Organic Chemistry Of Pharmacy Questions And Answers
No matter how intrinsically interesting students find the study of organic chemistry, there will usually be one or more hurdles that need to be overcome, the examinations.
Examination technique is undoubtedly a skill, and this chapter is aimed at developing such skills for organic chemistry. Throughout the book, we have tried to convince the reader that, by applying principles and deductive reasoning, we can reduce to a minimal level the amount of material that needs to be committed to memory.
This section contains a selection of typical examination questions, many of them based on real ones used at Nottingham, together with the answers. However, it is not so much the answers themselves that are important, but developing the skills to answer them.
We wish to show that, in many questions, the information given can actually direct us toward the answer. Thus, emphasis has been placed on what to look for in the question, how to approach the problem, and how to develop the answer in a logical manner.
We are not looking at questions that ask for essay-style answers, and thus require a lot of memorized material, but are concerned predominantly with those questions that start out ‘Propose a mechanism for ..’ or ‘Explain the following observations..’
Rather than having questions at the end of each chapter, we have purposely decided to put all these problems together in a separate chapter to help emphasize links and the integration of different ideas.
One of the disadvantages of the almost universal modular system as a study method is that particular themes become imprisoned in self-contained packages.
For real skill and understanding, it is necessary to be able to interrelate ideas from different modules. The problems used here are usually going to require the integration of knowledge from one or more chapters to provide the answer.
The source material required for each problem is broadly indicated so that they can be used as self-test examples after the appropriate chapters have been covered.
It may well be that these questions become more relevant towards the end of a particular course when examinations are no longer a distant consideration but are becoming an imminent prospect.
Examination Questions Useful Advice
Read the instructions very carefully. Make sure you are clear on how many questions you need to answer. Do not answer more questions than are required. You may be penalized, in that only those questions answered first will be marked; these may not be your highest scoring answers.
Read the entire paper. Choose the questions that you are going to answer and decide the order in which you will tackle them. Start with a question which you think you can answer well, to build up your confidence for the less-appealing questions, but do not spend more than the allocated time on this question.
Read each question carefully before beginning your answer. If the question is divided into parts, be clear about how many parts you must answer. Understand clearly what is being asked. Jot down headings to help you structure your answer. To be sure to get maximum credit, organize your answer, setting the solution neatly and legibly.
Cross out any rough work or mistakes. Take special care with multiple-choice questions, and do not make guesses if you are going to be penalized for wrong answers.
Answer the question asked. Easy marks are lost by not answering the question asked. Do not spend time giving information that is not required whilst missing out on other aspects that are clearly asked for. Do not try to copy out your lecture notes.
The examiner expects you to be selective and formulate a critical answer to the question.
Be quite clear you fully understand the meanings of the terms ‘discuss’, ‘evaluate’, ‘illustrate’, and ‘outline’, which are frequently used in examination questions.
You must appreciate the subtle differences, and provide the appropriate level of detail to answer the question effectively.
A title that includes ‘discuss’ or ‘evaluate’ requires you to sum up both positive and negative evidence or aspects. A title that says ‘illustrate’ or ‘outline’ is only asking for evidence or aspects that are consistent with the proposition.
Most organic chemistry questions do not require descriptive answers but can be adequately answered by generous use of structures and mechanisms. These can be annotated with pertinent points as appropriate.
Budget your time. Keep an eye on the clock, and do not spend more than the appropriate amount of time on any question. It may not be worthwhile trying to get a few extra marks on one question at the expense of gaining easy marks by starting another.
You must attempt to answer the full number of questions specified in the paper. Note any breakdown of marks allocated for each part of a question, and use this to apportion the time and effort spent on the component parts.
Check your answers. Try to finish a little early so that you can read through your answers. Make sure your answers are complete. It is easy to make simple errors.
Check for any missing answers to parts of questions, especially multiple-choice questions. Make sure your answers make scientific sense, and in numerical answers check that you have included units and considered significant figures.
Mechanisms or calculations that appear unnecessarily complicated are probably wrong and should be checked for obvious errors. Incorrect transcription of structures or figures is a common cause of problems.
How To Approach The Problem Propose A Mechanism
You will usually be provided with the reactants, reagents, conditions, and the products formed, though in some questions you may be required to predict the nature of the products.
- Carry out a quick analysis of the carbon skeletons of reactants and products to ascertain what the reaction represents.
- Is it merely a modification of substituents, for example, as in substitution or elimination?
- Is it the formation of a bigger skeleton, for example, as in an addition reaction?
- Is it the formation of a smaller skeleton, for example, as in a cleavage reaction?
- Is it a modification that has involved a rearrangement of the basic skeleton?
- Register this change, but do not try to accommodate the reaction at this stage. Instead, move on to the reagents and conditions, as follows.
- Consider the essential principles of mechanism. Look for nucleophiles, electrophiles, and leaving groups.
- Consider the conditions. If the reaction requires a catalyst, for example, acid or base, it is almost certain that this needs to be used in the first step. For example, acid may protonate an electronegative atom, making it a more reactive species.
- Thus, a protonated carbonyl becomes a better electrophile, and a protonated alcohol now has a better-leaving group. The base may remove an acidic proton to generate a better nucleophile, although in some reactions it may itself act as the nucleophile.
- At this stage, the mechanism should develop logically by interaction of a nucleophile with an electrophile, or by displacement of a leaving group.
If your mechanism is to make sense though, certain aspects, which are really rather obvious, need to be considered. You cannot sensibly use X– as a nucleophilic reagent under acid conditions, and there is not going to be any H+ under basic conditions. In the former case, you must use HX as the nucleophilic reagent.
In the latter case, a requirement for a proton, perhaps to finish off a mechanism, will probably be met by abstracting it from a solvent molecule, such as water or alcohol.
- There are stability considerations with carbocations, with tertiary carbocations being more stable than secondary ones; if your mechanism includes CH3+ or RCH2+ it is almost certainly wrong! Carbocation mechanisms are also going to be much more likely under acidic conditions (H+) rather than under basic conditions (HO–).
- Do not even consider radical reactions unless there is some obvious pointer from the reagents, such as a radical initiator such as a peroxide or electromagnetic radiation.
- After each step of the mechanism, write out the intermediate structure that will be produced. Do not try to include more than one such change per step.
- Always try to develop the mechanism logically, working forward so that the product you end up with turns out to be identical to that in the question.
- It is rather less satisfactory, and definitely more risky, to apply the reverse strategy, i.e. aiming for the product and trying to manipulate the reagents.
- If a proposed mechanism starts to get complex or overcomplicated, then stop – it is probably wrong. Check it through, first ensuring
- that the structure is transposed correctly at each stage. If this does not show up as an error, go back to the essentials – electronegativity, nucleophiles, electrophiles, leaving groups.
Worked Problems
These problems are typical examination questions, though perhaps longer than most, and have been graded as level 1 or level 2. Based on a 2-year period of study to cover most of the content of this book, level-1 questions might be answered after year-1 studies, whereas level-2 questions are more appropriate for year-2 studies.
Purists might criticize the avoidance of equilibrium arrows in the mechanisms shown. Some reactions, for example, hemiacetal formation or acid-catalysed ester hydrolysis, are undoubtedly reversible, yet we have shown them as proceeding only in the forward direction.
We believe it is more important to develop the skills for predicting a rational mechanism rather than remembering whether the reaction is reversible or not.
Unless there is any specific comment regarding reversible reactions, we should concentrate on the reaction in the sense given in the question.
Question 1. Taxol is an anticancer drug obtained from species of yew and has the partial structure shown. R represents a splendidly complex terpenoid group.
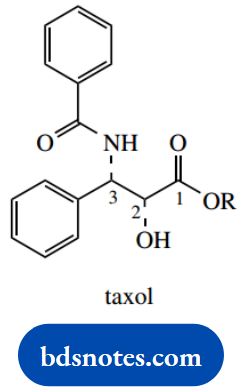
- From the following terms, indicate those which may be used to describe the chemical structure of taxol:
- Ketone;
- Lactone;
- Ester;
- Amine;
- Amide;
- Lactam;
- Acetal;
- Ether.
- Identify the chiral centres in taxol, and designate their configurations as R or S. Show the priorities assigned to the groups in establishing the configurations.
- Draw taxol in the form of a Newman projection looking down the 2-3 bond, and showing the stereochemically preferred conformation.
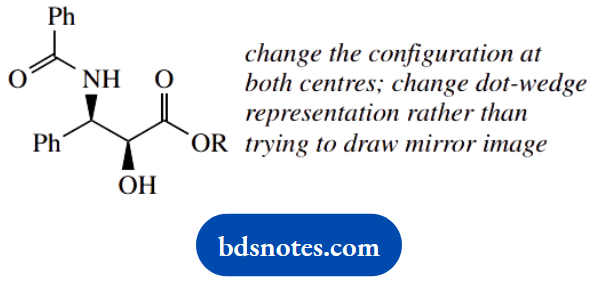
- Draw the enantiomer of taxol, using the wedge-dot convention.
- Give a mechanism for the reaction of taxol with acetic anhydride [(CH3CO)2O].
- What products would be produced if taxol were treated with hot, aqueous HCl?
Answer:
This is a relatively complex molecule, but these are simple questions requiring only standard procedures and reasoning.
1. Ester, amide
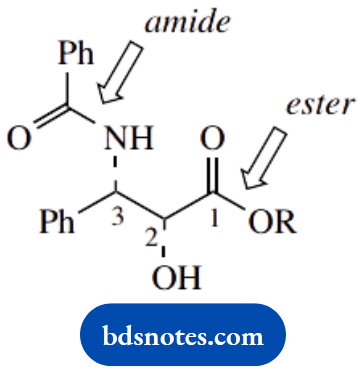
2. 2R, 3S

3.
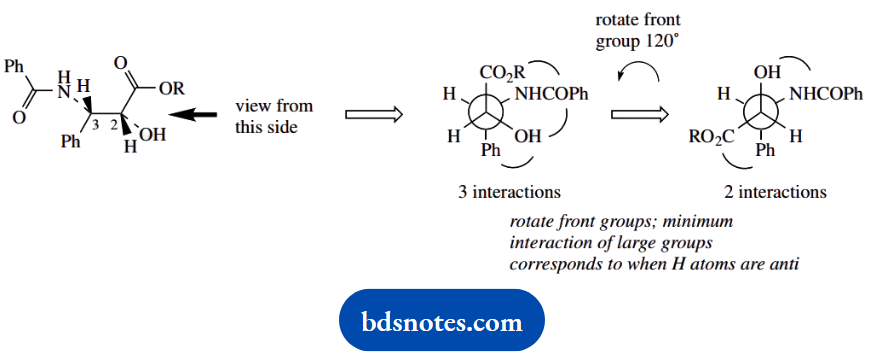
4. Enantiomer
5. Alcohol plus acetic anhydride gives acetyl ester.
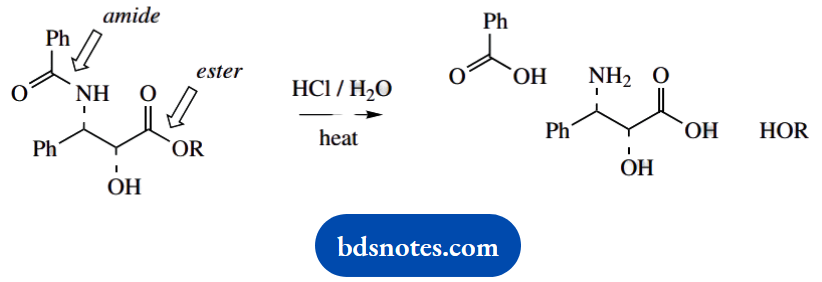
6. Note that ester and amide functions were identified in section (a); acidic hydrolysis (hot aqueous HCl) would lead to acid plus alcohol from ester, and acid plus amine from amide. There will be three products. You are not asked for any mechanisms.
Question 2. Fluvastatin is an inhibitor of the cholesterol biosynthetic pathway and may be used to reduce the risk of coronary conditions, for example, strokes and heart attacks, in susceptible patients. It has the structure shown, though drug material is supplied in the racemic form. A partial numbering system is given, and the rest of the molecule may be abbreviated to ‘aryl’ in answers.
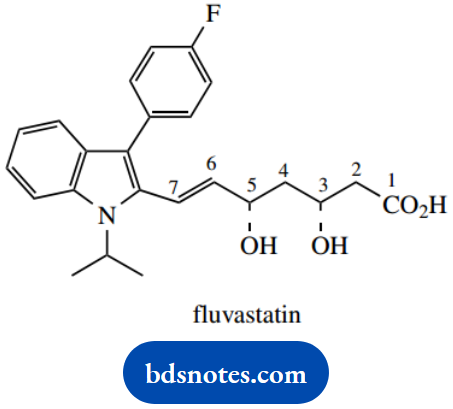
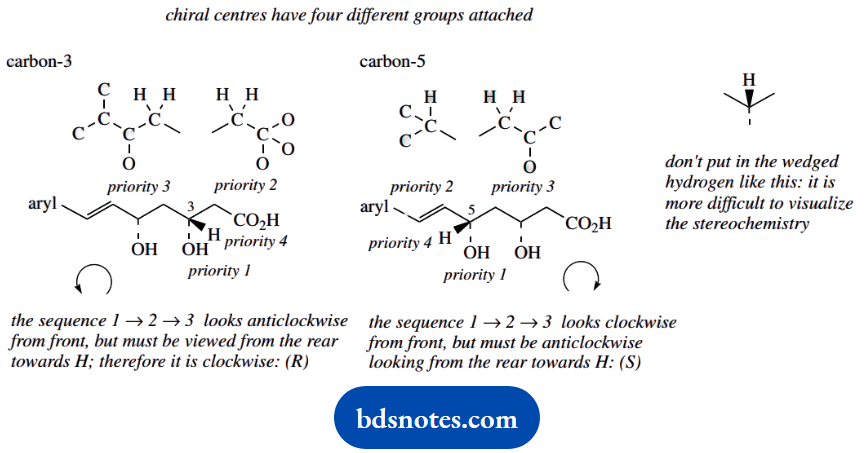
- Identify the chiral centres in fluvastatin, and designate their configuration as R or S. Show the priorities assigned to groups in establishing the configurations.
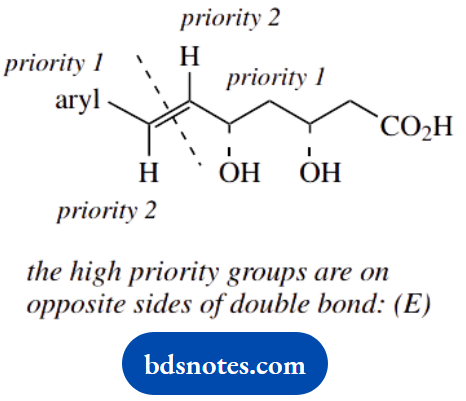
- Define the configuration of the double bond as E or Z. Show the priorities assigned to groups in establishing the configuration.
- Draw the structure of the other component of racemic fluvastatin.
- Draw the structure of a diastereoisomer of fluvastatin.
- Fluvastatin can easily form a cyclic ester (lactone):
- Show the structures, including stereochemistry, of two possible lactone products.
- Indicate, giving reasons, which of the two lactones would be favoured.
- With an appropriate drawing, predict the most likely conformation of the favoured lactone.
- Give a mechanism for the acid-catalysed formation of the lactone.
Answer:
This problem shows a fairly complex structure but asks relatively simple questions about it. It is important not to be discouraged by the structure; simply apply standard procedures and reasoning.
1. 3R and 5S
2. E
3. Another component of racemic fluvastatin is its enantiomer.
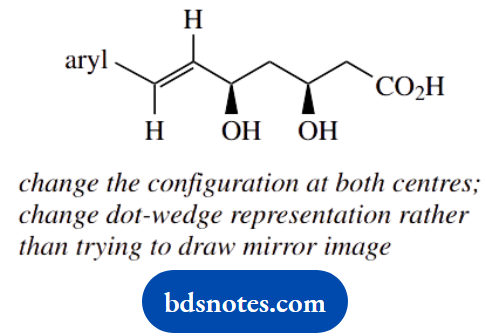
4. Diastereoisomers
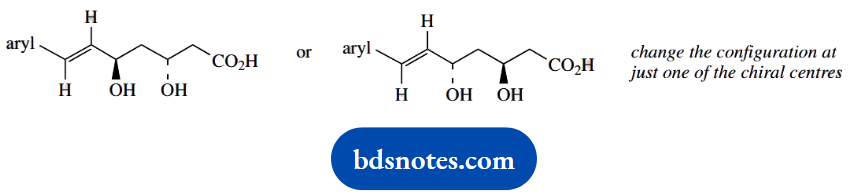
5. Make ester from acid plus one of the two alcohol groups
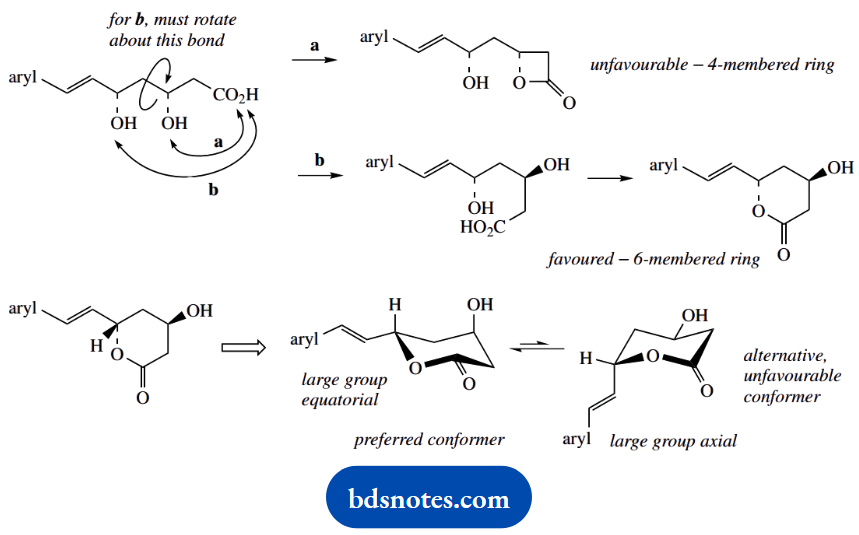
Acid Catalysed lactone formation.
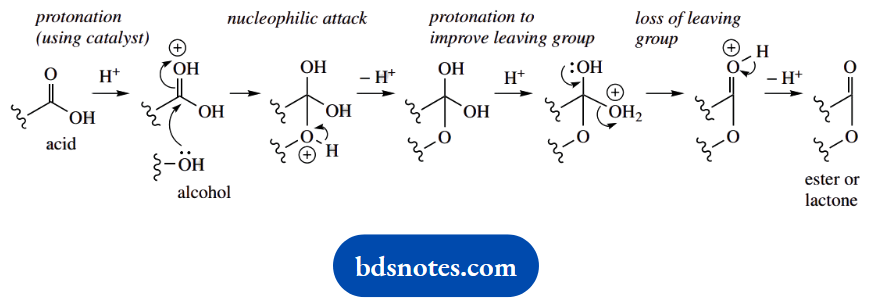
Question 3.
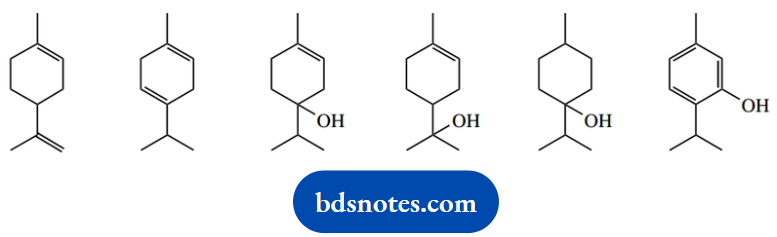
1. Indicate, with appropriate comments, which of the following compounds could exist in optically active form:
2. Draw the compound cineole to show the shape of its ring and other stereochemical features.
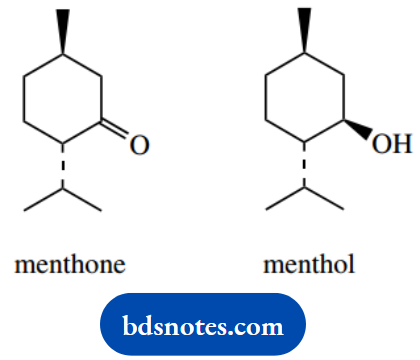
3. Reduction of menthone with sodium borohydride gives menthol and a second isomeric material, neomenthol. Give a mechanism for the reaction and suggest a structure for neomenthol.
What is the relationship of neomenthol to menthol? Is it a structural isomer, a configurational isomer, a diastereoisomer, an epimer, a meso compound, or a conformer? Several of these terms may apply.
Answer:
1. This is a ‘spot the chiral centre’ problem. A chiral centre has four different groups attached; we must also bear in mind that a meso compound has chiral centres, but the compound itself is optically inactive.
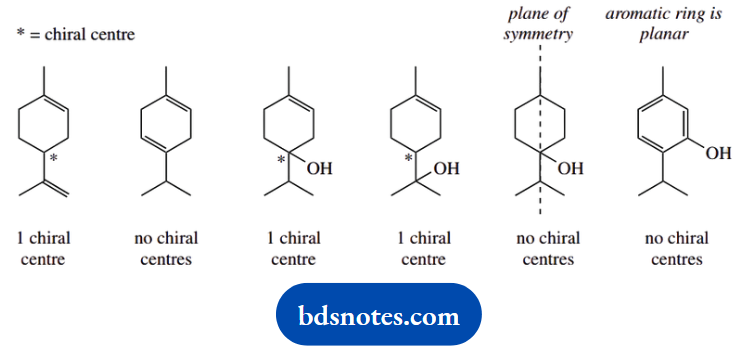
This arises because the molecule has a plane of symmetry, and optical activity conferred by one chiral centre is equal and opposite to that conferred by the other and is therefore cancelled out.
Only three of the compounds shown have chiral centres; these are indicated. These three compounds could, therefore, exist in optically active form. None of the compounds shown is a meso compound. Note that we have to consider part of the ring system as a ‘group’ attached to the centre in question.
Follow this around until a decision can be made. The last two are not exactly trick questions but require care. One has a plane of symmetry, and the carbon carrying the hydroxyl has two of its attached groups the same; the benzene ring is planar, so none of its carbons has the potential to be chiral.
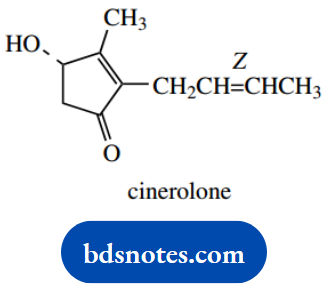
2. There are two parts of this molecule that require attention, the ring and the side chain. The latter is easier; we just have to interpret Z. In simple systems, as we have here, Z is the equivalent of cis.
Systematically, we need to consider the priorities of groups attached to the double bond. The Z configuration has groups of higher priority on the same side of the double bond, as shown.
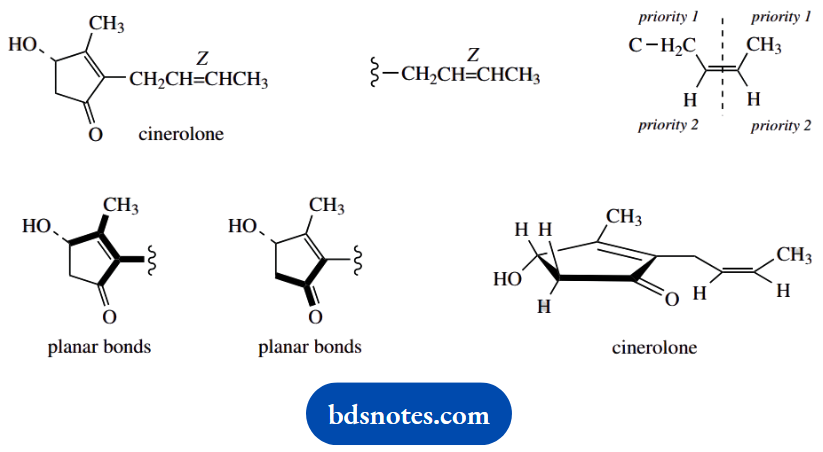
Now for the ring, and you are advised to make some deductions before you start thinking about the conformations of five-membered ring systems. We have a double bond and a ketone in this structure. These both confer planarity on adjacent atoms.
