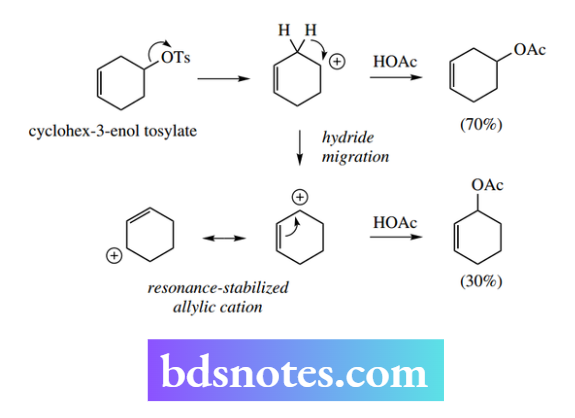
Although the predominant product is the corresponding acetate (one could formulate either SN1 or SN2 mechanisms for the formation of this product), about 30% of the alternative acetate is formed.
This can be rationalized as arising from a carbocation that is rearranged by hydride migration. This is favoured because the resultant carbocation is an allylic cation, and stabilized by resonance.
In most cases, the driving force for a rearrangement is the conversion of a secondary carbocation into a more stable tertiary carbocation.
Surprisingly, there are examples of where a tertiary carbocation is transformed into a secondary carbocation, but there needs to be some more powerful driving force to achieve this. Relief of ring strain is a particular case.
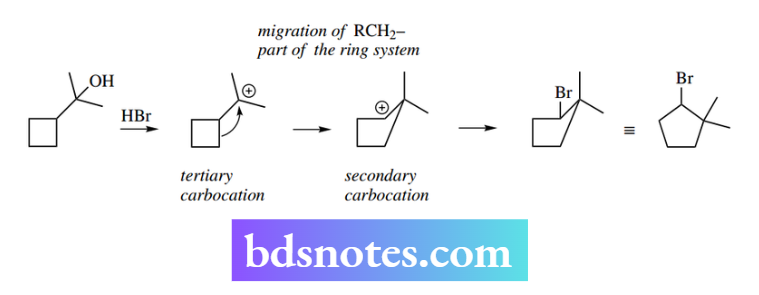
The cyclobutane-ring-containing alcohol can yield a tertiary carbocation, but the product from an SN1 reaction with HBr contains a cyclopentane ring.
Its formation is rationalized via a Wagner–Merwein rearrangement in which ring expansion occurs. This is represented as equivalent to a methyl migration, but the methylene group is part of the carbon chain.
There is significant relief of ring strain in going from a four-membered ring to a five-membered ring, which is more than enough to make up for the energy change in going from a tertiary carbocation to a less stable secondary carbocation. Carbocations also feature as intermediates in electrophilic addition reactions and Friedel–Crafts alkylations.
Rearrangements may also be observed in these carbocations if they have the appropriate structural features. It does not matter how the carbocation is produced, subsequent transformations will be the same as we have seen where rearrangements are competing reactions in nucleophilic substitution.
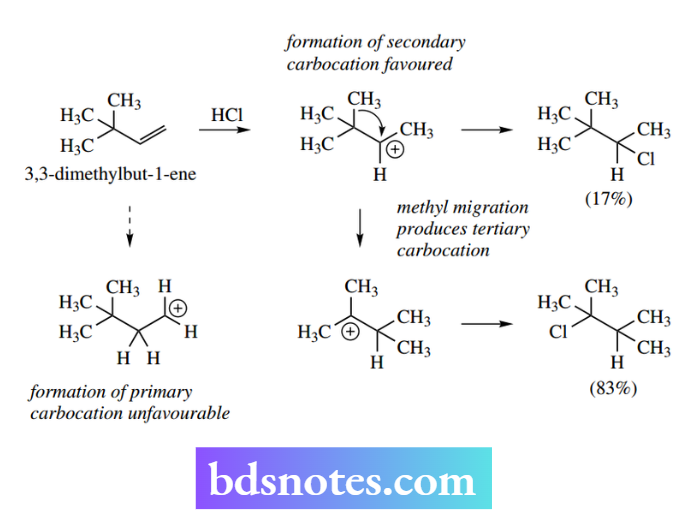
Thus, electrophilic addition of HCl to 3,3-dimethylbut-1- ene proceeds via protonation of the alkene, and leads to the preferred secondary rather than primary carbocation.