As a result, we must conclude that the carbon atoms of the ring must all be in one plane. Only the hydrogen and hydroxyl substituents will be out of the plane. The stereo drawing, therefore, requires a planar five-membered ring seen in perspective, as shown.
3. Sodium borohydride is a reducing agent that reacts with a ketone to give an alcohol through nucleophilic attack of hydride (it is not actually hydride that attacks, but we can formulate it as such).
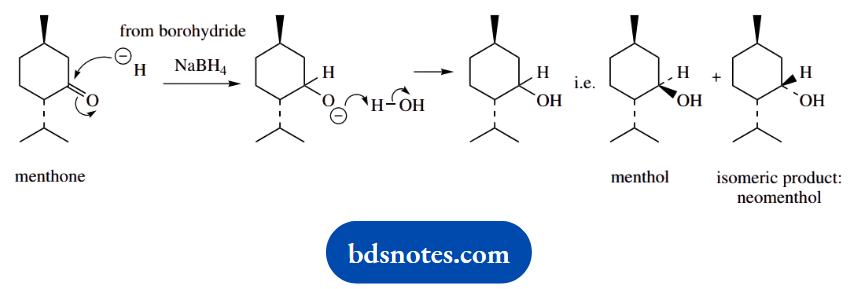
One of the products is menthol, and the second is an isomer, neomenthol. The carbonyl group is planar, so hydride can be delivered from either face. The isomer must be the alcohol in which the hydroxyl substituent has the alternative stereochemistry.
Neomenthol differs from menthol in having different stereochemistry at a single position. This makes it a configurational isomer, a diastereoisomer (there are two other chiral centres that are unchanged), and an epimer. The other terms are not applicable.
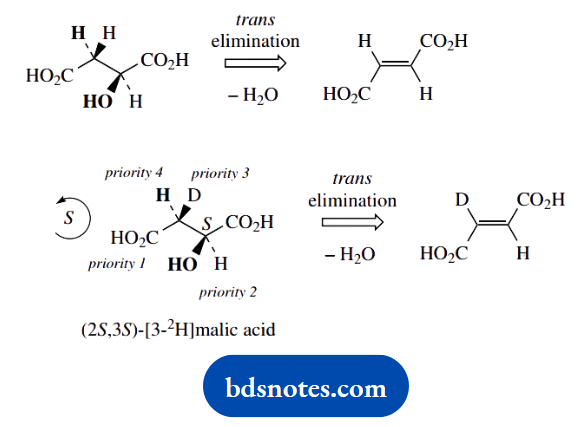
Question 4. The enzyme fumarase catalyses the stereospecific trans-addition of water to fumaric acid giving (S)-malic acid, and the reverse reaction, the trans-elimination of water from (S)-malic acid:
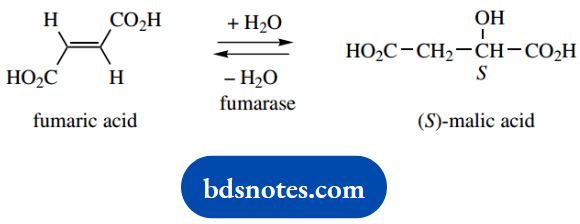
- Give systematic names with stereochemical descriptors for fumaric acid and malic acid.
- Show the stereochemistry of (S)-malic acid using wedge-dot conventions.
- Deduce the structure of the product from the action of fumarase on (2S,3S)-[3-2H]malic acid.
- The enzyme malate synthase also synthesises (S)-malic acid from glyoxylic acid (OHC-CO2H) and the thioester acetyl-coenzyme A (CH3CO.SCoA; this may be regarded as a source of the nucleophile – CH2CO.SCoA).
- Draw a mechanism for the addition reaction.
- Draw a mechanism for the hydrolysis of the intermediate thioester, assuming base catalysis.
- Indicate the stereochemistry implicit in the addition reaction. What face of the carbonyl group is attacked?
Answer:
Whilst this may look like an intermediary metabolism question, it requires no knowledge of biochemistry and is merely a simple chemical analysis of two reactions from biochemistry.
1. The nomenclature for a dicarboxylic acid follows from that of a monocarboxylic acid, for example, butanoic acid becomes butanoic acid. The 1,4-numbering is strictly redundant; the carboxylic acids must be at the ends of the chain. Stereochemical descriptors are (E) for the double bond in fumaric acid, and the (S) configuration stated for malic acid.
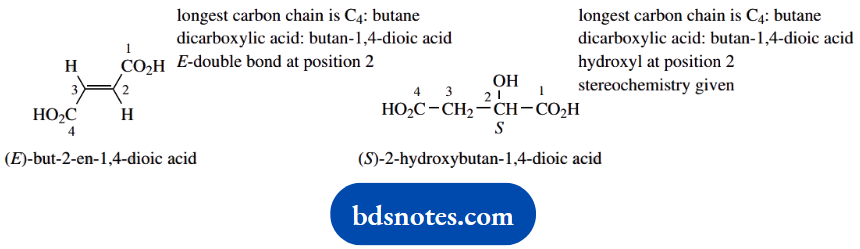
2. Draw the carbon chain in zigzag form, put in the large carboxylic acid groups anti, then the substituents. The configuration at C-2 is determined by trial and error.

3. The trans elimination of water from (S)-malic acid must remove the groups shown in bold. We then substitute one of the hydrogens at position 3 with 2H (deuterium; D) to get the 3S configuration by the trial-and-error procedure in (b).
(2S,3S)-[3-2H]malic acid is thus labelled as shown, and trans elimination of water must result in the deuterium label ending up as shown in fumaric acid.
4. As soon as you have written down the reagents, you will find the reaction follows quite readily. We have a simple nucleophilic addition to an aldehyde.
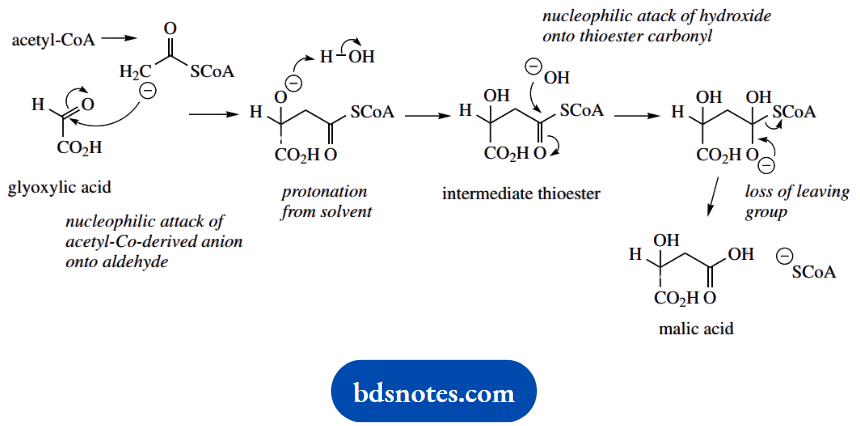
5. The intermediate thioester is then hydrolysed by the base as included above.
6. Now for the stereochemistry. Draw the malic acid product with the 2S configuration. It is also possible to turn around the structure obtained in part (b). You can only get this configuration if the planar aldehyde is attacked from the face shown.
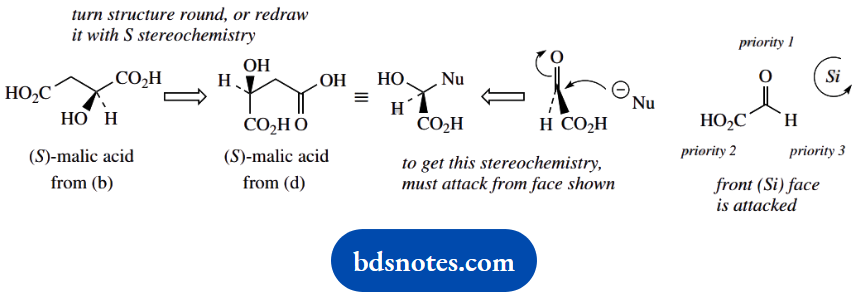
This is the Si face, using the priority rules for enantiotopic faces, which are an extension of the Cahn-Ingold-Prelog R and S system.
Question 5. In each of the following reactions, an optically active starting material is converted into a product that is optically inactive. Suggest mechanisms for the reactions and explain the loss of optical activity.
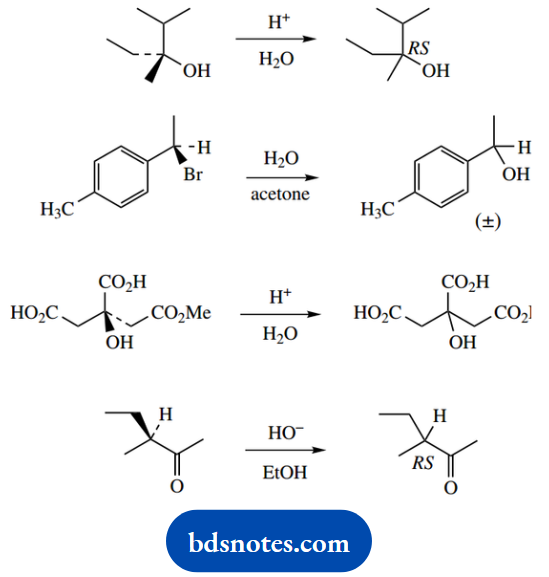
Answer:
Whilst this may appear a problem in stereochemistry, it is actually mechanism-based and depends upon the nature of the reaction intermediates. The loss of optical activity is because we form a racemic product, i.e. a mixture of enantiomers, or produce a compound that is no longer chiral.
1. The first reaction converts a single enantiomer into a racemic product; it is a racemization. The logical way of achieving any racemization is to transform the starting material into a planar intermediate, which can then be attacked equally from either side.
Such an intermediate might be a carbocation, as in this example. Although this might appear a non-reaction, it is actually SN1 chemistry where the leaving group and nucleophile are the same, namely water.
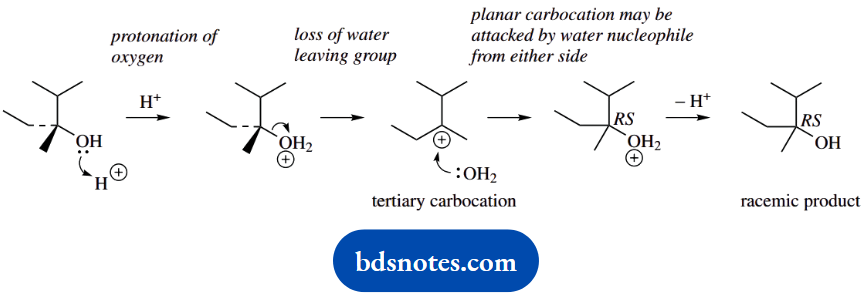
The reaction occurs because a favourable tertiary carbocation is generated. Since the carbocation also has three different substituents, the nucleophilic attack of water forms a chiral centre, and thus enantiomeric products.
2. This reaction differs little from (a), though the leaving group and nucleophile are no longer the same; it is a standard SN1 reaction. We show the alternative way of depicting a racemic product: RS or (±)- may be used for racemates.
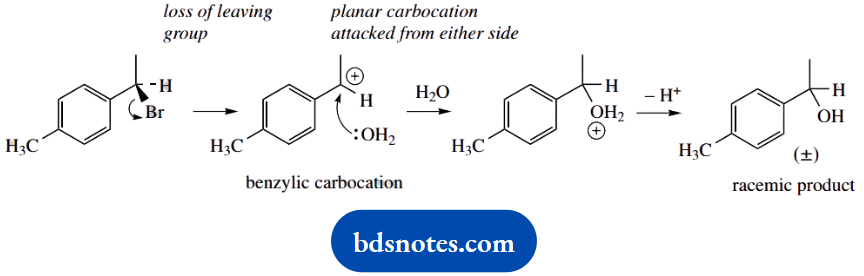
The intermediate carbocation initially appears secondary, but it is also benzylic and, therefore, resonance stabilized by the aromatic ring.
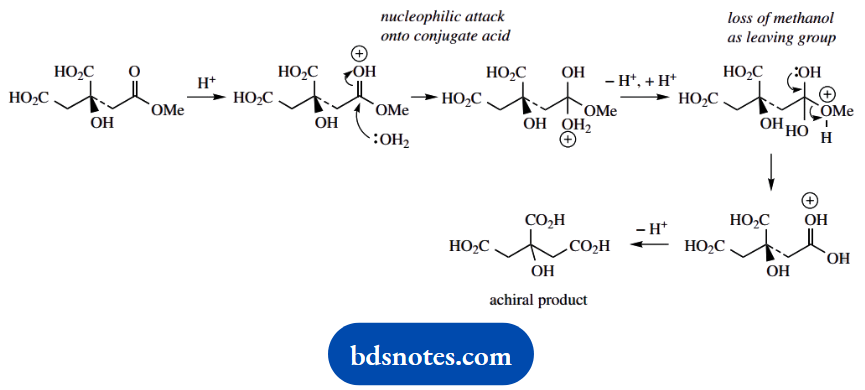
3. There is no need even to consider the chiral centre in this problem; hydrolysis of the ester grouping to an acid generates a molecule that is achiral because it has two equivalent substituents. The answer is simply acid-catalysed ester hydrolysis.
4. This is a base-catalysed racemization that is dependent upon the formation of an enolate anion. The resonance form shown first is planar, so deprotonation can occur from either face, producing the racemic ketone.
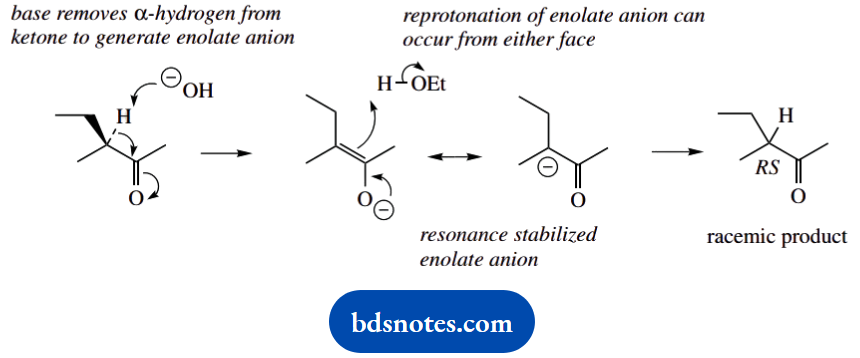
This is an equilibrium reaction, and it raises a couple of points. First, there are two a-positions in the ketone, so what about the COCH3-derived enolate anion? The answer is that it is formed, but since the CH3 group is not chiral, proton removal and deprotonation have no consequence.
Racemization only occurs when we have a chiral a-carbon carrying a hydrogen substituent. Second, the enolate anion resonance structure with charge on carbon is not planar, but roughly tetrahedral. If we reprotonate this, it must occur from just one side.
Yes, but both enantiomeric forms of the carbanion will be produced, so we shall still get the racemic mixture.
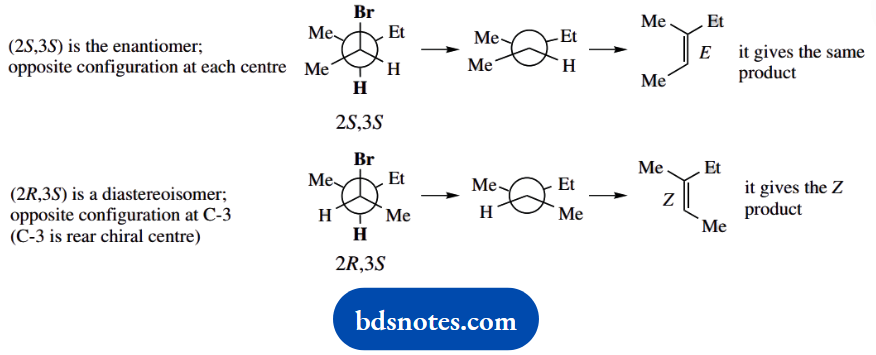
Question 6. Treatment of (2R,3R)-2-bromo-3-methylpentane with sodium ethoxide in ethanol gives (E)-3-methylpent-2-ene as the major product. What would be the major product from the similar base treatment of (25,35)- and (2R,35)-2- bromo-3-methyl pentane? Hint: Newman projections will be useful here.
Answer:
(2R,3R)-2-bromo-3-methylpentane \(\quad\underrightarrow{\mathrm{NaOEt}}\quad\) (E)-3-methylpent-2-ene
Draw out the structures with stereochemistry. Since it is not always easy to visualize a stereoisomer with the required configuration, draw one tentative arrangement, work out the chirality, and then adjust if necessary. It is always easiest to put the group of lowest priority (usually H) away from you, i.e. dotted bond.
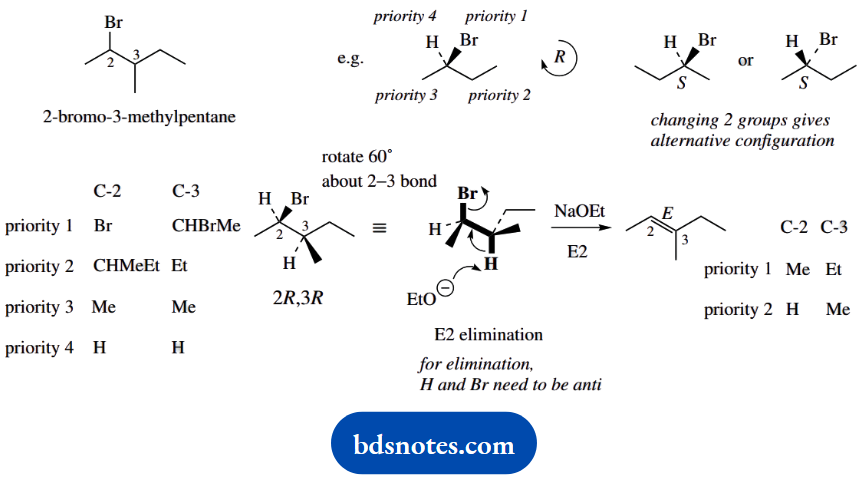
Draw the starting material as a Newman projection
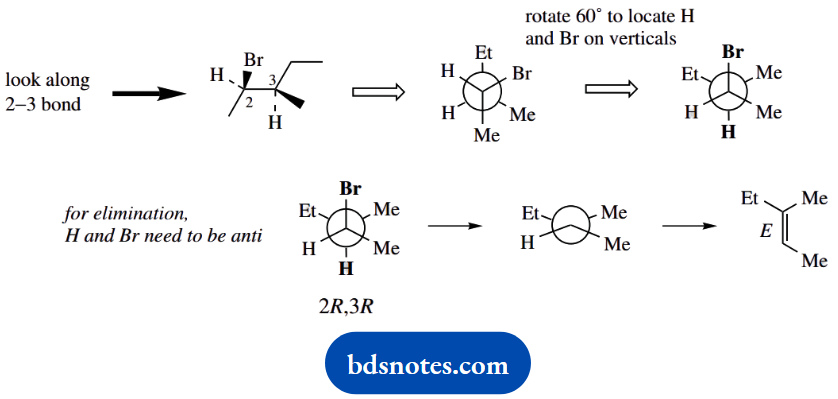
The major products from similar base treatment of (2S,3S)- and (2R,3R)-2-bromo-3-methylpentane can be deduced as below
Question 7. In the following four transformations, give detailed mechanisms for the steps indicated.
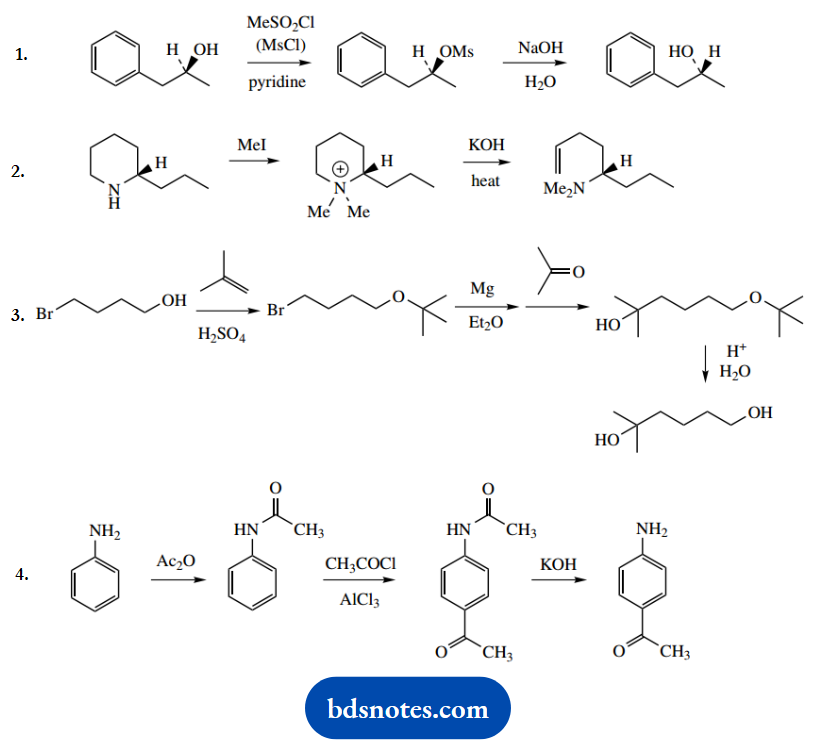
For each sequence, explain why the intermediary steps are advantageous in obtaining the product shown.
Answer:
1. This achieves a change in stereochemistry and, therefore, requires an SN2 reaction. An alternative SN1 mechanism is likely to lead to a racemic product, so may be discounted.
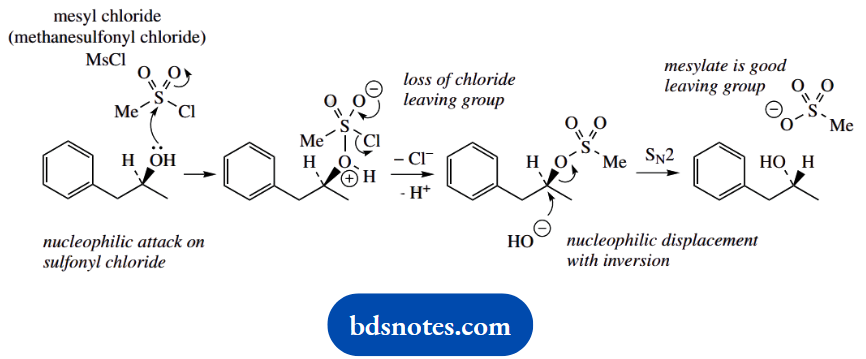
The purpose of the first reaction with methane sulfonyl chloride (mesyl chloride) is to form a mesylate ester, a derivative with a better-leaving group than hydroxide.
Then, the SN2 reaction can proceed more favourably. Esterification with mesyl chloride is mechanistically analogous to esterification with acetyl chloride.
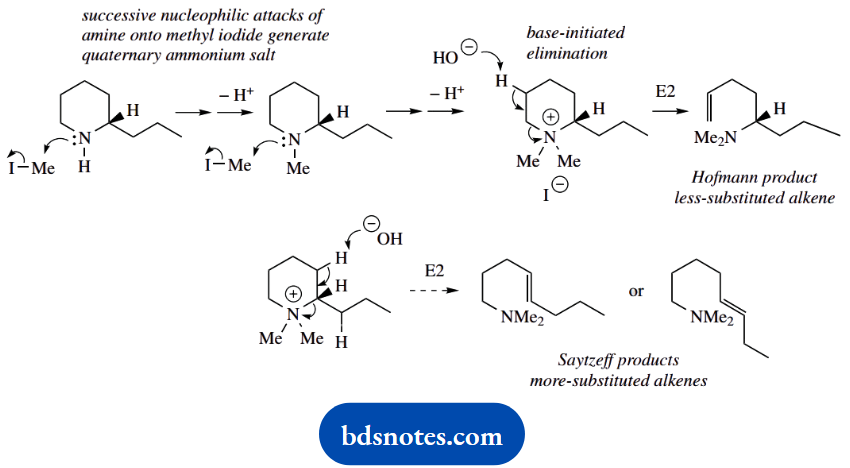
2. The extra steps here are also to improve the leaving group characteristics. We require a base-initiated E2 elimination reaction, and by forming a quaternary ammonium salt we produce a better leaving group. Since we are starting with a cyclic amine, the amine leaving group remains as part of the product.
In this particular example, we also need to consider the direction of elimination. Alternatives are possible, as shown. We actually obtain the Hofmann-type less-substituted alkene as a major product, since the elimination involves a relatively large leaving group, the quaternary ammonium cation.
3. The overall reaction is the generation of a Grignard reagent from the alkyl bromide, followed by nucleophilic addition of this carbanion equivalent onto the ketone acetone.
However, there is also a hydroxyl group in the starting material, and we must use a protecting group to ensure this does not react with and destroy the OH-sensitive Grignard agent. It is protected here through the formation of an ether.
This is an electrophilic addition reaction, using a tertbutyl carbocation formed by the protonation of an alkene.
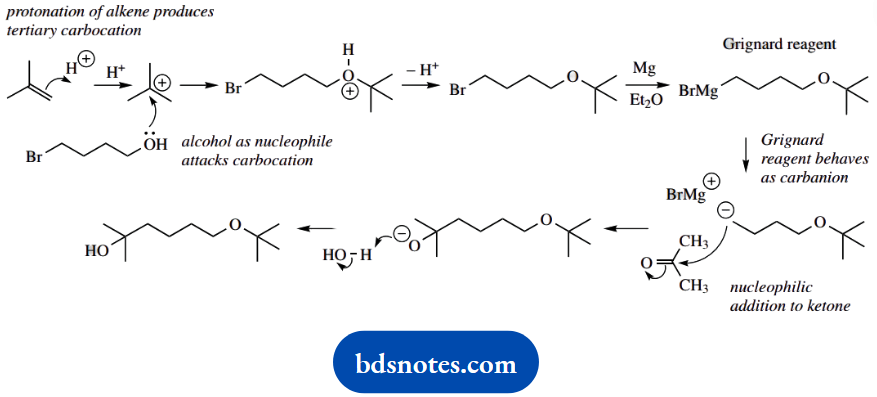
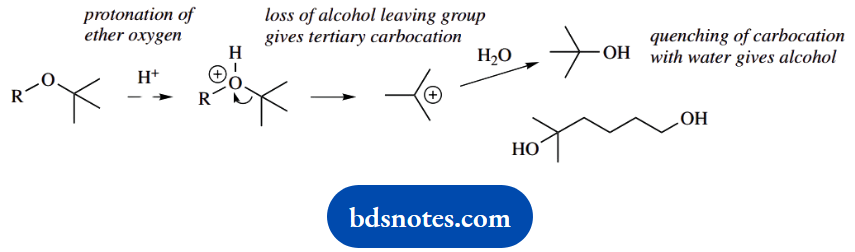
With the alcohol protected, preparation of the Grignard reagent can proceed, and this can then react with the ketone carbonyl in a nucleophilic addition.
The protecting group can then be removed by treatment with acid, to restore the hydroxyl function. This also involves a tertiary carbocation that is subsequently quenched with water.
4. In this sequence of reactions, we achieve Friedel-Crafts acylation of the aromatic amine. This could not be performed directly. Although an amino group is ortho and para directing and should give mainly the para product because steric considerations favour this over the ortho product, this is because the amine group is an electron donor.
As such, this means it will preferentially complex with the Friedel-Crafts reactive intermediate, the acylium cation, preventing further reaction.
Converting the amine to an amide stops this behaviour. Amides are non-basic and poor electron donors; this is because the nitrogen lone pair is utilized with the carbonyl group to provide resonance stabilization. The N-acetyl is thus a protecting group for the amine.
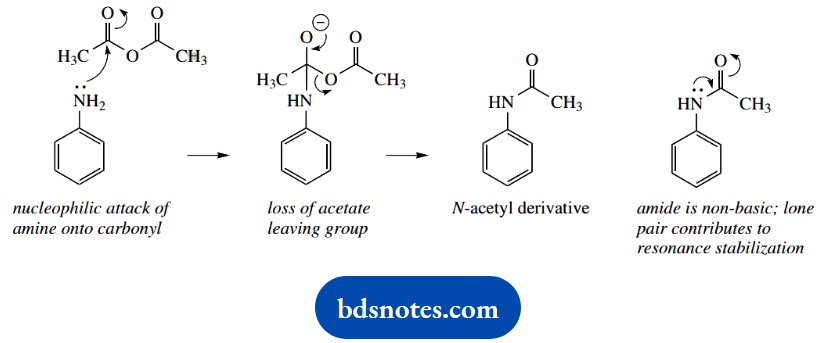
The sequence requires first simple N-acetylation with acetic anhydride. This product is then the substrate for the Friedel-Crafts reaction. The acylium cation is generated from acetyl chloride and aluminium chloride.
You may perhaps remember that the acylium cation is the acylating species or, alternatively, you can deduce that the acyl chloride interacts with the Lewis acid AlCl3, and is most likely to give AlCl4–. The acylium cation then emerges as the other part. This is resonance stabilized, as shown.
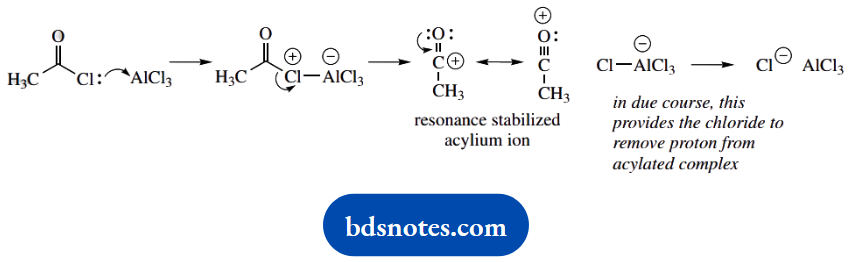
Now the electrophilic attack of the aromatic system onto the acylium cation. The product is para-substituted, so go for that site of attack. We then need the removal of a proton, and chloride is the obvious base, but since this is complexed as AlCl4–, we need the latter to decompose to chloride and AlCl3.
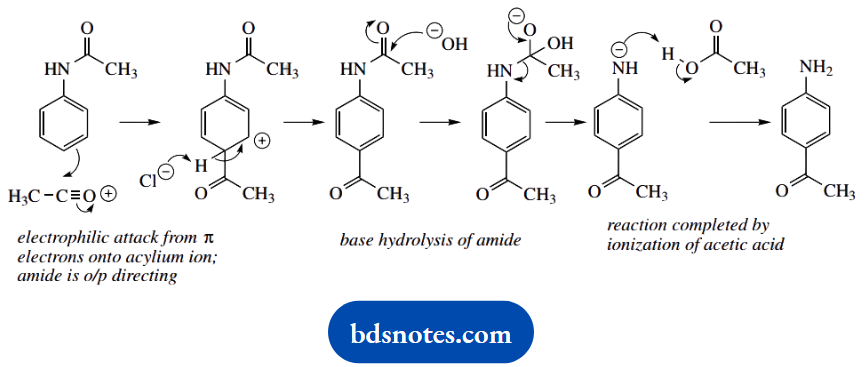
The sequence is completed by base hydrolysis of the amide and removal of the protecting group. This is much the same as an ester hydrolysis, and needs to include as the last stage the ionisation of acetic acid; since RNH– is a poor leaving group, it is this ionisation that allows the reaction to proceed.
Question 8.
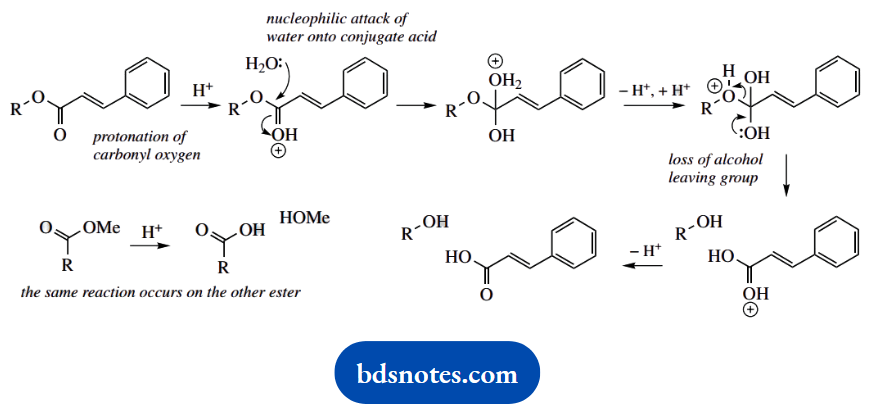
1. The natural alkaloid diester cinnamoyl cocaine can be converted into the local anaesthetic and much-abused drug cocaine by the sequence shown.
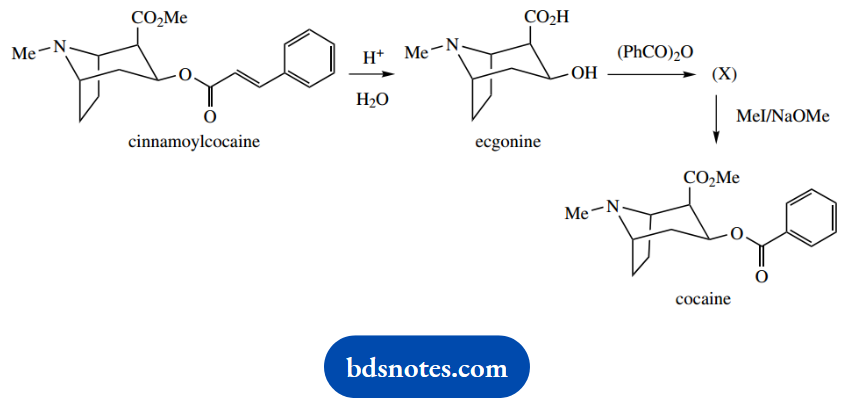
Give a mechanistic interpretation for each of the reactions, and give a structure for the intermediate (X).
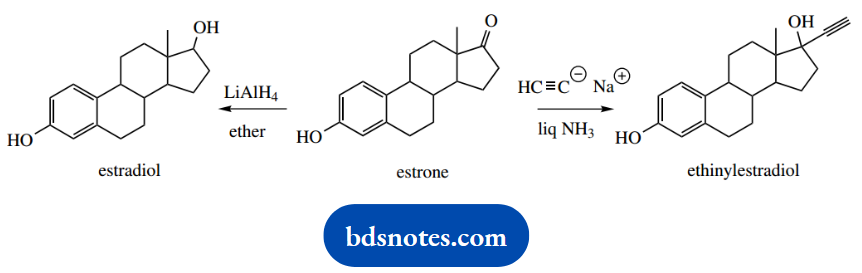
2. Give mechanisms for conversion of estrone into the oestrogen drugs estradiol and ethinylestradiol.
Answer:
Do not be put off by the structures; instead, you should be looking at the functional groups and the changes involved. The reactions are relatively simple.
1. You are told cinnamoylcocaine is a diester. The first reaction is merely acid-catalysed hydrolysis of these two groups. Abbreviate groups as necessary; and provided you make things clear, you will not be required to give the same mechanism for the second group.
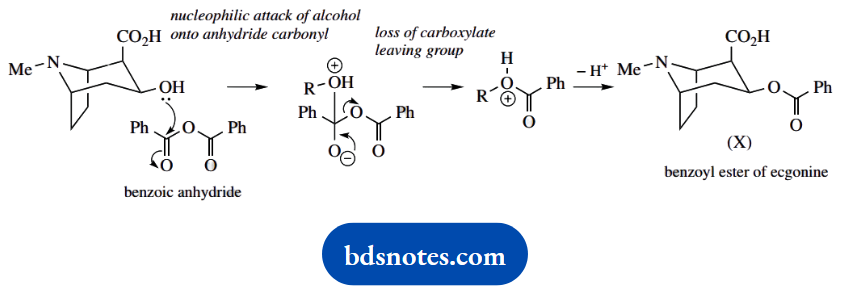
Ecgonine now reacts with (PhCO)2O to give (X). This reagent is benzoyl anhydride, an analogue of acetic anhydride. The alcohol group reacts, displacing benzoate as the leaving group and forming (X), the benzoyl ester of ecgonine.
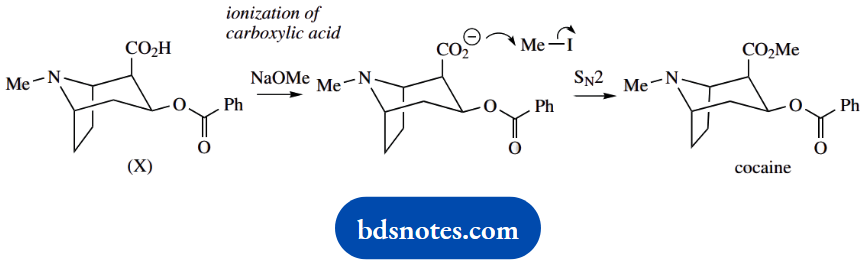
The next step is an unusual approach to making an ester via an SN2 reaction. We usually make esters by the reaction of alcohols with activated carboxylic acid derivatives like acyl chlorides, or anhydrides as we have just seen.
We do not particularly want to have to convert the carboxylic acid (X) into a more reactive derivative, and we can not just react with methanol and acid because that would be likely to transesterify the existing ester.
The clue is in the use of base; this will form the carboxylate anion. The reaction then becomes a base-initiated SN2 reaction with MeI; the carboxylate anion is the nucleophile.
2. This problem provides two examples of nucleophilic addition to a ketone; the ketone is the only reacting functional group in this complex steroidal structure.
Estradiol is formed by lithium aluminium hydride reduction of the ketone. We can formulate this simply as hydride acting as the nucleophile, though hydride delivery by LAH is strictly more complex than this. Unless you are specifically asked for details, treat LAH as a source of hydride ions.
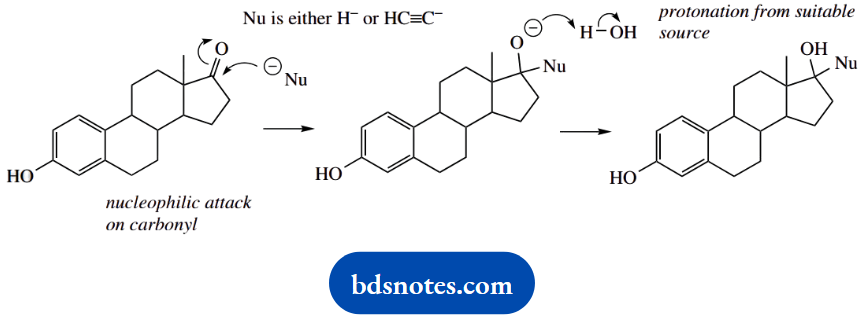
Similarly, acetylide is the nucleophile in the synthesis of ethinylestradiol. In each case, the reaction is finished off by protonation from a suitable source, for example, water.
Question 9.
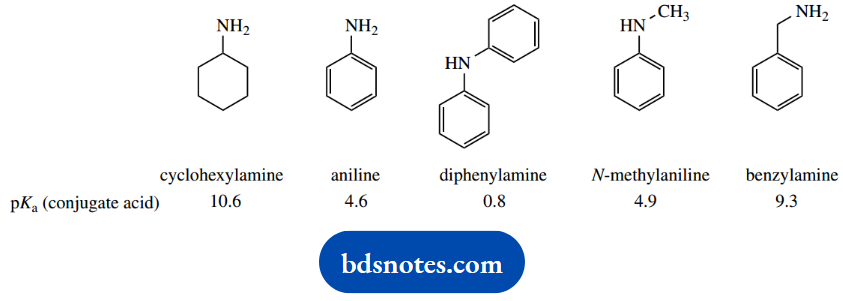
1. Provide a reasoned explanation for the relative pKa values determined for the five amines shown.
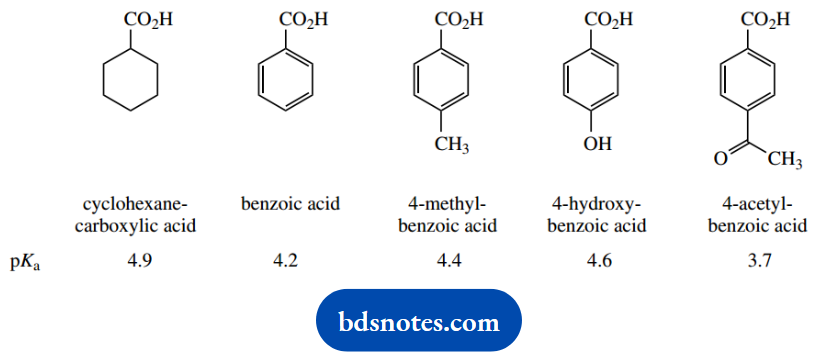
2. Comment on the relative pKa values determined for the following five carboxylic acids.
Answer:
The relative acidities or basicities of a range of similar organic compounds can usually be rationalized quite readily by considering the implications of electron-withdrawing and electron-donating substituents in the molecule.
Consideration of inductive and resonance effects is usually adequate, and although other effects, for example, electronegativity or hybridization, may come into play, they should not form the first line of reasoning.
Remember also that, where inductive and resonance effects are in opposition, the resonance effect is usually of greater magnitude and will prevail.
One of the compounds shown should be used as the point of reference from which comparisons can be made. There is no way we would encourage memorising of pKa values, but two easily remembered figures can be valuable for comparisons.
These are pKa around 5 for a typical aliphatic carboxylic acid, and pKa around 10 for a typical aliphatic amine. These then allow us to consider whether the compound in question is more acidic, more basic, etc.
For acids, the predominant consideration should be the stabilization or destabilization of the conjugate base. The stability of negatively charged conjugate bases will tend to be enhanced by electron-withdrawing substituents that can help delocalize the charge.
Electron-donating substituents will do the opposite, and thus reduce the likelihood of conjugate base formation.
Conversely, we can reason that the formation of conjugate acids from bases will be favoured by electron-donating substituents and inhibited by electron-withdrawing groups.
However, the feature of bases is that they have a lone pair of electrons that are able to coordinate with a proton. Sometimes, this lone pair may feed into the molecule via a resonance effect, and this can stabilize the free base and inhibit conjugate acid formation.
With bases, therefore, we normally consider two approaches, either stabilization of the conjugate acid, which increases basicity, or stabilization of the free base, which decreases basicity.
Most substituents are easily recognized as potentially electron withdrawing or electron donating, for example, halogens, carbonyls, amines, and phenols. We also need to remember that phenyl groups provide an electron-withdrawing inductive effect (a consequence of sp² hybridization), whilst alkyl groups are weakly electron donating.
1. We have here a mix of aliphatic and aromatic amines, and two benchmark compounds, cyclohexylamine and aniline. Remember, of course, that the pKa values refer to the acidity of the conjugate acids, and not to the acidity of the amines themselves, which could be relevant in different circumstances.
Nevertheless, it is usually acceptable to talk of the pKa of aniline, when we should strictly say the pKa of the conjugate acid of aniline.
The pKa of cyclohexylamine is typical of an aliphatic amine. Aniline is a much weaker base (the smaller the pKa, the weaker the base) than cyclohexylamine.
This is a combination of an inductive effect and a resonance effect, which reinforce each other. The phenyl ring provides an electron-withdrawing inductive effect that destabilizes the conjugate acid. More important, though, is the resonance effect that stabilizes the uncharged amine.
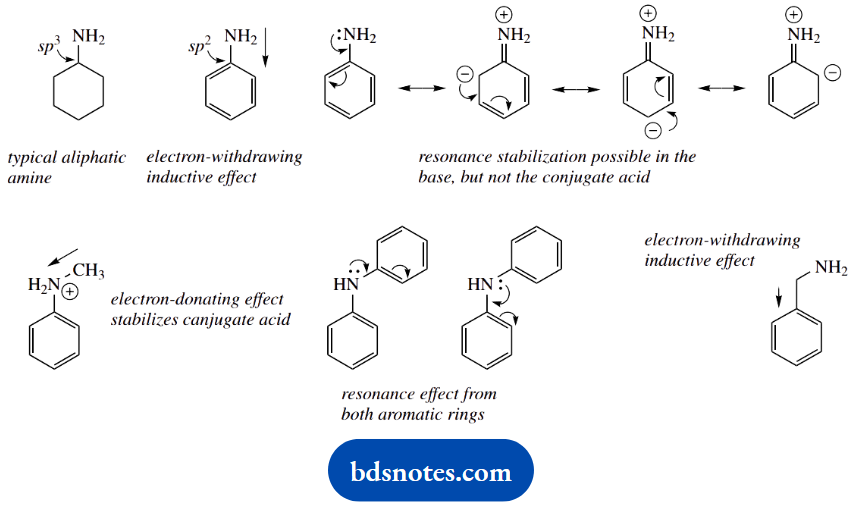
As we move to N-methylaniline, we see only a modest change in pKa. This is undoubtedly due to the electron-donating effect of the methyl group, and this would be expected to stabilise the conjugate acid, increasing the observed basicity.
There is a modest increase in basicity, but it is apparent that the resonance effect, as in aniline, is also paramount here, and this compound is also a weak base. However, diphenylamine (N-phenylaniline) is an extremely weak base; this can be ascribed to the resonance effect allowing electron delocalization into two rings.
On the other hand, benzylamine (pKa) is a much stronger base than aniline, somewhat weaker than cyclohexylamine. Benzylamine is merely an aliphatic amine, with an electron-withdrawing aromatic ring separated by one carbon from the nitrogen.
There is no chance of a resonance effect as in aniline, and the inductive effect is smaller than if the phenyl were directly bonded to nitrogen. The net result is a small lowering of charge on nitrogen with destabilization of the conjugate acid, and hence a lower basicity than cyclohexylamine.
2. The reference point here is the acidity of cyclohexanecarboxylic acid (pKa), a typical aliphatic acid. Benzoic acid is a stronger acid (the smaller the pKa, the stronger the acid), and this can be ascribed to the hybridization-derived electron-withdrawing inductive effect of the phenyl ring.
This helps to delocalize the charge on the conjugate acid and is a stabilizing factor. Inductive effects in the opposite direction destabilize the conjugate base, and we see a weakening of acidic strength. This is why 4-methyl benzoic acid is not as strong an acid as benzoic acid, though the inductive effect of the methyl is small.
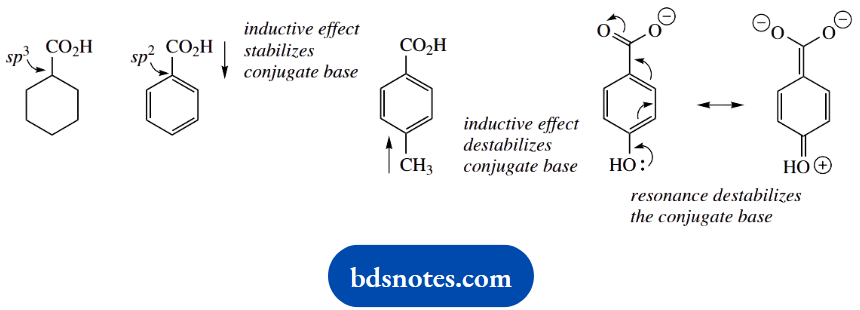
The inductive effect in 4-hydroxybenzoic acid is electron withdrawing, but this is a modest effect compared with the electron-donating resonance effect.