However, this carbocation may then undergo a methyl migration to produce the even more favourable tertiary carbocation. Finally, the two carbocations are quenched by a reaction with chloride ions. The product mixture is found to contain predominantly the chloride from the rearranged carbocation.
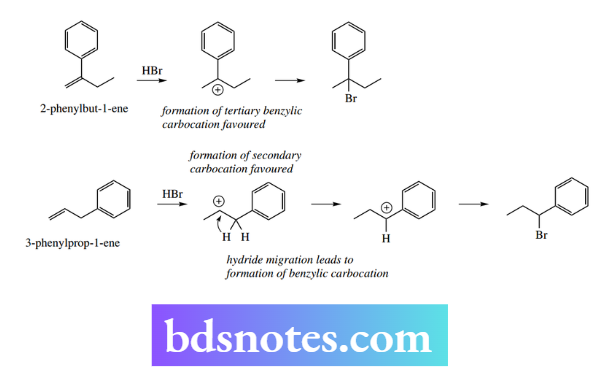
The enhanced stability of benzylic carbocations is nicely illustrated by the addition of HBr to the two alkenes shown below. In the case of 2-phenylbut-1- ene, protonation of the alkene leads to a carbocation that is both tertiary and benzylic and is significantly favoured over an alternative primary carbocation. Quenching with the bromide nucleophile gives the tertiary bromide.
On the other hand, 3-phenyl prop-1- ene is protonated to a secondary carbocation. In this case, rearrangement by hydride migration leads to a more favourable benzylic carbocation, and a benzylic bromide is the observed product.
Rearrangements seem to provide us with an unexpected complication to ruin our carefully thought-out plans for interconverting chemicals.
It is sometimes difficult to predict when they might occur, but we should recognize occasions when they might become a nuisance, e.g. look at the structure of any proposed carbocation intermediate.
In most cases, we shall be more concerned with rationalizing such transformations, rather than trying to predict their possible occurrence.
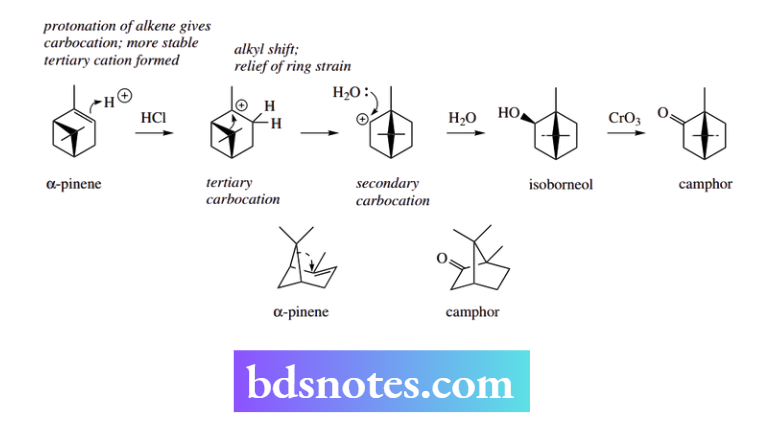
Carbocation rearrangements: synthesis of camphor from α-pinene
Although the monoterpene camphor occurs naturally, substantial amounts are produced semi-synthetically from α-pinene, a component in turpentine. Treatment of α-pinene with aqueous HCl protonates the double bond by an electrophilic addition (see Section 8.1.1) and generates the more favoured tertiary carbocation. Rather than simply being attacked by a nucleophile, this carbocation rearranges.
The tertiary carbocation contains a strained four-membered ring, and an alkyl shift allows relief of ring strain, generating five-membered rings and a secondary carbocation.
It would appear that the relief of ring strain more than compensates for the loss of tertiary character in the carbocation.
Thus, it is the secondary carbocation that interacts with a nucleophile. In this case, the nucleophile is water, the major component of the aqueous HCl. The product is thus isoborneol.
Camphor is then obtained from isoborneol by oxidation of the secondary alcohol to a ketone.