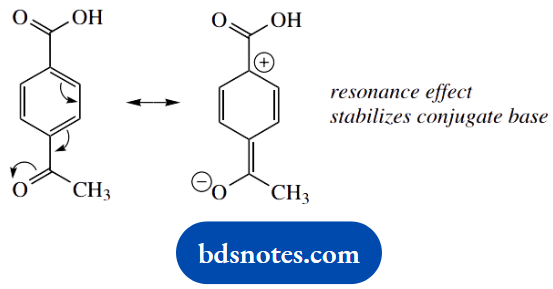
This significantly destabilizes the conjugate base, and 4-hydroxybenzoic acid (pKa) is less acidic than benzoic acid. The last example, 4-acetylbenzoic acid, is the most acidic of the group, and this is primarily the result of an electron-withdrawing resonance effect, though there is also a favourable inductive effect.
Question 10.
- Ignoring possible dilution effects, calculate the weight of sodium acetate (MW 82.03) that should be added to 1 litre of an aqueous solution containing 3.85 g of acetic acid (MW 60.05) to produce a solution of pH 4.80. The pKa of acetic acid is 4.75.
- Calculate the pH of the solution following the addition of 1 ml of 1.0 M aqueous solution of sodium hydroxide.
- Calculate the pH of a solution containing 6.50 g of dimethylamine (MW 45; pKa 10.8) in 600 ml distilled water.
- Calculate the percentage ionized of an acidic drug (pKa 5.70) dissolved in an aqueous buffer solution of pH 4.90.
Answer:
Calculations of this type require the application of standard equations that should be committed to memory unless you feel up to derive them from first principles.
The first is the Henderson–Hasselbalch equation:
pH = \(\mathrm{p} K_{\mathrm{a}}+\log \frac{\text { [base] }}{\text { [acid] }}\)
The second pair of equations relates pH to pKa for weak acids and weak bases, though they are actually variants of the Henderson–Hasselbalch equation:
for acids \(\mathrm{pH}=\frac{1}{2} \mathrm{p} K_{\mathrm{a}}-\frac{1}{2} \log [\mathrm{HA}]\)
for bases \(\mathrm{pH}=\frac{1}{2} \mathrm{p} K_{\mathrm{w}}+\frac{1}{2} \mathrm{p} K_{\mathrm{a}}+\frac{1}{2} \log [\mathrm{B}]\)
It follows that we use the Henderson–Hasselbalch equation when both acid and base concentrations are applicable.
With these types of calculations, two general points should be made. Make sure your result makes sense. For example, do not forget the painfully obvious fact that bases should yield pH values >7, and acids <7.
The second point relates to the number of significant figures you present in your result.
Do not write down every figure your calculator provides, but round up to an appropriate level, for which the question will give guidelines. If a pH or pK is given to two decimal places, do likewise in your answers.
This is the preparation of a buffer solution, using acetic acid (acid) and sodium acetate (base). Putting values into the Henderson–Hasselbalch equation.
pH = \(\mathrm{p} K_{\mathrm{a}}+\log \frac{\text { [base }]}{\text { [acid] }}\)
we get \(4.80=4.75+\log \frac{\text { [base] }}{\text { [acid] }}\)
so that \(\log \frac{[\text { base }]}{\text { [acid] }}=4.80-4.75=0.05\)
and \(\frac{[\text { base ] }}{[\text { acid] }}=10^{0.05}=1.12\)
The [acid] is known; it is \(\left[\text { acid] }=\frac{3.85}{60.05}=0.0641 \mathrm{~m}\right. \text {. }\)
Therefore, [base] = 1.12× 0.0641 = 0.0718 M.
Amount of sodium acetate required = 0.0718 × 82.03 = 5 .89 g.
2. We again use the Henderson–Hasselbalch equation; we are adding an extra base, which in turn removes some acid, so the [base]/[acid] ratio will alter accordingly.
The amount of base added is 1 ml of 0.1 M NaOH = 0.001 M; this will also remove 0.001 M of acid
pH = \(\mathrm{p} K_{\mathrm{a}}+\log \frac{[\text { base }]}{[\text { acid }]}\)
so pH = \(4.75+\log \frac{0.0718+0.001}{0.0641-0.001}\)
= \(4.75+\log \frac{0.0728}{0.0631}\)
pH = \(4.75+\log 1.15=4.75+0.06=4.81\)
New pH=4.81, a change of just 0.01 pH units
3. The pH of a solution of a weak base may be determined from the equation:
pH = \(\frac{1}{2} \mathrm{p} K_{\mathrm{w}}+\frac{1}{2} \mathrm{p} K_{\mathrm{a}}+\frac{1}{2} \log [\mathrm{B}]\)
[base] = \(\frac{6.50}{45} \times \frac{1000}{600}=0.241 \mathrm{~m}\)
pH = \(\frac{1}{2}\)(14) + \(\frac{1}{2}\)(10.8) + \(\frac{1}{2}\) log(0.241) = 7 + 5.4 + \(\frac{1}{2}\)(−0.618)
pH = 7 + 5.4 − 0.31 = 12.09
pH of solution = 12 .09
4. This again requires the use of the Henderson–Hasselbalch equation; we have a partially ionized acid, so the ionized fraction represents [base] and the non-ionized fraction represents [acid].
Let us call the fraction ionized I; then the fraction non-ionized is 1 − I.
pH = \(\mathrm{p} K_{\mathrm{a}}+\log \frac{\text { [base] }}{\text { [acid] }}\)
4.90 =5.70 + v log \(\frac{I}{1-I}\)
so that \(\log \frac{I}{1-I}=4.90-5.70=-0.80\)
and \(\frac{I}{1-1}=10^{-0.80}=0.158\)
from this I = \(0.158-0.158 I\)
and I = \(\frac{0.158}{1.158}=0.136\)
This means the percentage ionized is 13.6%.
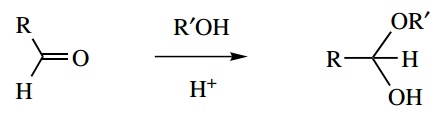
Question 11. Give a mechanism for the acid-catalysed formation of a hemiacetal:
1. Give a mechanism for the acid-catalysed formation of a hemiacetal:
The heart drug digoxin contains in its structure three molecules of digitoxose as the trisaccharide (digitoxose)3:
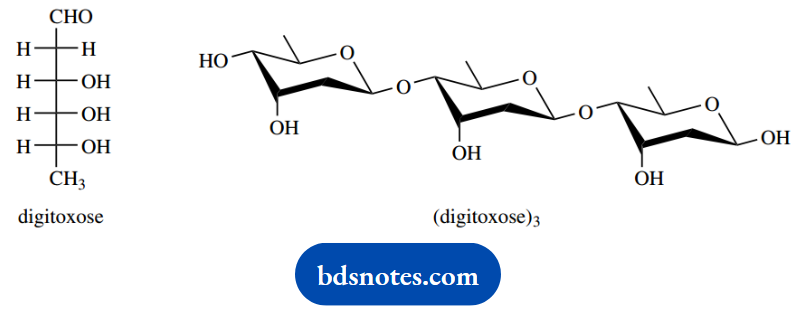
2. Explain how a digitoxose molecule is capable of forming a six-membered cyclic form as seen in the trisaccharide with the stereochemistry shown.
3. Indicate what information is conveyed by the four underlined parts of the nomenclature (+)-β-D- digitoxopyranose for the digitoxose units in the trisaccharide.
4. Give a mechanism to account for the acid-catalysed formation of the dimer (digitoxose)2 from two molecules of digitoxose (cyclic form). It may be assumed that no other functional groups in the molecules interfere with this reaction.
5. Explain mechanistically the following two observations:
- Digitoxose reacts with HCN to give a mixture of two diastereoisomeric products;
- Sodium borohydride reduction of digitoxose gives a single product.
Answer:
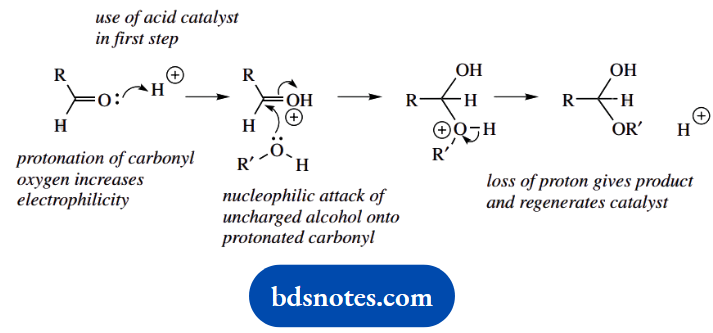
1. This multi-part question starts with a standard general mechanism. It is the lead-in to other questions based on this mechanism. This is a textbook reaction; but, rather than remembering it in detail, work it out.
The acid catalyst is used in the very first step to produce the conjugate acid of the carbonyl compound, increasing its electrophilicity. The standard nucleophilic addition is followed by proton loss, giving the product and regenerating the catalyst.
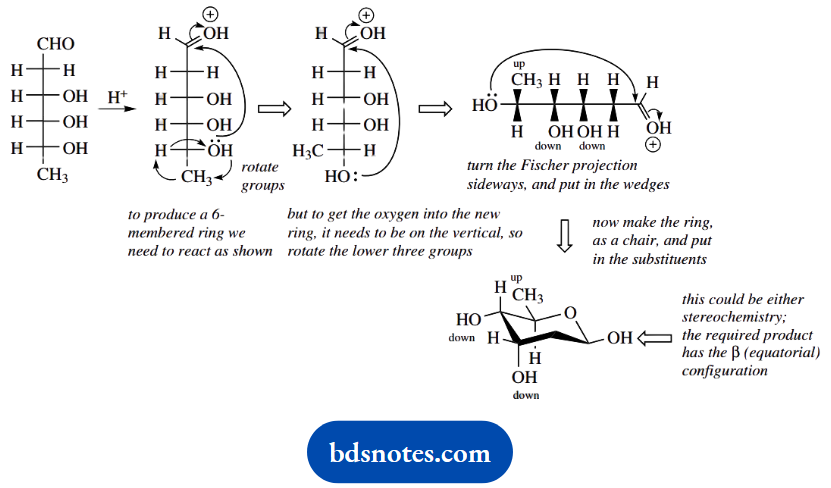
2. The sugar digitoxose is shown in the form of a Fischer projection. The cyclic form is merely a hemiacetal; we need to react with one of the hydroxyls with the carbonyl to form a six-membered ring.
We must also manipulate the Fischer projection to deduce the stereochemistry in the product, which should end up the same as shown in the question.
Note the approach here: deduce the stereochemistry rather than trying to obtain the stereochemistry shown. We are only asked for a single digitoxose molecule, despite the structure shown being the trimer.
3. The underlined parts in the name (+)-β-D-digitoxopyranose convey the following information:
(+) means the compound is in one enantiomeric form and is dextrorotatory; P defines the configuration at the new chiral centre formed during hemiacetal formation (the anomeric centre) and it has the OH group equatorial; D conveys the configuration at the highest numbered chiral centre (C-5); pyranose means the hemiacetal ring is a six-membered oxygen heterocycle.
4. The mechanism for the formation of the dimer (digitoxose)2 from two molecules of cyclic digitoxose is the transformation of a hemiacetal into an acetal.
We are told that no other functional groups interfere in the reaction, so let us miss them out of the structures.
It is a textbook reaction, the continuation of part (a), but on specified molecules. We need to join position 1 of one molecule to position 4 of a second, and the reaction is acid-catalysed.
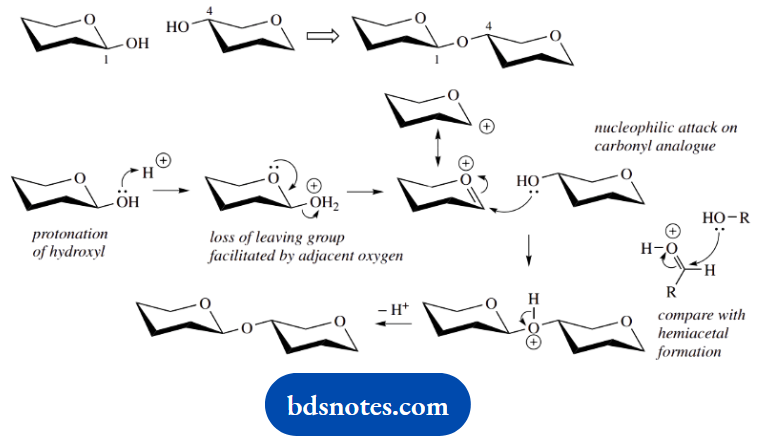
The reaction starts off with a protonation – use the catalyst. Resist the urge to protonate the 4-hydroxyl, but go for the one at a position that has the added functionality of the hemiacetal linkage. It is going to be the more reactive one.
Protonation is followed by loss of water as the leaving group. The intermediate oxonium cation shown is actually a resonance form of the simpler carbocation; now you can see the role of the adjacent oxygen. The reaction is completed by attack of the nucleophile, the 4-hydroxyl of another molecule.
This is not special but is merely another version of the hemiacetal synthesis done in part (a).
5. This looks more complicated than it is. HCN reacts with an aldehyde to give a cyanohydrin through a nucleophilic attack of cyanide; NaBH4 reacts to give alcohol through a nucleophilic attack of hydride (it is not actually hydride that attacks, but we can formulate it as such).
Furthermore, an attack on the planar carbonyl group may be from either face. Now just follow that through.
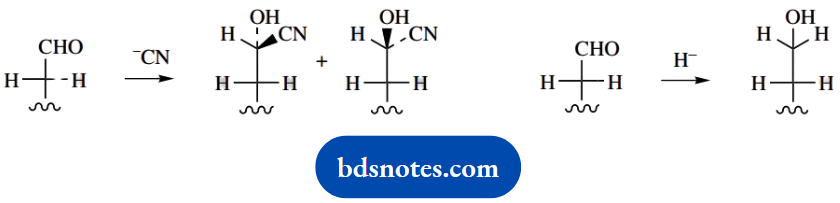
It can be seen that the addition of cyanide creates a new chiral centre, whereas the addition of hydride does not. Therefore, cyanide reacts to give two epimeric products.
We already have chirality in the rest of the digitoxose chain, so the cyanide reaction must yield two diastereoisomers. On the other hand, borohydride reduction gives just a single product.
Problem 12.
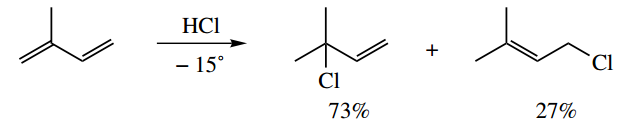
1. Explain why the addition of HCl to the conjugated diene at low temperature gives two main products, in the proportions shown.
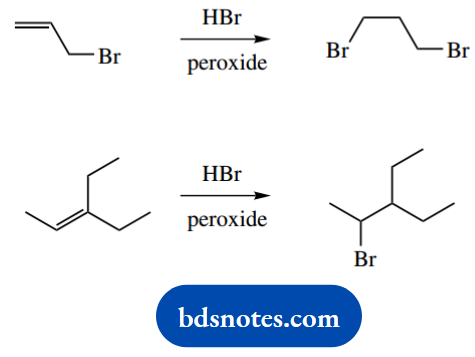
2. Suggest mechanisms for the following two reactions:
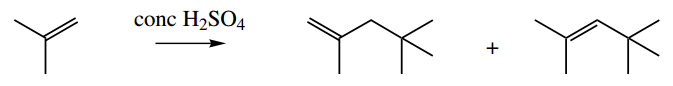
3. Give a mechanism to explain the formation of two products in the following reaction:
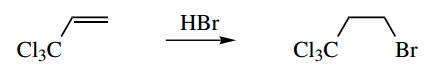
4. Explain why HBr adds to the trichlorinated alkene with anti-Markovnikov regiochemistry.
Answer:
We have here a mixture of electrophilic and radical addition reactions to alkenes. Remember the guidelines that radical reactions are characterized by the inclusion of radical initiators, such as light or peroxides.
In the absence of such initiators, consider only the alternative electrophilic mechanisms.
1. Electrophilic addition to conjugated dienes is complicated by the concepts of kinetic and thermodynamic control. Briefly, you should think of kinetic control being related to the stability of the intermediate carbocations, whereas thermodynamic control is dependent on the stability of the products.
Here, we have low-temperature conditions, so we need to consider the protonation of the alkene, and the relative stability of the carbocations formed. Draw the mechanisms for the two possible scenarios where we protonate either end of one double bond.
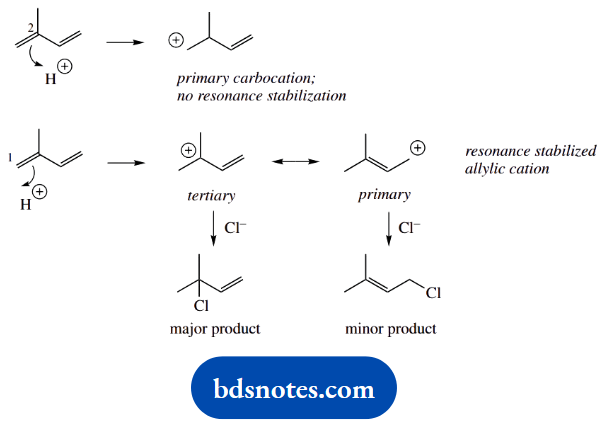
Protonation on C-2 gives an unfavourable primary carbocation. On the other hand, protonation on C-1 gives a favourable resonance-stabilized allylic carbocation.
The two products are then formed by the capture of chloride in a ratio that reflects the relative contribution of the limiting structures for the allylic carbocation; one is tertiary and the other is primary.

You may think that is the end of the problem; but, since we have an unsymmetrical diene, it is also necessary to consider protonation of the other double bond. Protonation on C-4 also gives a favourable resonance-stabilized allylic carbocation, this time with primary and secondary limiting structures.
Protonation on C-3 gives an unfavourable primary carbocation with no resonance stabilization. Since the products formed are related to initial protonation at C-1, it is apparent that, despite the stability associated with an allylic cation, a tertiary limiting structure is formed in preference to that with a secondary limiting structure.
2. The inclusion of ‘peroxides’ indicates a radical reaction. Jot down the general sequence for radical reactions involving peroxides and HBr. This requires homolysis of a general peroxide, hydrogen abstraction from HBr, and generation of a bromine radical.
Remember to use the fish-hook curly arrows here, representing the movement of a single electron. You can use pairs of arrows if you prefer, or single ones as in a two-electron mechanism. Now we can attack the double bond, and the carbon attacked is the one that leads to the more stable radical, in this case, a secondary radical.
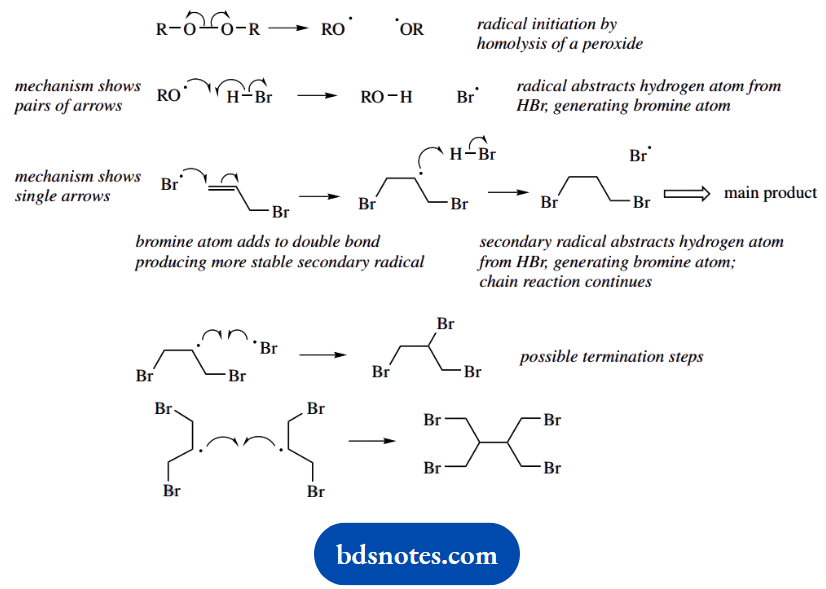
This new radical then abstracts hydrogen from another molecule of HBr, and the chain reaction can continue. You may wish to show typical chain termination steps.
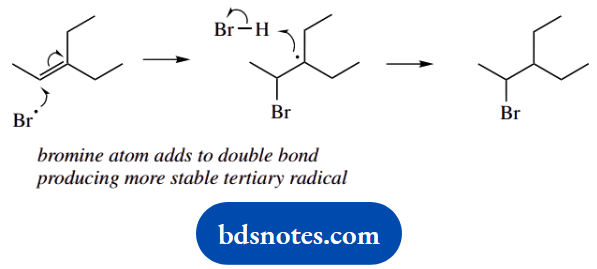
The main product in the second reaction is easily rationalized, in that the addition of a bromine atom to the alkene gives the favourable tertiary radical rather than the less attractive secondary radical.
3. The obvious change here is that two molecules of the alkene are combining under acidic conditions. We should consider an electrophilic reaction initiated by the protonation of the alkene to a carbocation.
Protonation is governed by the stability of the carbocation; this is straightforward here, in that we could get a favourable tertiary carbocation or a very unfavourable primary one.
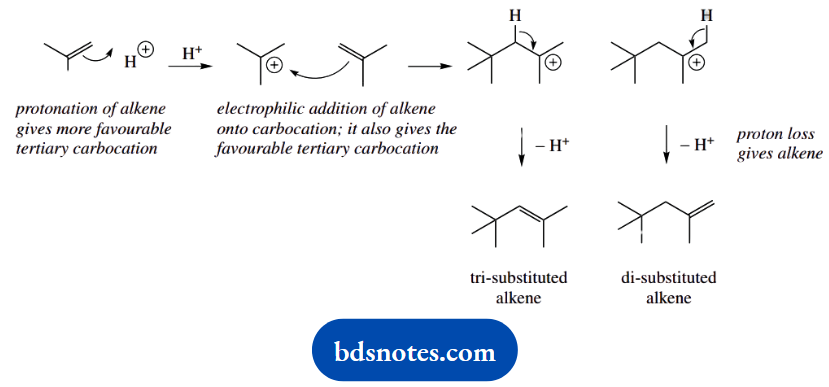
The next step is merely a repeat; instead of an electrophilic reaction with a proton, we have an electrophilic reaction with a carbocation. The reasoning is the same; we get the tertiary carbocation intermediate.
What then follows is the loss of a proton to give an alkene, and there are two possible products, depending upon which proton is released. Both products are formed, though we would expect the more-substituted alkene to predominate. We are not asked for, or given, any information about product ratios.
4. According to the reasoning we have so far used, protonation of the double bond in this alkene should give the secondary carbocation rather than the alternative primary carbocation.
Based on the information given in the question, it does not. We must get the primary carbocation, which is then quenched by bromide, i.e. anti-Markovnikov addition.
It is not necessary to remember what Markovnikov or anti-Markovnikov additions mean, we just need to consider the carbocation intermediates and their relative stability.
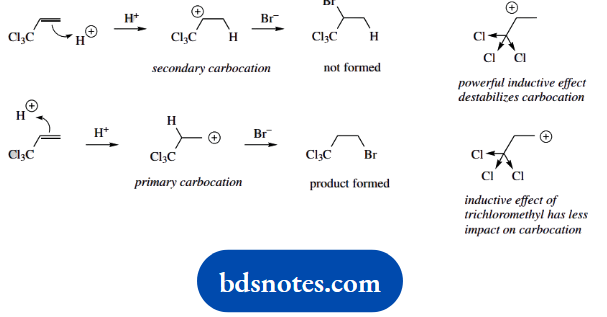
We can assume the trichloromethyl group is to blame. What do we know about this? It has three very electronegative chlorine atoms, so there will be a powerful inductive effect, withdrawing electrons. Now alkyl groups, which are weakly electron donating, help to stabilise a carbocation; and the more there are, the more stabilization we get.
At the other extreme, electron-withdrawing groups destabilize a carbocation. It appears from this reaction that the primary carbocation is more favourable than one adjacent to the powerful electron-withdrawing trichloromethyl group. Hence the observed regiochemistry.
Question 13. For the transformations shown, provide the following information:
- A detailed mechanism for the reaction;
- Resonance structures for the organic reactive intermediate formed during the reaction;
- The structure of an alternative product that might be formed in the reaction, with a brief explanation for its formation.
Answer:
1. This should be recognized as the acylation of an aromatic ring, and from the use of aluminium chloride it is a standard Friedel-Crafts acylation reaction.
You may remember that the acylium cation is the acylating species; alternatively, you can deduce that the acyl chloride interacts with the Lewis acid AlCl3, and is most likely to give AlCl4–. The acylium cation then emerges as the other part. This is resonance stabilized as shown.
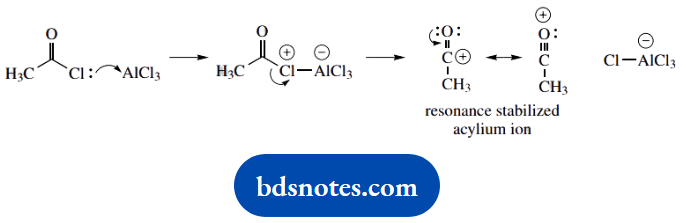
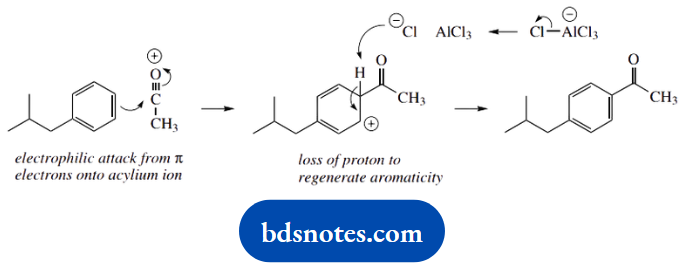
Now the electrophilic attack of the aromatic system onto the acylium cation. The product is para-substituted, so go for that site of attack.
We then need the removal of a proton, and chloride is the obvious base, but since this is complexed as AlCl4–, we need the latter to decompose to chloride and AlCl3.
We should also show resonance stabilization of the addition cation, delocalizing the charge around the ring; this helps with the alternative product question.
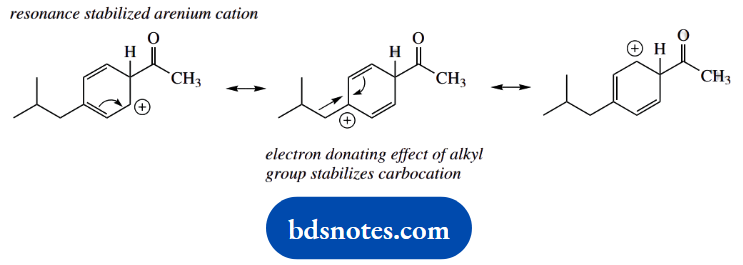
An alternative product might be the ortho-substituted analogue. Alkyl groups are ortho and para directors for further electrophilic substitution. This follows from the stabilization of one of the resonance forms by the electron-donating effect of an alkyl group.
This is seen in the para-substitution case, and extrapolation to ortho substitution shows a similar stabilization.
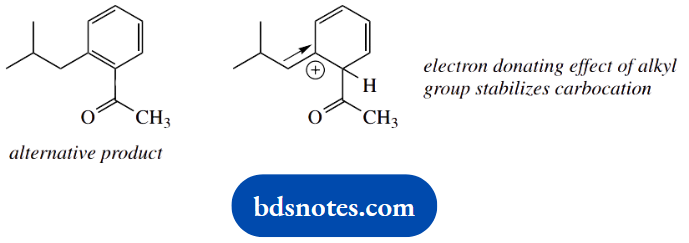
Though not asked for, it might be useful to indicate that, because we have relatively bulky substituents in this example, the para product will be formed in preference to the ortho product.
2. This is another electrophilic aromatic substitution, though a different reagent, and a different type of stabilization for intermediate carbocations. The active species in nitration is the nitronium ion.
We are introducing the nitro group NO2, so the involvement of NO+2 is logical. How it is formed is rather unusual, but aromatic nitration is such a routine transformation that it is worth remembering the basics.
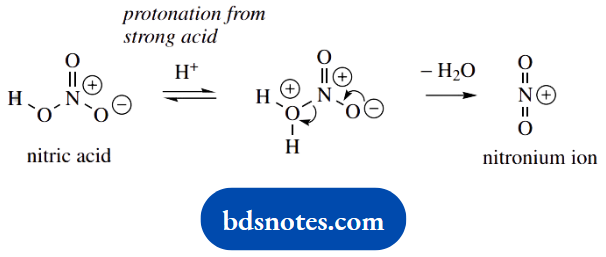
The electrophilic attack is much the same as in the Friedel-Crafts reaction above; note that we have to write the nitro group with charge separation in line drawings.
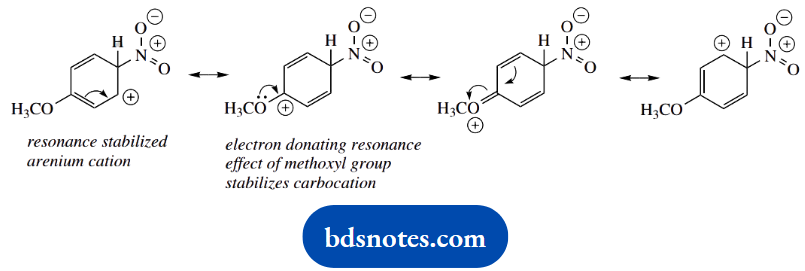
Again, we can draw resonance structures for the intermediate uranium cation. The extra stabilization this time comes from an electron-donating resonance effect involving the lone pair electrons of the methoxyl substituent.
This is substantially greater than the electron-donating effect from an alkyl group; phenols and their ethers react more readily towards electrophilic reagents than do alkyl benzenes.
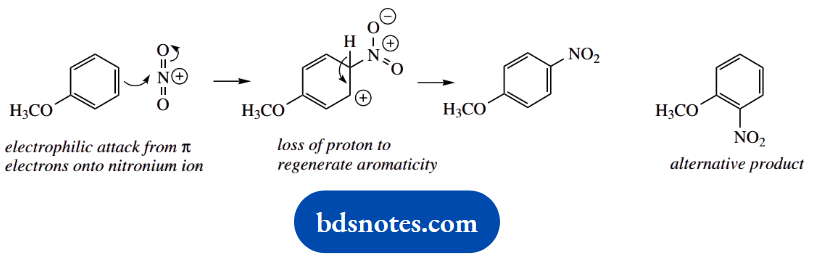
By the same reasoning as in part (a), an alternative product is likely to be the corresponding ortho derivative. The intermediate carbocation can be stabilized in the same way.
3. This is a radical reaction. The pointer is that the reaction also requires light energy. Accordingly, we need to formulate a sequence starting from the homolytic fission of a chlorine molecule to chlorine atoms, then a chain reaction stemming from these reactive species.
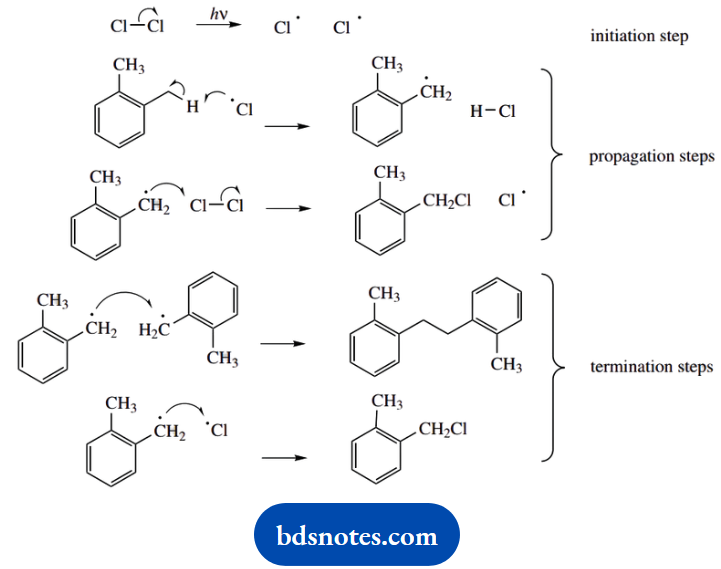
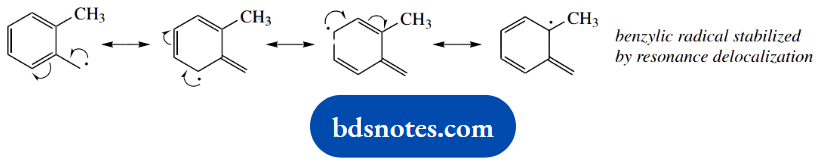
The propagation steps involve the removal of a hydrogen atom from one of the methyl substituents on the benzene ring. Abstraction from the methyl group is favourable because it generates a resonance-stabilized benzylic radical, in which the unpaired electron can be delocalized into the aromatic ring system.
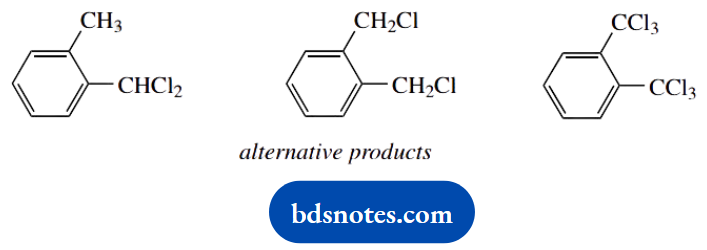
Two possible termination steps are shown, one of which produces the same product as the chain reaction. Recombination of two chlorine radicals is feasible but less likely. Either of the termination products could be suggested as an alternative product; but, in practice, what we are going to get is derivatives with more than one chlorine substituent.
It is difficult to control a radical process once the chain reaction is underway, so we usually get a mixture of products. Note particularly that further substitution is on the original or alternative methyl group, and not on the ring.
Again, the formation of a resonance-stabilized benzylic radical directs the reaction. Eventually, we might get the hexachloro compound shown.
4. This is a simple SN2 displacement of a bromide-leaving group by the phenolate anion. Phenols are relatively acidic, and treatment with base generates the resonance-stabilized conjugate base.
The phenoxide anion with a charge on the electronegative oxygen is the preferred nucleophile. The reaction is then completed by displacement of bromide to produce the ether.
Note the use of the reagent NaOH in the first step. Do not consider hydroxide as the nucleophile; the immediate consequence of mixing the reagents is the ionization of the phenol.
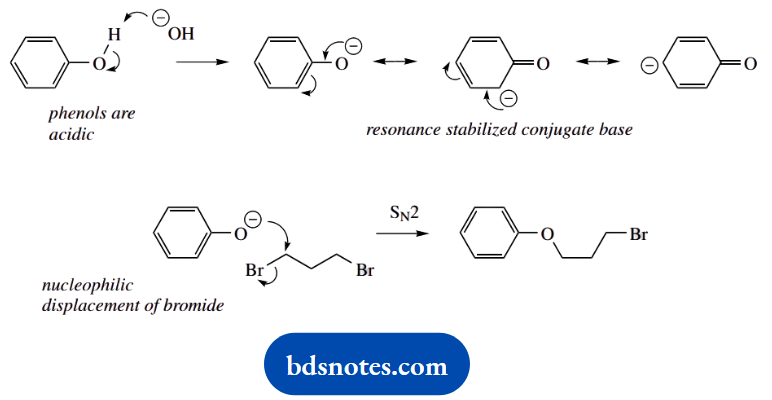
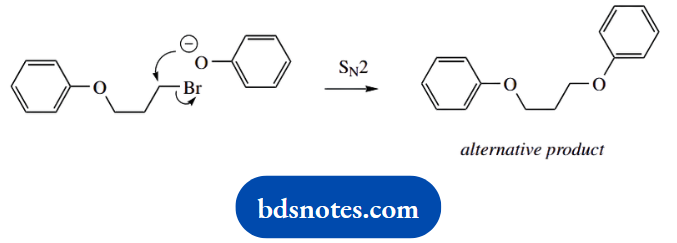
And what about an alternative product? There are two lines of thought, and the most obvious is that the reaction is repeated since we are using a dibromide as substrate.
Alternatively, we could consider one of the other resonance forms of the phenolate anion as a nucleophile. This would generate a C-alkylated phenol. In the majority of cases, C-alkylation is not observed, in that the preferred resonance structure has a charge on the electronegative oxygen.
Question 14. Provide mechanistic explanations for the following observations:
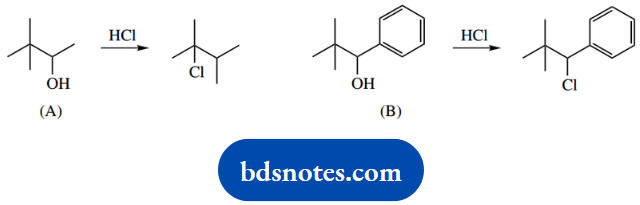
1. Treatment of 3,3-dimethylbutan-2-ol with hot aqueous HCl gives principally 2-chloro-2,3-dimethylbutane; similar treatment of 3-methylbutan-2-ol gives principally 2-chloro-2-methylbutane.
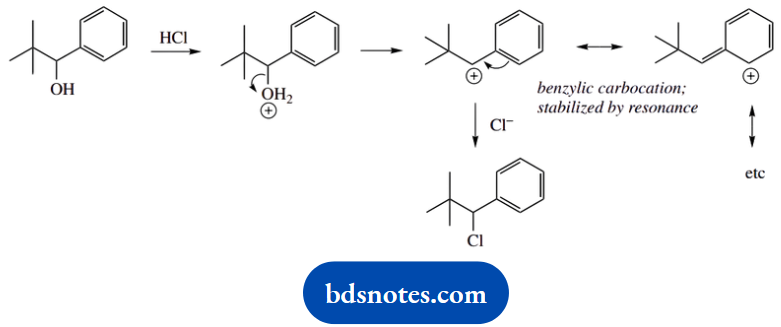
2. During nucleophilic substitution, the alcohol (A) undergoes rearrangement. Under the same conditions, the alcohol (B) does not suffer rearrangement.
3. Treatment of 3,3-dimethylbutan-2-ol with hot sulfuric acid gives three products: 3,3-dimethylbut-1-ene (3%), 2,3-dimethylbut-2-ene (61%), and 2,3-dimethylbut-1-ene (31%).
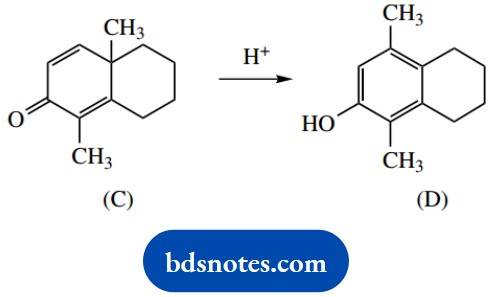
4. Under acidic conditions, the unsaturated ketone (C) rearranges to a phenol (D).
Answer:
These are all examples of carbocation rearrangements. The approach here is to spot the potential for carbocation formation, then see if an alternative carbocation might be formed by migration of an alkyl group, or perhaps hydride, to produce a more favourable carbocation.
Favourable carbocations are typically tertiary, allylic, or benzylic.
1. Draw out the structures, which are both secondary alcohols. Draw out the products and observe that, at least in the first example, we must have a rearrangement reaction because the carbon skeleton is different.
There is also something unusual about the second reaction; the nucleophile is not attached to the same carbon that had the potential to leave the group.
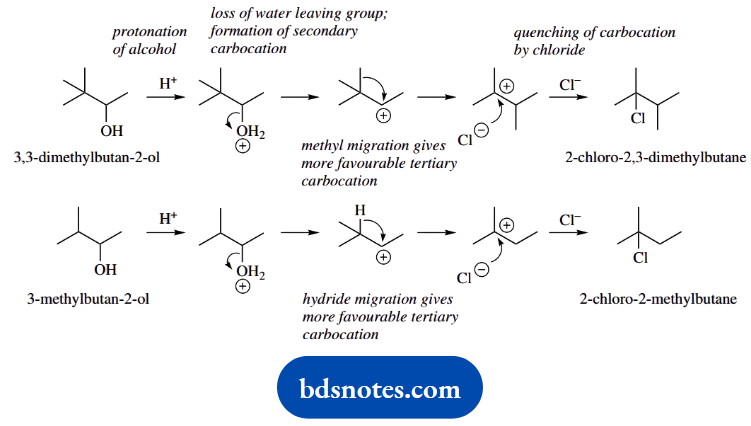
Since we can spot a rearrangement in the first example, formulate a mechanism for that first. The sequence is quite simple, protonation of the hydroxyl to make a better leaving group, loss of water and generation of a secondary carbocation.
Now we should be able to see that a methyl migration would produce a tertiary carbocation, a rearranged skeleton, and following through we get the required product.
Now do the same for the second compound, and keep thinking about rearrangements. We can formulate this in the same way once we see that a hydride migration would achieve the secondary to tertiary carbocation change.
2. The first example is actually the same as we have discussed in part (a), though here we are given the structure. What is different about the behaviour of the second compound? Draw out the same type of mechanism as far as the secondary carbocation.
Why doesn’t this secondary carbocation rearrange? Why doesn’t it want to become tertiary? There must be something extra that is stabilizing it so that there is no need for rearrangement. There is, and it is the aromatic ring.
This carbocation is benzylic and stabilized by resonance. There is no need for rearrangement to acquire improved stability; accordingly, chloride attacks at the same carbon the leaving group was attached to, an SN1 reaction.
3. The starting material is one of those from part (a), but because of the conditions, probably the high temperature, we are getting eliminations rather than substitutions. On drawing out the products, we can see two of them are produced as a result of rearrangements.
The first product is not rearranged and is a simple elimination. Formulate it as E1, because the rearrangements are carbocation related.
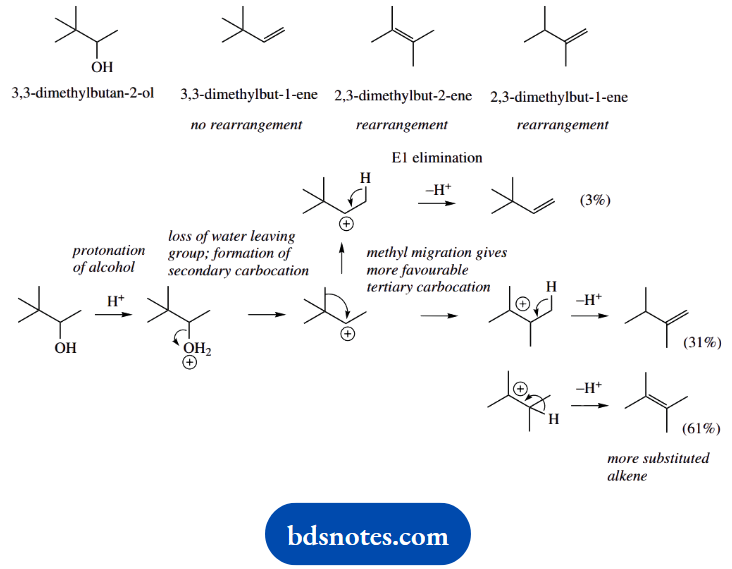
Methyl migration as in part (a) converts the first-formed secondary carbocation into a more favourable tertiary carbocation, and we see proton loss from this to give the other two products. These are unexceptional; there are two possible sites for proton loss.
Now for the relative proportions of products. Only 3% of the unrearranged product shows just how unfavourable the secondary carbocation is compared with the rearranged tertiary carbocation.
The relative proportions of the other two alkenes are explained by the increased thermodynamic stability of the more- -substituted alkene, though this is not sufficient to produce just the single product.
4. This is clearly a rearrangement reaction; the methyl has migrated to the adjacent carbon in the transformation. There is no leaving group to generate a carbocation.
We might consider protonation of the alkene; but be realistic, acid is going to protonate the carbonyl oxygen rather than the alkene. Let us see where that might lead.
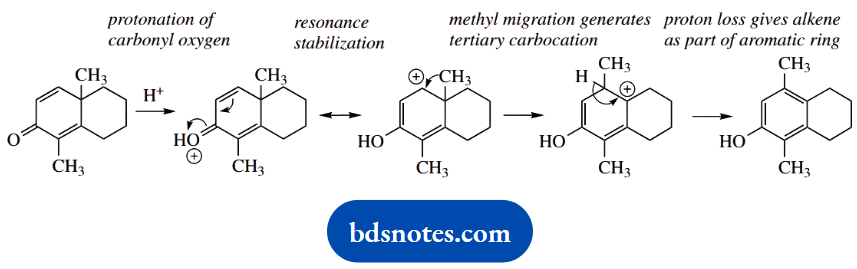
Protonation of the carbonyl oxygen gives the conjugate acid, and because there is a double bond conjugated with the carbonyl, we are able to draw resonance structures.
The pertinent resonance structure is the one that puts the positive charge adjacent to the migratory methyl group. Methyl migration then leads to a tertiary carbocation, and proton loss introduces further stability through the generation of an aromatic ring.
Question 15. Give stereodrawings for the natural products illustrated, in order to indicate their molecular shape.
Answer:
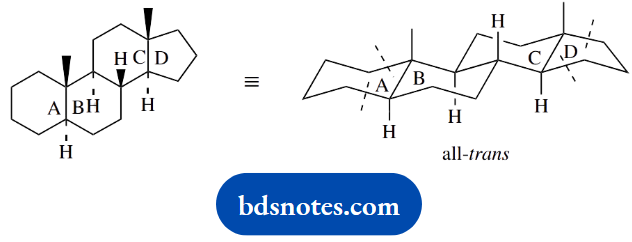
The shape of fused ring structures can be deduced readily by extrapolation from a standard ring system. The most useful starting point is the all-trans-fused steroid system. The approach is to modify this according to requirements.
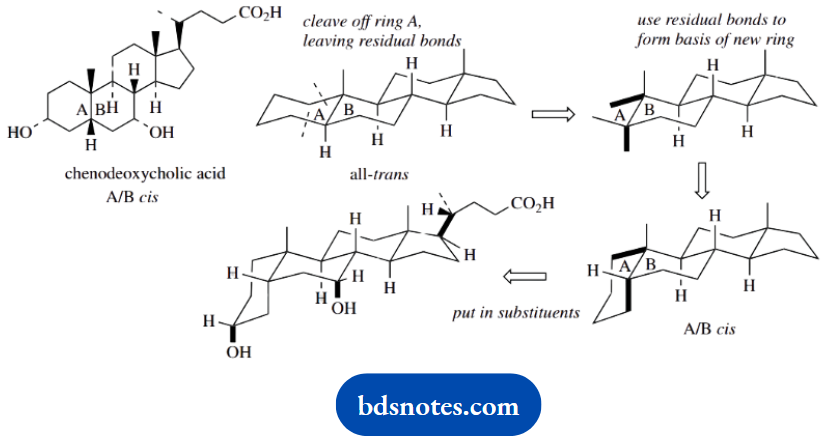
Chenodeoxycholic acid typifies a steroid with A/B cis-fusion; the remaining rings are the same as the all-trans model.
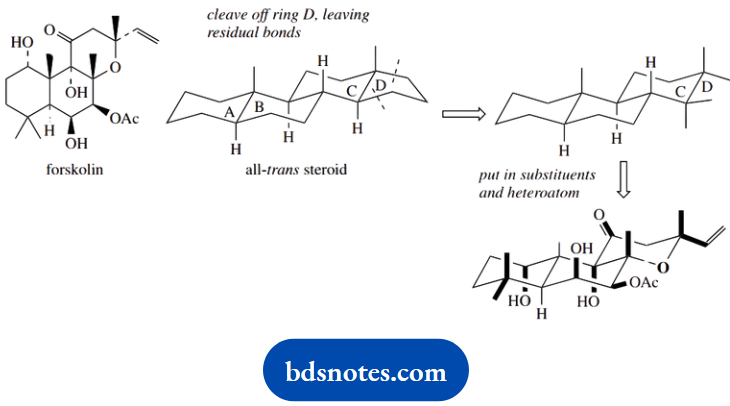
Forskolin is not a steroid; however, if we disregard substituents and that one ring is a heterocycle, then all we have is the first three rings A, B, and C of an all-trans steroid.