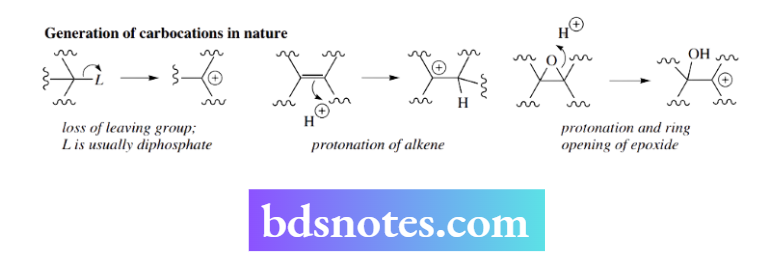
Carbocation rearrangements in nature: b biosynthesis of lanosterol Many examples of carbocation rearrangements can be found in nature, particularly in the biosynthesis of terpenoids and steroids.
Nature generates carbonation in three main ways. The first of these is the loss of a leaving group, with diphosphate being the most common leaving group.
Protonation of an alkene also produces a carbocation, and, as we would predict, this tends to form the more substituted and thus more stable carbocation. Also encountered is the ring opening of an epoxide group, which may be considered to be acid-initiated.
Perhaps the most spectacular of the natural carbocation rearrangements is the concerted sequence of 1,2-methyl and 1,2-hydride Wagner–Meerwein shifts that occur during the formation of lanosterol from squalene. Lanosterol is then the precursor of the steroid cholesterol in animals.
Carbocation formation is initiated by an epoxide ring opening in squalene oxide, giving a tertiary carbocation, and this is transformed into the four-ring system of the protosteryl cation by a series of electrophilic addition reactions.
The resultant protosteryl cation has a tertiary carbocation in the side chain, and a hydride shift generates another tertiary cation. A second hydride shift follows, then two methyl shifts, each time generating a new tertiary cation.
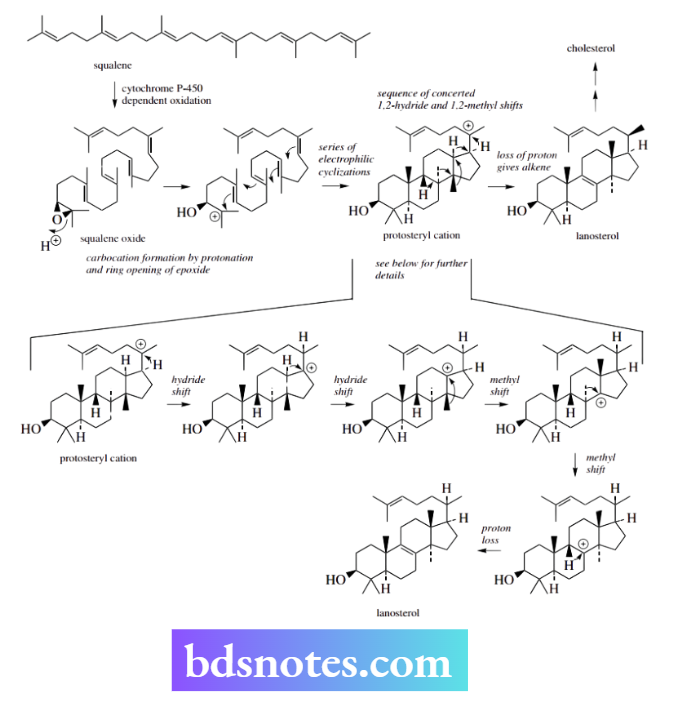
Lastly, the positive charge is neutralized via the loss of a proton, giving the alkene lanosterol.
There is no obvious energy advantage in such tertiary-to-tertiary cation changes, but it must be appreciated that this is an enzyme-catalysed reaction, and the enzyme plays a crucial role in the reactions that occur.
These hydride and methyl migrations do occur, as demonstrated by isotopic labelling studies.
Further, it is noted that most of them involve inversion of stereochemistry at the particular centre, a feature of the concerted nature of these rearrangements, so that as one group leaves another approaches from the rear. Thus, we have the features of SN2 reactions in a carbocation mechanism.
This is a complicated series of reactions but includes impressive examples of carbocation rearrangements. The electrophilic cyclization sequence is also quite striking.
Leave a Reply