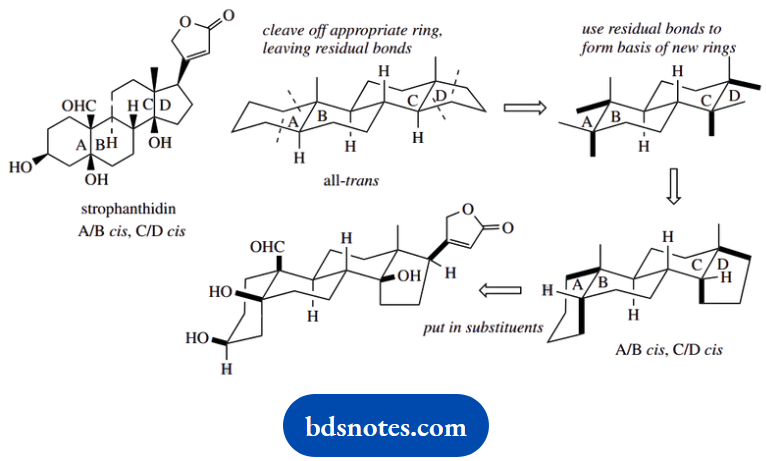
Strophanthidin is a steroid with rings A/B ds-fused, and rings C/D also cis-fused. The latter feature is approached in the same way as the A/B cis-fusion shown with chenodeoxycholic acid above.
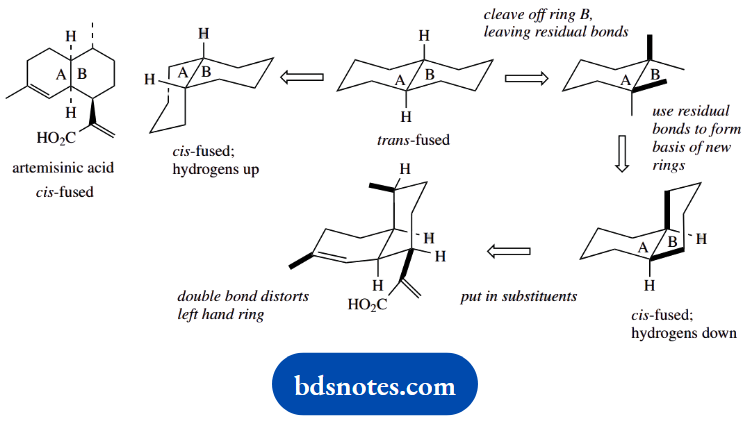
Artemisinic acid is a ds-fused decalin; we can still deduce its shape from the steroid approach. Note that both hydrogens at the ring fusion are down, whereas we have been looking at systems where the ring fusion substituents are up. We shall need to change ring B rather than ring A.
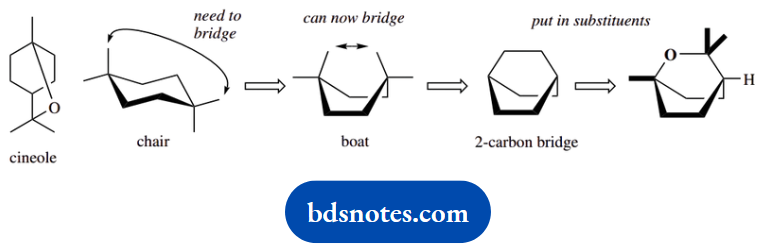
Cineole provides an example of a bridged ring system. The essence of this problem is to spot that we cannot bridge the opposite carbons of a cyclohexane chair conformation; it is necessary to have the cyclohexane in a boat form. Then the answer is readily obtained.
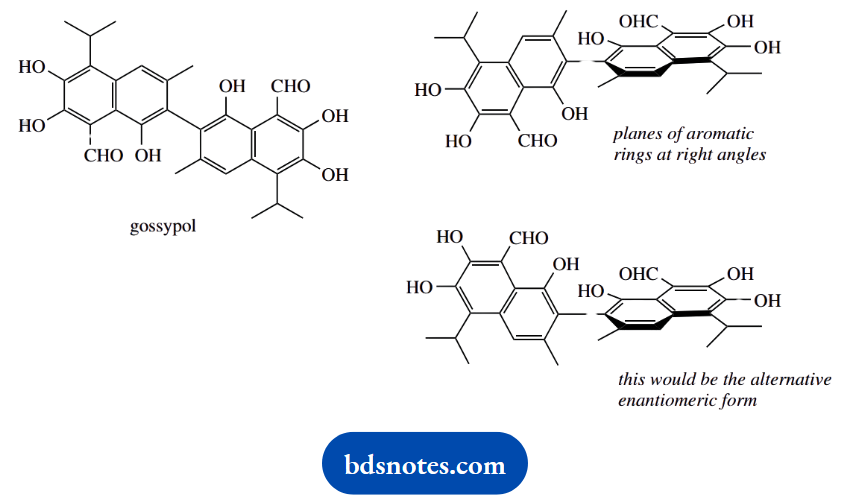
Gossypol is a biphenyl system with ortho substituents, so there is restricted rotation about the bond joining the aromatic rings, creating torsional asymmetry and the existence of enantiomers without a chiral centre.
We are only asked about the shape, and this will have the planar aromatic systems at right angles to minimize interaction between the ortho substituents.
Question 16. Treatment with acid converts steroid (A) into its isomer (B).
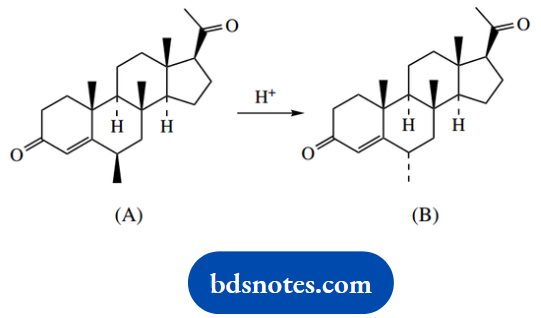
Use a stereo drawing to indicate the molecular shape of (A). Give a mechanistic explanation for the acid-catalysed isomerization of (A) into (B)
Answer:
The shape of fused ring structures can be deduced readily by extrapolation from a standard ring system. The most useful starting point is the all-trans-fused steroid system. The approach is to modify this according to requirements.
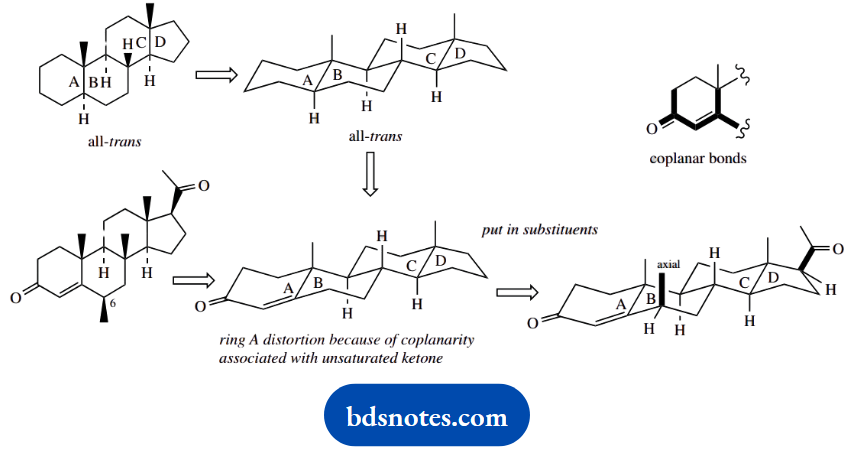
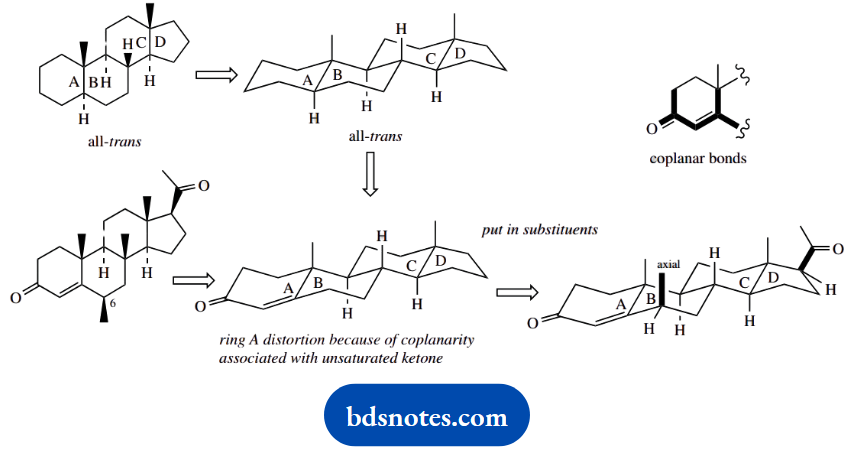
The steroid skeleton in question differs from the all-trans-fused system only with respect to ring A, which has an unsaturated ketone. Because of the coplanarity of bonds in this system, there will be some distortion in ring A, which can be represented (approximately) as shown.
We then need to put in the substituents. The critical one to note is the methyl in ring B because this must be axial. In the following part of the question, the stereochemistry at this position changes, so that the methyl becomes equatorial.
This is not a conformational change, but a configurational change, so requires appropriate chemistry. We should be formulating a proposal where this centre first becomes planar, and then reverts to tetrahedral, but with the substituent then taking up the more favourable equatorial position.
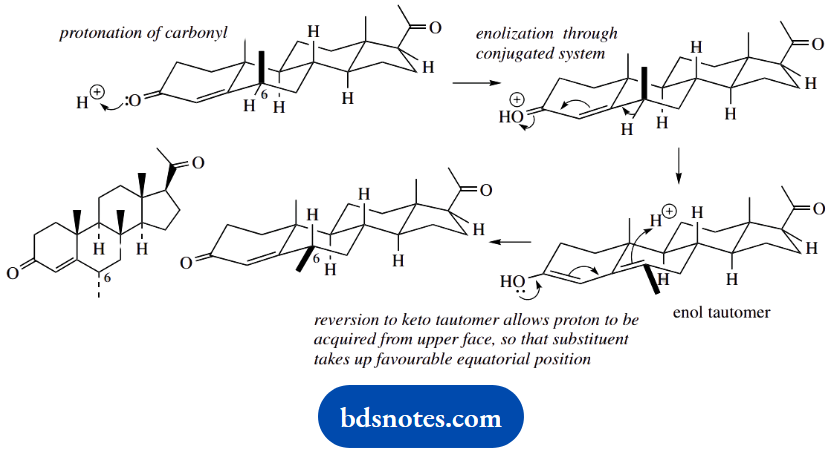
The functional group in ring A is a ketone; a planar system that takes in carbons adjacent to a ketone is the enol tautomer. We are using acid catalysis here, and that is appropriate for enolization.
If we first protonate the carbonyl and then try moving electrons, we shall soon find that the proton at position 6 (standard steroid numbering) can be lost in generating a conjugated enol tautomer.
In a reversal of this process back to the ketone, we can pick up a proton at position 6 from either face. In this case, protonation on the upper face allows the methyl substituent to take up the more favourable equatorial position.
Question 17. Provide mechanistic explanations for the following observations:
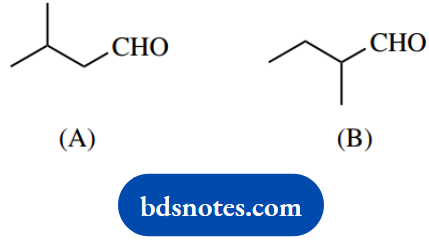
1. In a base-catalysed aldol reaction, compound (A) yields an α, β-unsaturated aldehyde, whereas compound (B) gives a β-hydroxyaldehyde.
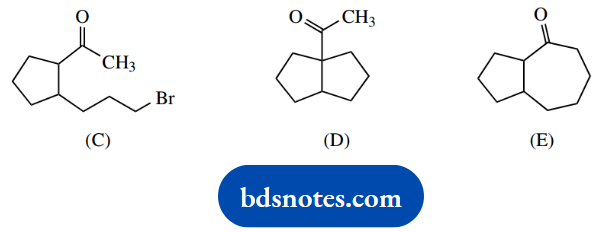
2. Two possible products (D) and (E) can be considered when bromoketone (C) is treated with base. Product (D) is favoured when the base is potassium tertbutoxide in tertbutanol, and compound (E) is produced using lithium di-isopropylamide in tetrahydrofuran.
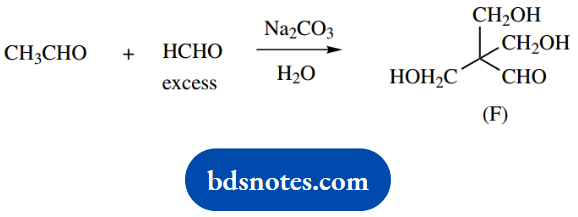
3. Treatment of acetaldehyde with an excess of formaldehyde under basic conditions produces the trihydroxyaldehyde (F).
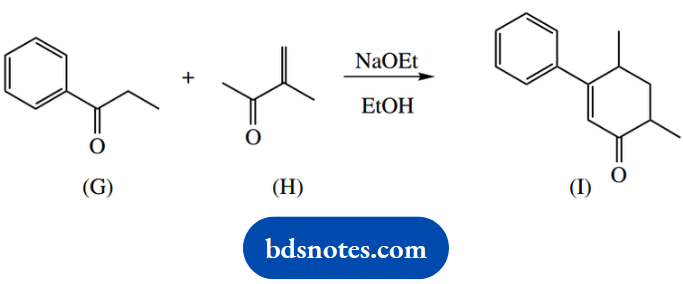
4. The cyclic ketone (I) is formed when ketones (G) and (H) are treated with base.
Answer:
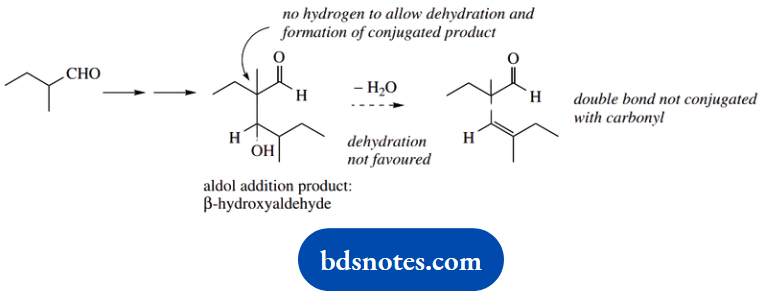
1. A base-catalysed aldol reaction involves an enolate anion acting as a nucleophile and adding to another carbonyl compound. In each of these cases, the enolate anion derived from the aldehyde will react with a second molecule of the same aldehyde.
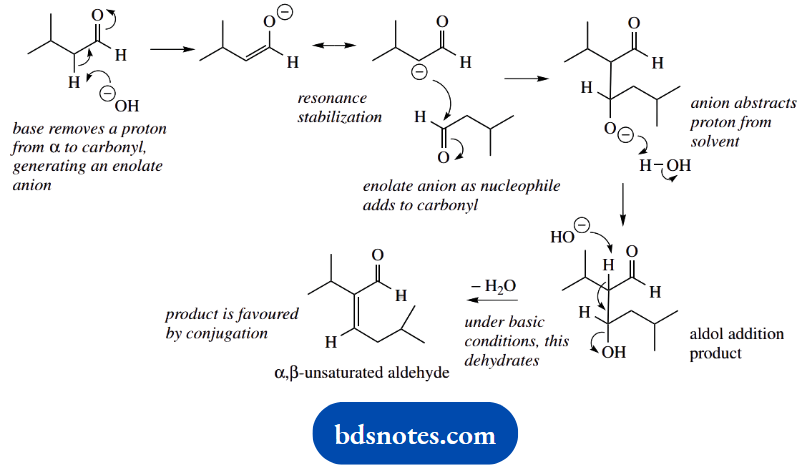
Aldehyde (B) also has hydrogen a to the carbonyl and can generate an enolate anion; the aldol reaction follows as with the first aldehyde to give the product shown. However, dehydration of the β-hydroxyaldehyde is not favoured.
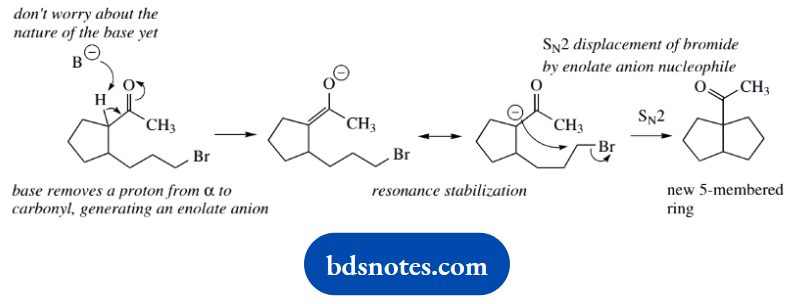
2. Structural analysis suggests we have intramolecular reactions with displacement of bromide and formation of a new ring. Since it is a base-initiated reaction, we should consider enolate anions as the nucleophiles.
There are two possible sites to remove protons in the starting material, and the likely substitution reactions follow.
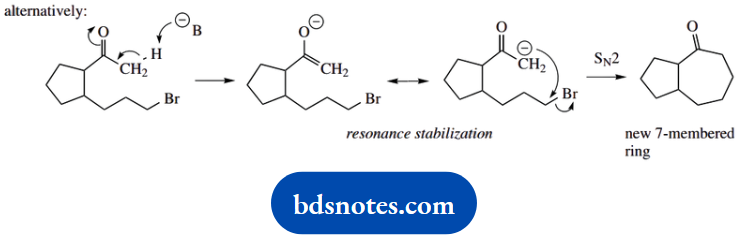
Now LDA is a very strong base, and it is also a large base. We deduce that it can only remove a proton from the less-hindered position, i.e. the COCH3, and this leads to seven-membered ring formation.
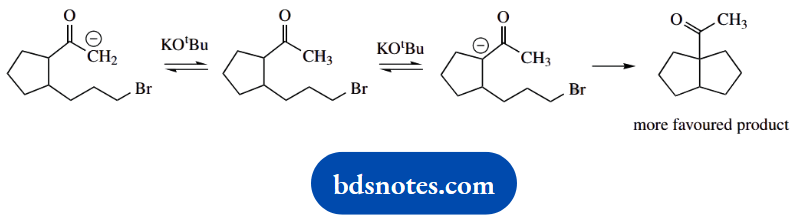
To say tert-butoxide is a smaller base and removes a proton from the alternative site is not sufficient. If it can remove a proton from the hindered position, then it can also remove one from the less-hindered position, so we ought to get a mixture of the two possible products.
Here, we need to remember that although LDA forms the enolate anion in an irreversible manner, enolate anion formation using tert-butoxide is an equilibrium and, therefore, reversible. Therefore, we see the formation of the more favourable product, i.e. that with the five-membered ring.
3. The obvious change here is that three molecules of formaldehyde react with one molecule of acetaldehyde. Formaldehyde is in excess; the reaction is initiated by the mild base, sodium carbonate. The combination of these compounds under basic conditions suggests enolate anion chemistry.
Only acetaldehyde can form an enolate anion; the less-substituted formaldehyde is a better electrophile than acetaldehyde, and it is also present in excess.
In the product, we can visualize the original acetaldehyde fragment carrying three CH2OH substituents. The product is readily rationalized in terms of three similar aldol reactions, utilizing all three of the acidic hydrogens in acetaldehyde.
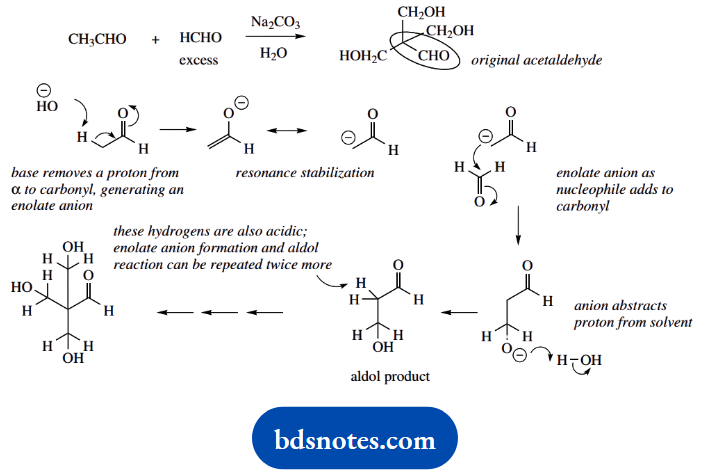
4. Redrawing this equation allows us to see how the two starting materials are required to combine. We can also think in terms of enolate anion chemistry to achieve the necessary bonding; we have a carbonyl compound and basic conditions.
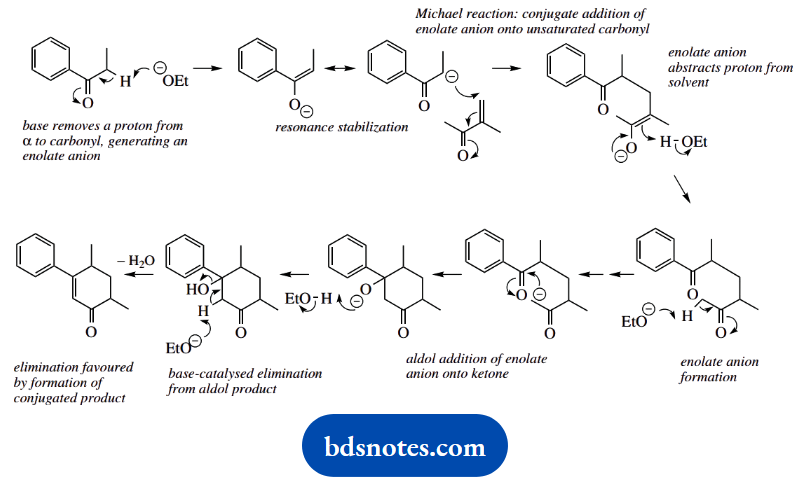
The reaction is considered a combination of a Michael reaction, the conjugate addition of an enolate anion onto an unsaturated carbonyl compound, plus an aldol reaction followed by the elimination of water.
The irreversible elimination drives the reversible aldol reaction and gives a favourable conjugated ketone in a favourable six-membered ring. On paper, one could also draw an acceptable mechanism in which the order of events was reversed.
This is not so neat and would require generating an enolate anion γ to the α,β-unsaturated ketone formed by the first aldol-dehydration sequence.
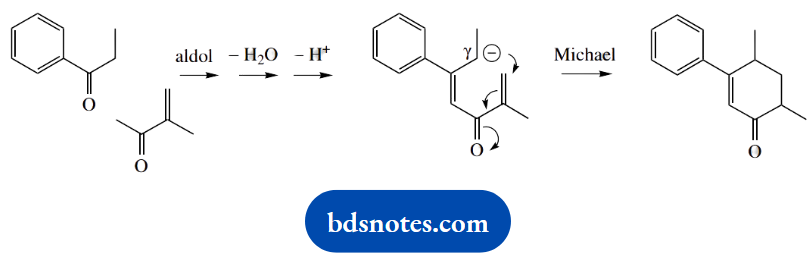
Question 18. Provide mechanistic explanations for the following observations:
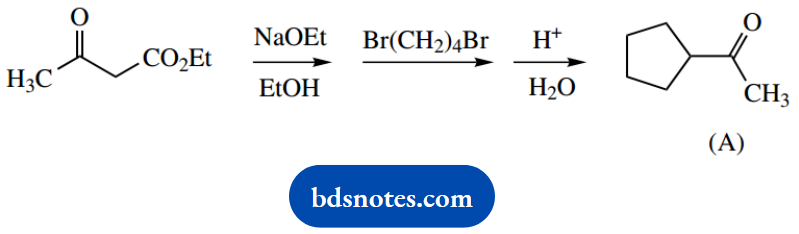
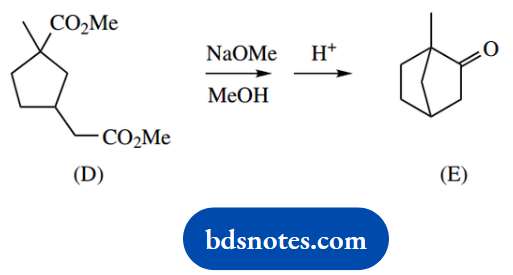
1. The ketone (A) may be synthesized from ethyl acetoacetate by base-catalysed reaction with 1,4- dibromobutane, followed by heating with aqueous acid.
2. The diketone (B) is transformed into the bicyclic ketone (C) on treatment with a mild base.

3. Base-catalysed reaction, followed by acid hydrolysis converts the diester (D) into the cyclic ketone (E).
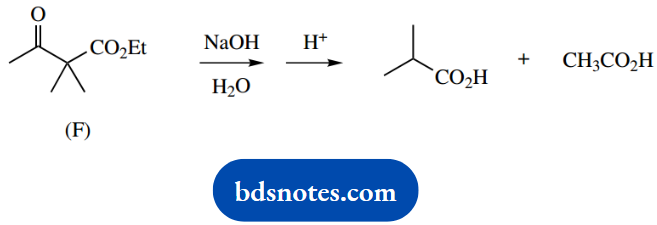
4. The ketoester (F), on heating with an aqueous base, is converted into a mixture of two acids.
Answer:
These provide examples of enolate anion chemistry. This can be surmised from the variety of bond-forming reactions on ketone or ester substrates all achieved under basic conditions.
This shows a three-step sequence; do not be put off, but consider the implications of the first two. We start with ethyl acetoacetate, a β-keto ester.
Under the basic conditions, this can give an enolate anion that presumably then reacts with the halide. No halogen is in the product, so think about SN2 displacement; since we actually use a dihalide, we might consider two such reactions.
Put this down, and you will see we are well on the way.
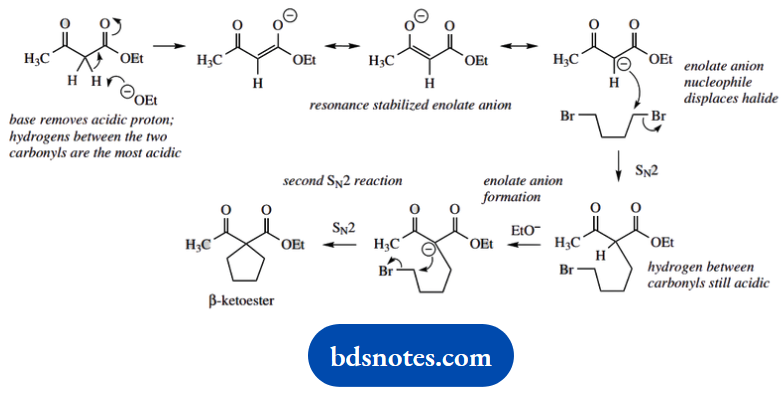
The methylene hydrogens between the two carbonyls are the most acidic, so this is where enolate anion formation occurs. Now follows an SN2 reaction with the dibromide reagent.
It is soon apparent that this sequence of enolate anion formation and SN2 displacement can be repeated since the substrate still contains acidic hydrogen. We soon end up with an alkylated ketoester.
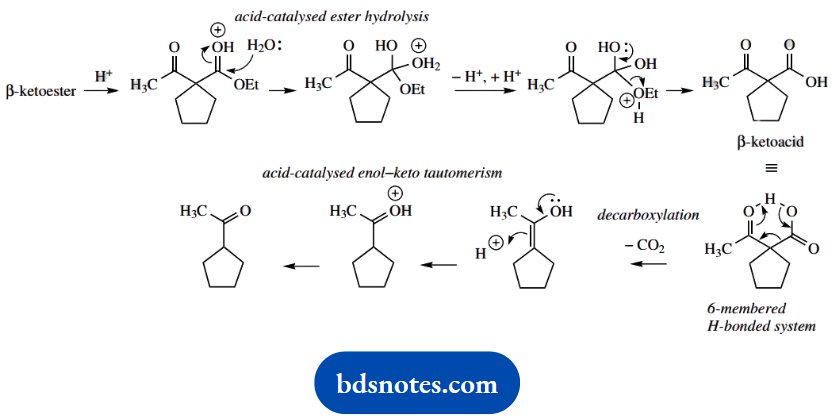
To get the final product we need to lose the ester function. This is a standard combination of acid-catalysed ester hydrolysis followed by heating. The β-ketoacid forms a hydrogen-bonded six-membered ring that facilitates decarboxylation.
2. The base used in this reaction is quite mild, namely sodium carbonate, but this does not change the approach through enolate anions. Overall, we have modified a system with two ketone functions, lost one, and produced an alkene.
This is an aldol reaction followed by dehydration, but it is an intramolecular reaction. Do not worry about the five-membered/seven-membered ring combination; that will fall into place as we consider the mechanism.
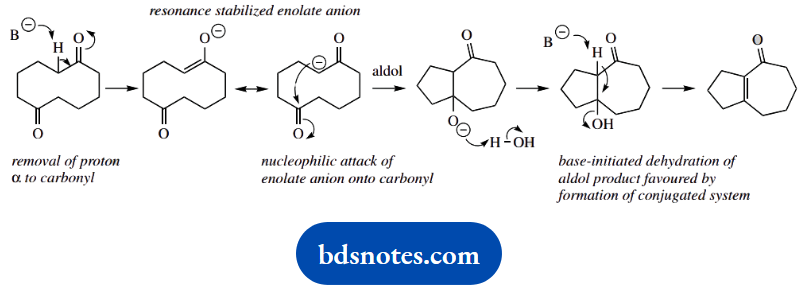
Base removes a proton from adjacent to a carbonyl group. We can use a general B– to represent the base; however, we could use hydroxide, since that is released upon dissolving sodium carbonate in water.
It does not matter which a-position we choose; they are actually all equivalent in this symmetrical substrate. The nucleophilic attack of the enolate anion onto the second carbonyl is followed by base-initiated dehydration so that a favourable conjugated ketone is the product.
As you can see, the size of the ring systems is automatically defined by the reaction.
3. This looks much worse than it is. Analyse the starting material, the conditions, and the functional group changes. A diester under basic conditions leads us to consider enolate anions. From there, we should be thinking about the Claisen reaction, the product of which is a β-ketoester.
The loss of ester functions is accommodated by typical follow-up reactions of hydrolysis and decarboxylation, leading to a ketone. And that is what we get here. As in (b), the ring systems will be automatically defined and do not require special consideration.
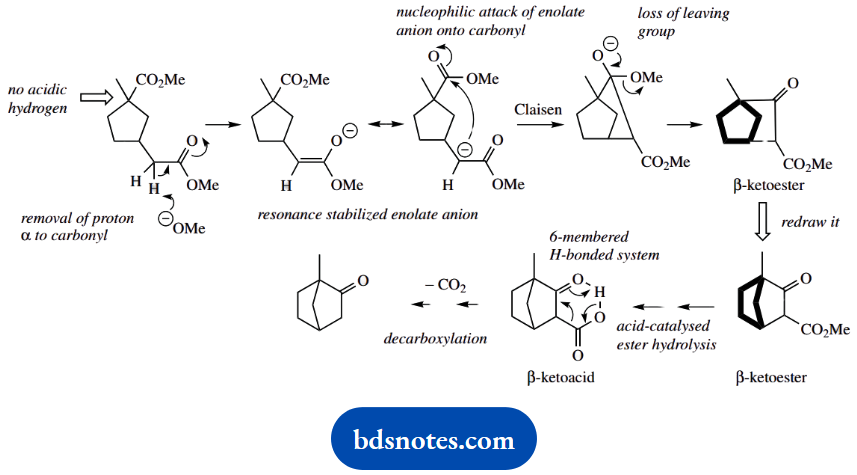
Only one of the ester functions has hydrogen, so only one enolate anion is possible. This attacks the other ester in a Claisen reaction, leading to the expulsion of the leaving group.
This product will need a bit of redrawing to get it into the format of the final product; you see it is exactly right and follows from the mechanistic approach.
All that remains is acid-catalysed hydrolysis of the ester and decarboxylation of the P-ketoacid; this is exactly as in part (a), so is not shown again. It involves a hydrogen-bonded cyclic intermediate.
4. This little reaction is a reverse Claisen reaction on a β-ketoester. We normally think of a Claisen reaction occurring between two esters; the products here are acids, but this is because the aqueous basic conditions also lead to ester hydrolysis.
When you are confronted with a reverse reaction, it is a good idea to write down the forward reaction, which you are probably more familiar with, and then just reverse the mechanistic sequence.
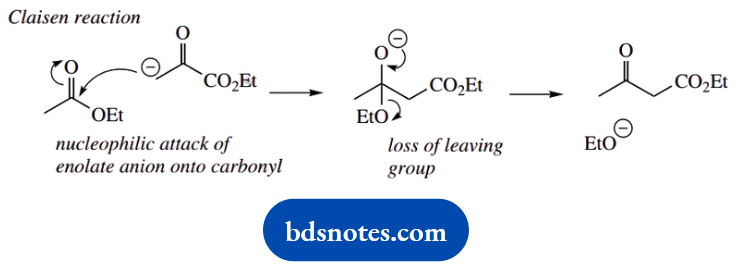
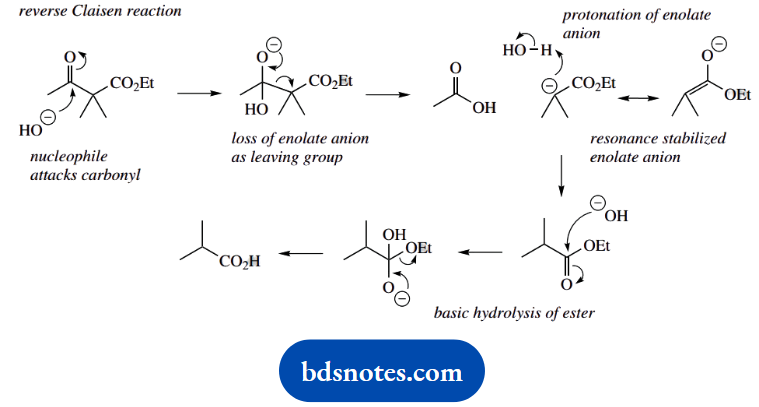
In this case, we formulate the Claisen reaction between two ester molecules as enolate anion formation, nucleophilic attack, and then loss of the leaving group. Now reverse it.
Use hydroxide as the nucleophile to attack the ketone carbonyl, then expel the enolate anion as the leaving group. All that remains is the protonation of the enolate anion and base hydrolysis of its ester function.
Question 19. Provide mechanistic explanations for the following observations:
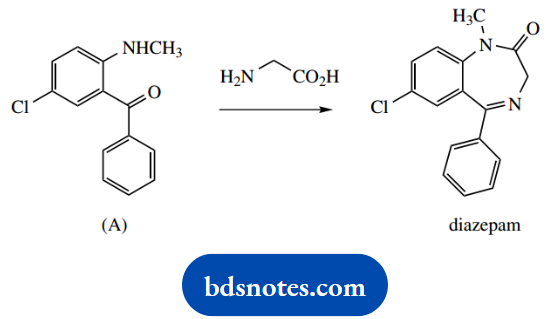
1. The antidepressant diazepam may be synthesized by the reaction of the aminoketone (A) with glycine.
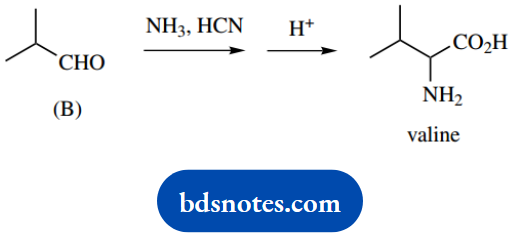
2. The amino acid valine may be synthesized by the reaction of aldehyde (B) with ammonia and HCN, followed by acid hydrolysis.
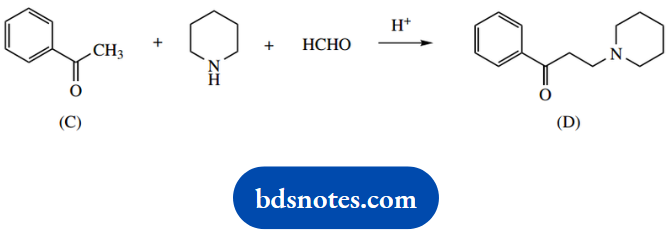
3. The aminoketone (D) is produced when ketone (C) is reacted with piperidine and formaldehyde under mild acidic conditions.
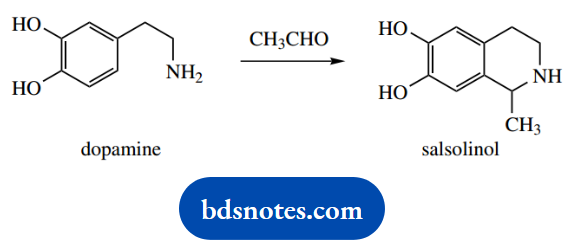
4. Dopamine reacts readily with acetaldehyde to produce the cyclic amine salsolinol.
Answer:
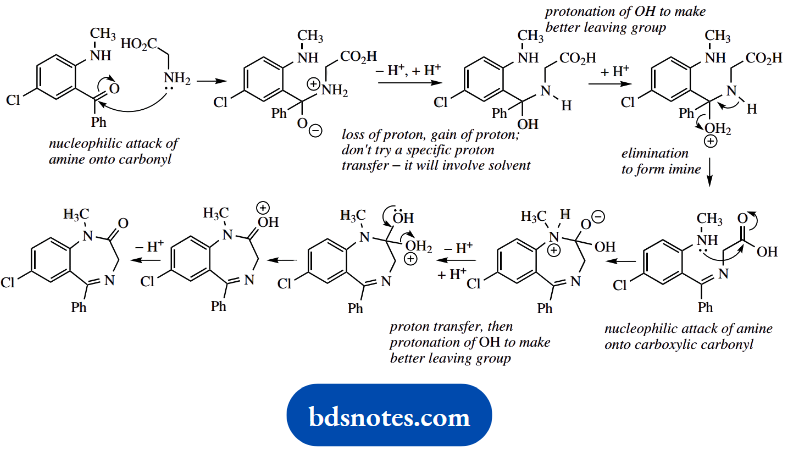
1. Do a quick comparison of the reagents and the product; it should be apparent that we need to synthesize a new amide bond and a new imine bond. Both reactions involve a nucleophilic attack of an amino group onto a carbonyl.
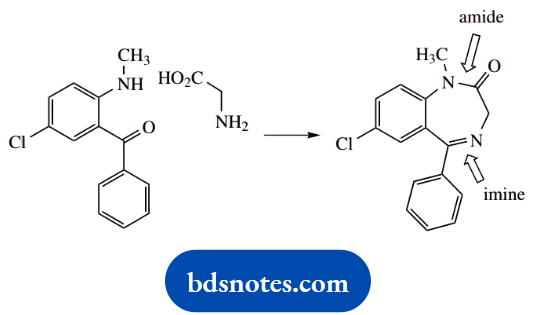
These are standard reactions, and it does not matter which you do first. Resist the urge to do both at the same time; it’s unlikely to occur, and will certainly complicate your structures and mechanisms. Note that we have to include a number of protonations to make the mechanisms work.
This is quite legitimate, and although the question has not specified any conditions, you can assume there will be a source of protons. In any case, you will observe that protons are first released to the solvent and then transferred back to an alternative site in the molecule.
2. This is a standard Strecker synthesis of an amino acid; it is not important that you remember the name in ‘named’ reactions. An analysis of the transformation should lend some clues.
You are informed it is a two-stage process; acid hydrolysis is a necessary part, and you can relate this to hydrolysis of a nitrile, supplied by the cyanide in the first part. The initial reaction is between the aldehyde and the ammonia to form an imine.
This then acts as a carbonyl analogue for the attack of the cyanide. Alternative thoughts starting with aldehyde plus cyanide giving a cyanohydrin are unproductive, in that you then wonder how to use the ammonia.
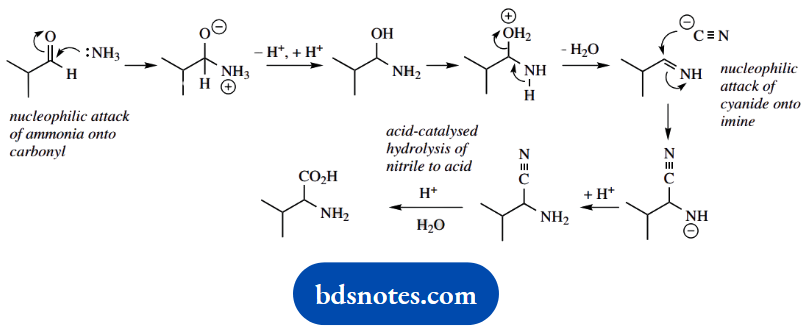
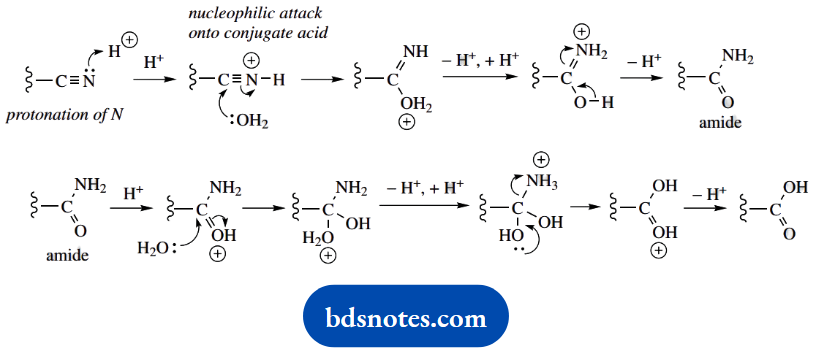
The initial part of the reaction is the same as in part (a) and leads to the imine intermediate. Nucleophilic attack by the cyanide onto this carbonyl analogue provides the next intermediate, an aminonitrile.
The last part of the synthesis is hydrolysis of the nitrile function. It involves another intermediate, an amide, and this also has to be subjected to acid hydrolysis.
As a result, it all adds up to a lengthy mechanism that can become rather tedious, because it contains so many repetitive steps. It is shown in general terms below.
3. This is another ‘named’ reaction, the Mannich reaction. Three reagents join together in a two-stage process.
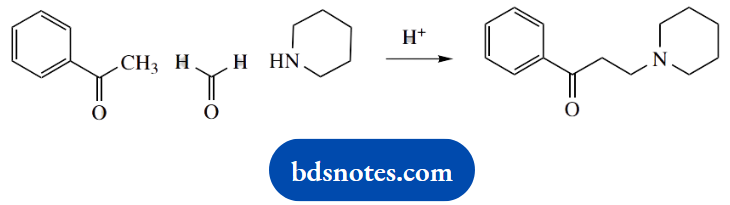
We have an aldehyde, an amine, and a ketone. As in part (b), the amine reacts first to give an imine, and this behaves as a carbonyl analogue, which in the Mannich reaction is then the electrophile for an enolate anion equivalent.
How can we remember the sequence of events? The most common mistake is to react the aldehyde and ketone via an aldol reaction, but this then leads to an alcohol and one is faced with a substitution reaction to incorporate the amine.
It is the mild acidic conditions that help us to avoid wrong proposals. Such conditions are not especially conducive to aldol reactions and substitution reactions.
Instead, they are favourable for imine formation. And which of the carbonyl reagents reacts? That is easy, it is the most reactive electrophile, the aldehyde.
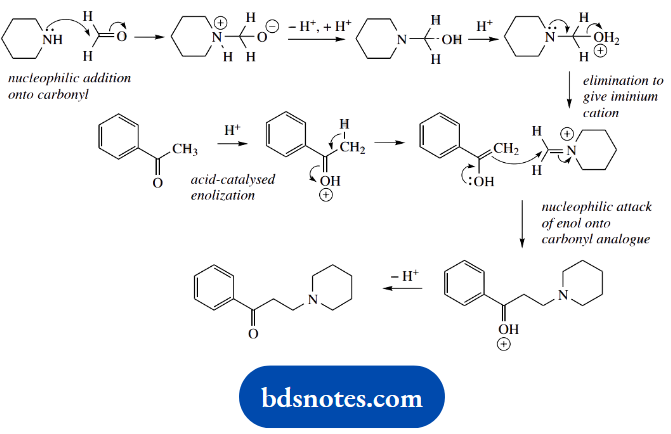
We thus have standard imine formation; in this case, the secondary amine leads to an iminium cation. This is attacked by the ketone nucleophile. This cannot be an enolate anion because of the mild acidic conditions under which the reaction proceeds, so we formulate it as involving the enol tautomer.
Accordingly, we need to include an acid-catalysed enolization step.
4. Yet another ‘named’ reaction, this time the Pictet-Spengler tetrahydroisoquinoline synthesis. It is really a Mannich-like reaction as in part (c), with the enol nucleophile being a phenol, and achieving a ring cyclization
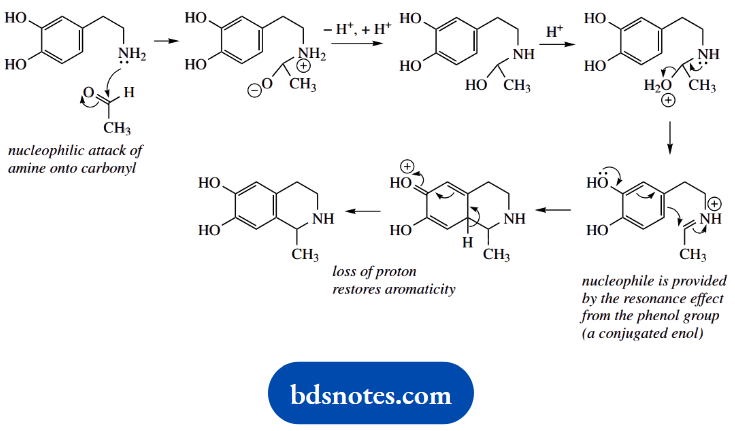 .
.
The mechanism of imine formation is standard, as seen in the other examples. The cyclization reaction is then like the Mannich reaction, the attack of an enol on to the iminium cation. This time though, the nucleophile is provided by the resonance effect from the phenol system.
Question 20. A synthetic pathway to vitamin A involves the following partial sequence:
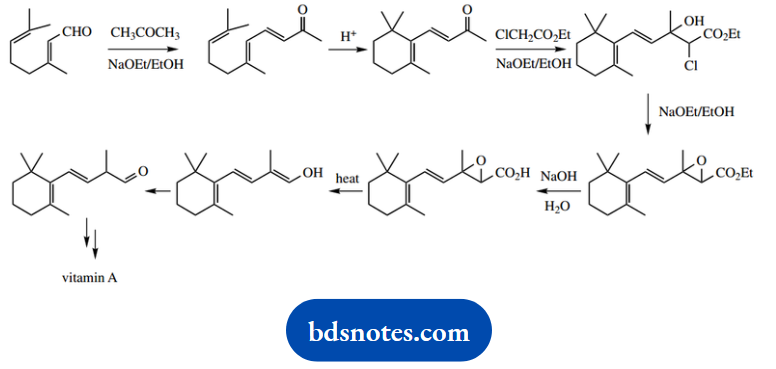
Give detailed mechanisms for the reactions in this sequence.
Answer:
This problem covers a reaction sequence and a variety of different reactions, some easier than others. This one includes enolate anions, electrophilic cyclization, nucleophilic substitution, and simple carboxylic acid chemistry.
The first reaction involves a ketone reaction with an aldehyde under basic conditions, so enolate anion chemistry is likely. This is a mixed aldol reaction; the acetone has acidic a-hydrogens to form an enolate anion, and the aldehyde is the more reactive electrophile.
The reaction is then driven by the ability of the intermediate alcohol to dehydrate to a conjugated ketone.
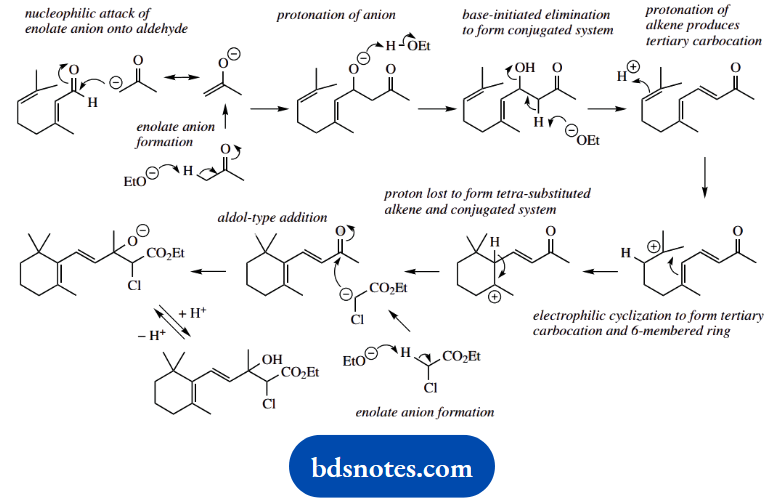
The next transformation is an electrophilic cyclization. Protonation of the terminal alkene produces the more favourable tertiary carbocation, then ring formation occurs by electrophilic addition to the neighbouring alkene.
Again, this produces a favourable tertiary carbocation. The loss of a proton gives the required alkene. Note that potentially three different carbons could lose a proton.
The reaction shown generates the most stable product; this has the maximum number of alkyl substituents and also benefits from extended conjugation.
We then get another aldol-type reaction. The enolate anion is produced from the ethyl chloroacetate, and simple addition yields an anion that is subsequently protonated.
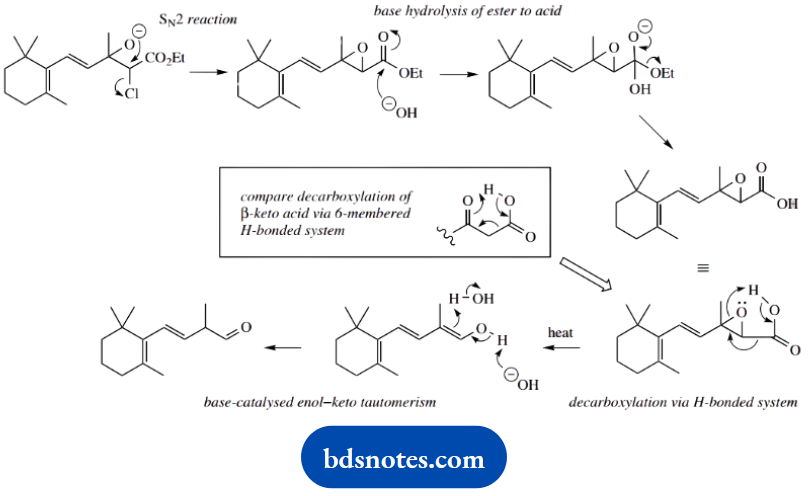
However, the same anion has to be regenerated for the next reaction, in which an SN2 displacement leads to epoxide formation. Base hydrolysis of the ester produces the carboxylic acid, which decarboxylates on heating.
The mechanism for this reaction should be suggested by the structural change (loss of carbon), and this is achieved merely by heating.
What other systems do this? It is actually analogous to the thermal decarboxylation of a β-keto acid, a process where we invoke a cyclic mechanism within a hydrogen-bonded system.
Think laterally to see whether we can do the same here. We can, though any hydrogen bonding must involve a five-membered ring rather than a six-membered ring. The sequence is completed by base-catalysed tautomerism of the enol to a ketone.
Question 21. Propose a mechanism for all steps involved in the following synthesis of the ester of chrysanthemum acid, a component of the natural insecticides, the pyrethrins:
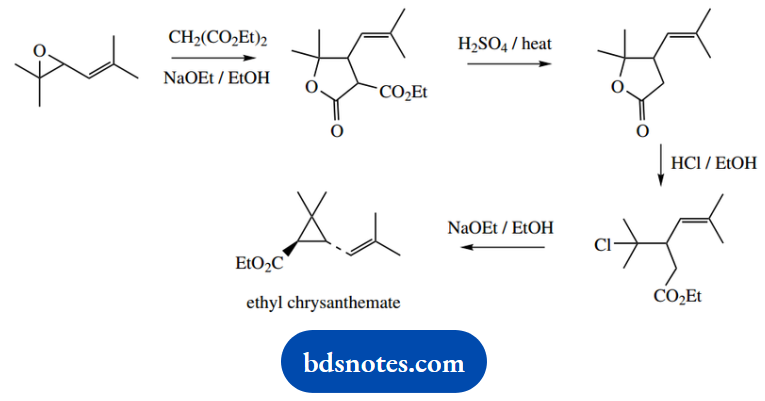
Answer:
This problem brings in a wide range of reactions. It includes enolate anions as nucleophiles, with SN2 reactions opening an epoxide ring, and displacing halide.
There is a simple SN1 reaction and the decarboxylation of a β-keto acid through a cyclic hydrogen-bonded system. Most of the remaining steps involve ester formation and hydrolysis.
Although a lactone intermediate can be isolated in the sequence, it need not be specifically implicated in the mechanism; it is formed under acidic conditions from a hydroxy acid, but we could utilize the hydroxy acid in the sequence. Some of the steps might then occur in a different order from that shown.
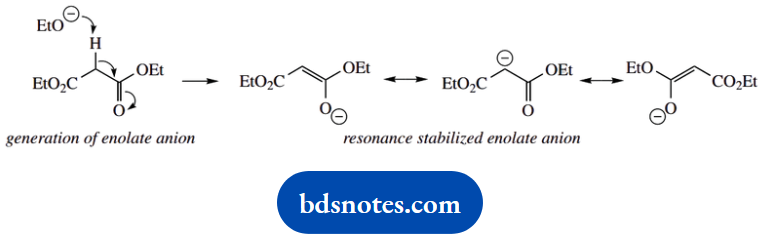
The mechanistic steps can be deduced by inspection of structures and conditions. Enolate anion formation from diethyl malonate under basic conditions is indicated, and that this must attack the epoxide in an SN2 reaction is implicated by the addition of the malonate moiety and disappearance of the epoxide.
The subsequent ring formation follows logically from the addition of an anion and is analogous to the base hydrolysis of an ester. Ester hydrolysis followed by decarboxylation of the β-keto acid is then implicated by the acidic conditions and structural relationships.
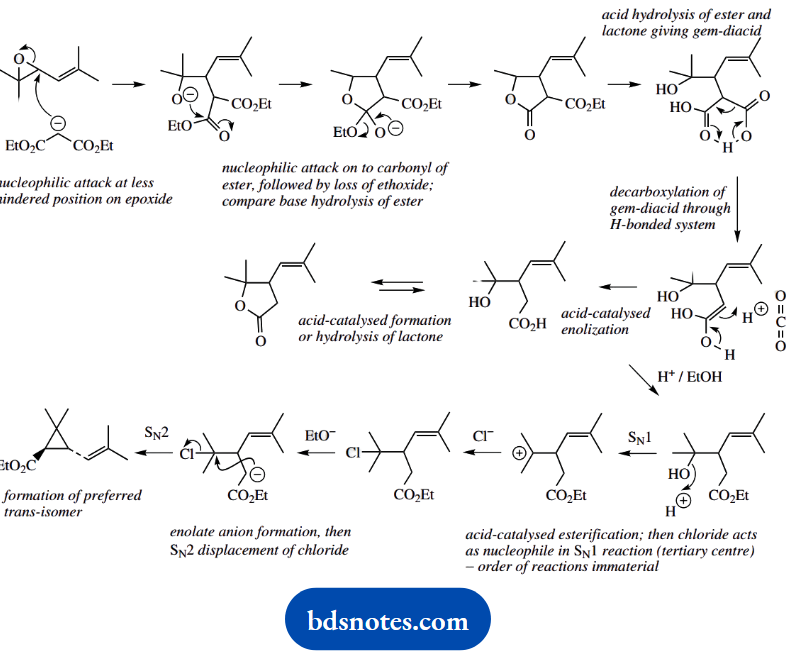
The next step is not immediately obvious. The generation of an ethyl ester from a lactone can be accommodated by transesterification (we might alternatively consider esterification of the free hydroxy acid).
The incorporation of chlorine where we effectively had the alcohol part of the lactone leads us to nucleophilic substitution. That it can be SN1 is a consequence of the tertiary site.
Cyclopropane ring formation from an SN2 reaction in which an enolate anion displaces a halide should be deducible from the structural relationships and basic conditions.
Question 22. pKa values for the most basic group in the following compounds have been determined to be as shown:
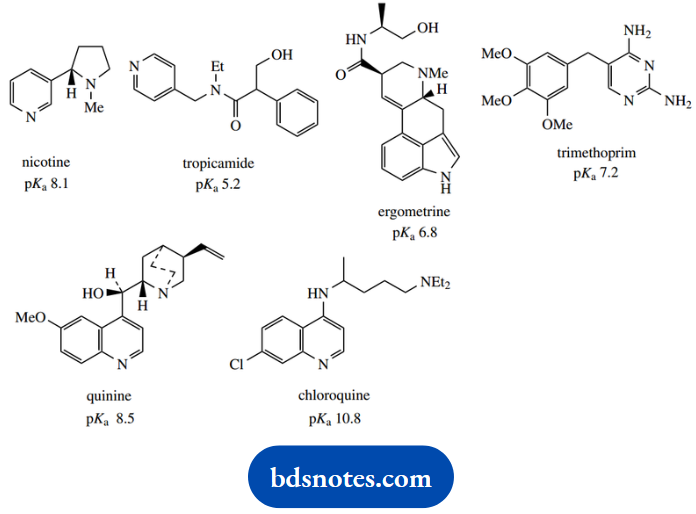
For each compound, predict the site of protonation that this pKa value represents. Give reasons for your choice.
Answer:
The relative basicity of an aromatic nitrogen heterocycle is dictated by the ring size, the presence of any other heteroatoms, and possible effects from substituents.
It is potentially a rather more complex problem than with, say, simple amines, and should be approached logically and systematically. In practice, these examples do not present particularly difficult problems.
There is no way we would encourage the memorising of pKa values, but two easily remembered figures can be valuable for comparisons. Conjugate acids of typical aliphatic amines have pKa values around 10, whereas for pyridine the figure is around 5.
These then allow us to consider whether the compound in question is more or less basic, etc. The smaller the pKa, the weaker the base.
Remember, of course, that the pKa values refer to the acidity of the conjugate acids, and not to the acidity of the amines themselves, which could be relevant in different circumstances.
Nevertheless, it is usually acceptable to talk of the pKa of the base, when we should strictly say the pKa of the conjugate acid of the base.
Nicotine has two nitrogen atoms, one as a cyclic tertiary amine and one in a pyridine ring. The basicities are easily distinguished, in that a pyridine system is much less basic than a simple amine.
This is essentially a hybridization effect, the nitrogen lone pair in pyridine being held in a sp2 orbital. This means the lone pair electrons are held closer to the nitrogen, and are consequently less available for protonation than in a sp3– hybridized aliphatic amine.
Hence, as mentioned above, pyridine has a pKa of approximately 5. It follows that pKa 8.1 is more appropriate for the pyrrolidine nitrogen.
Tropicamide has pKa appropriate for a pyridine ring. The second nitrogen is part of an amide group and is thus a very weak base. Amides are stabilized through resonance, a stabilization that is not possible in the conjugate acid.
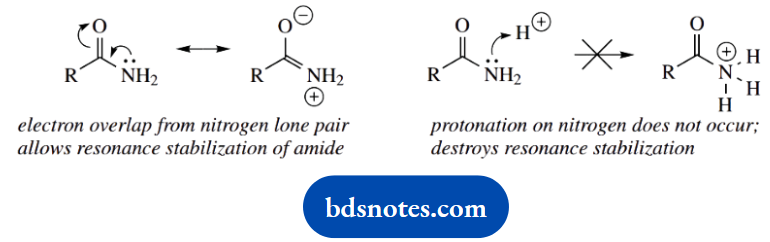
Ergometrine has three nitrogen atoms, a cyclic tertiary amine, an amide (very weak base as just described for tropicamide), and one in an indole ring.
Indoles, like pyrroles, are very weak bases, with negative pKa values for the conjugate acid. This is because nitrogen has already used its lone pair electrons by contributing to the aromatic sextet.
Consequently, protonation on nitrogen destroys aromaticity in the five-membered ring. In fact, like pyrrole, indole preferentially protonates on carbon rather than nitrogen.
It follows that pKa 6.8 for ergometrine is not appropriate for the indole nitrogen or the amide nitrogen, but must be the figure for the cyclic tertiary amine.
It is somewhat less basic than we might predict for a cyclic tertiary amine, and this must relate to its position in a fused-ring system.
Trimethoprim is a diaminopyrimidine derivative. It is reasonably basic (pKa 7.2) and we should remember here that amino substituents are able to utilize their lone pairs and provide resonance stabilization to a conjugate acid.
Consequently, aminopyrimidines protonate on a ring of nitrogen. If we consider the protonation of the two ring nitrogens separately and then think about potential resonance stabilization, we can predict the site of protonation.
If we protonate on N-3, we can predict resonance stabilization involving either of the adjacent amino functions. In each case, we see charge distribution over three atoms using an N-C=N system.
However, pro-tonation on N-1 allows similar resonance stabilization, but increased charge dispersal emanating from the 4-amino substituent. We do not need to consider any further resonance structures.
Effects involving the 2-amino substituent will be the same whether N-1 or N-3 is protonated. We can predict that protonation occurs on N-1.
Quinine has two potentially basic centres, a cyclic tertiary amine at a ring junction, and one in a quinoline ring system. pKa 8.5 is reasonably basic, and this is most likely from the aliphatic tertiary amine. We need to convince ourselves that the quinoline nitrogen is less basic. This is true.
As far as reactivity is concerned, a quinoline ring system behaves as two separate parts, either pyridine or benzene, depending upon the reagent. Thus, quinoline has pKa very similar to that of pyridine, i.e. around 5.
Chloroquine is more basic than quinine, and the pKa suggests we must be looking at an aliphatic amine. There are two; so which? Note though that one of the amine functions is para to the quinoline nitrogen, so we may expect resonance stabilization when the quinoline nitrogen is protonated.
This will increase the basicity of the quinoline system from about pKa 5, but almost certainly not as far as represented by pKa 10.8. However, it solves our problem, since it means the 4-amino substituent is donating its lone pair into the aromatic ring system and is not, therefore, available for bonding to a proton.
This site is going to be less basic than a tertiary amine. pKa 10.8 must represent the terminal -NEt2.
Question 23.
1. During porphyrin biosynthesis, porphobilinogen is produced from two molecules of 5-aminolaevulinic acid and is subsequently converted into uroporphyrinogen I. Formulate a chemical mechanism to account for the transformations.
2. The Reissert indole synthesis is shown below. Propose rational mechanisms to account for the first and third steps. Explain why the nitro group is essential for the success of the first step.
Answer:
1. Ignore the fact that this is a biosynthetic sequence. We are required to formulate a mechanism that treats it as a chemical transformation.
The first step is the formation of a pyrrole ring system from two identical aminoketones. It is actually a Knorr pyrrole synthesis, but we do not need to identify it as such, just approach it logically.
In fact, if we look back at the Knorr pyrrole synthesis, we shall see that, under chemical conditions, the reagents used here are not sufficiently reactive for the pyrrole synthesis; we need a more activated compound, like ethyl acetoacetate.
Furthermore, we could not possibly proceed without masking the carboxyls as esters. This underlines how a biosynthetic sequence might differ somewhat from a purely chemical synthesis.
We shall consider the sequence as firstly imine formation (an abbreviated form of this mechanism is shown), followed by imine-enamine tautomerism. This provides a nucleophilic centre and allows a subsequent aldol-type reaction with enamine plus ketone. The pyrrole ring is produced by proton loss and dehydration.
The linear tetrapyrrole has methylene bridges between the pyrrole rings; we start from porphobilinogen that has either -H or -CH2NH2 as the ring substituents at these positions.
Since the nitrogens are lost, we should consider an elimination, and this is assisted by the pyrrole nitrogen. We can consider protonation of the amine to facilitate the elimination. The product is an electrophilic methylidene pyrrolium cation.
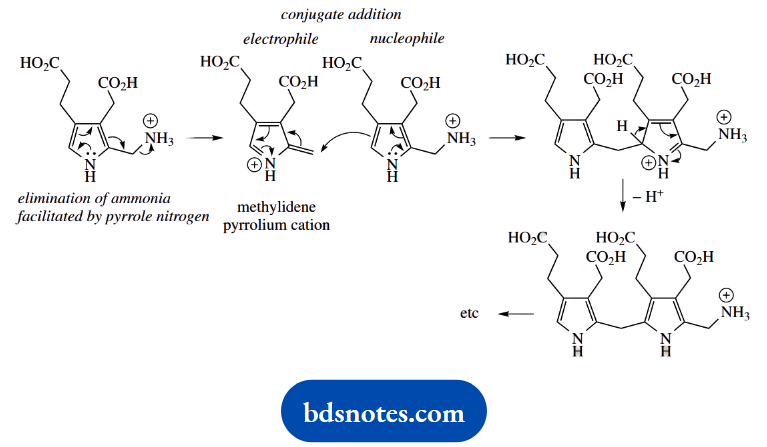
Pyrrole is very reactive towards electrophiles; charge distribution from the nitrogen makes either C-2 (or C-3) electron-rich. Thus, a second porphobilinogen acts as the nucleophile towards the methylidene pyrrolium cation in a conjugate addition reaction.
It is now possible to see that two further identical steps will give us the required linear tetrapyrrole and that one more time will then achieve ring formation.
2. It is not necessary to know the Reissert indole synthesis; this is a rational application of known reactivity. The second step is catalytic reduction of a nitro group to an amine, and no details are required.
Step 1 has all the hallmarks of enolate anion chemistry. It is carried out under strongly basic conditions, and a diester (diethyl oxalate) suffers displacement of an ethoxide in a Claisen-like reaction. We thus need to generate an ArCH–2 anion.
Toluene is more acidic than, say, ethane, but not that acidic for proton removal by ethoxide. This is where the last part of the question comes in: why is the nitro group essential for the success of the first step? It probably relates to generating the anion; and indeed, we can make a case for the nitro group stabilizing the conjugate base by resonance.
Later on, the nitro is reduced to an amine; an amino group would inhibit the first step since it reduces the acidity of the methyl substituent via its electron-donating resonance effect.
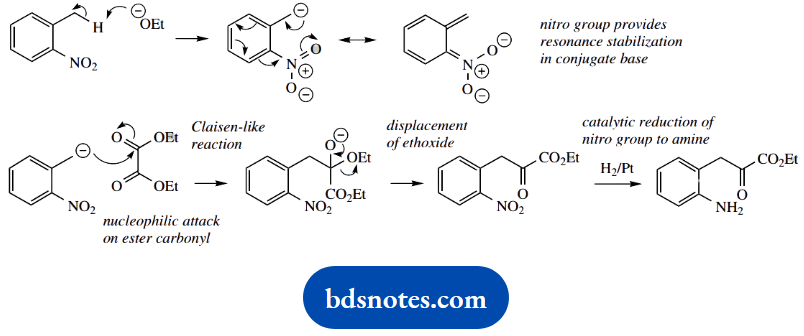
Finally, indole ring formation is via condensation of the amino and keto functions. This is analogous to imine formation, as seen in part (a), but dehydration produces the aromatic pyrrole ring rather than an imine.
Alternatively, one could write imine formation followed by tautomerism to the aromatic enamine.
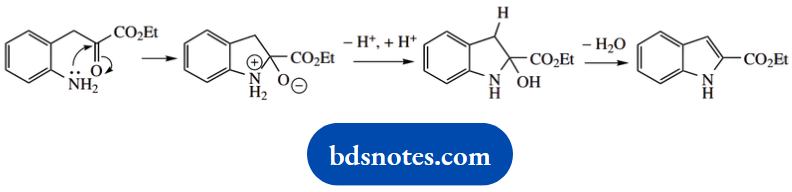
Question 24.
1. Treatment of 2-methylpyridine with benzaldehyde under basic conditions gives the compound stilbazole. Give a mechanism for the reaction and predict the product formed when 2,3-dimethylpyridine is reacted with benzaldehyde under the same conditions.
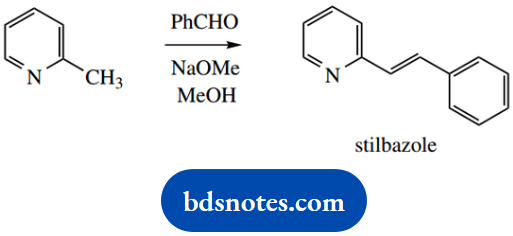
2. What product would you expect to obtain if 2-chloropyridine was treated with sodium methoxide in methanol? What product would be obtained upon similar treatment of 3,4-dihydropyridine?
3. Give mechanisms to account for the following two reactions:
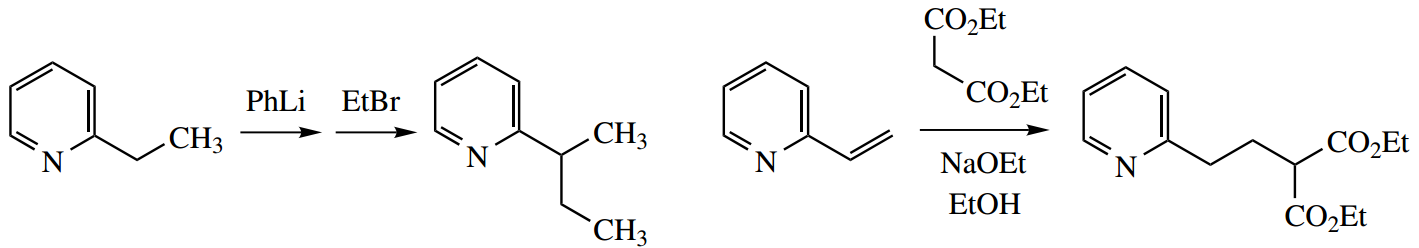
4. Treating indole with the unsaturated ketone shown (mesityl oxide) under acid conditions gives a carbazole derivative. Formulate a rational mechanism for the transformation, which proceeds through an iodonium cation intermediate (not isolated).
Answer:
1. The basic conditions point to the initial generation of an anion that is subsequently used in reaction with benzaldehyde. Since the product is an alkene conjugated to the aromatic rings, an addition-elimination sequence seems a likely proposal.
To generate the anion, the 2-methylpyridine needs to lose a proton from the methyl substituent. This is feasible; the resultant anion is resonance stabilized and behaves much like an enolate anion.
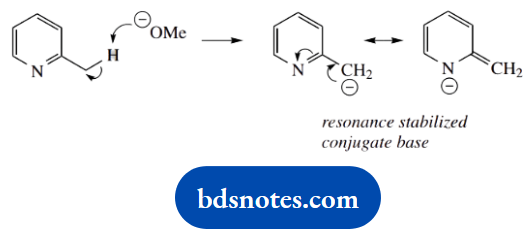
Then follows an aldol-like reaction with subsequent dehydration, favoured by the conjugation in the product.
Removal of a proton from 3-methylpyridine does not provide the same resonance stabilization in the anion. The charge cannot be dispersed to the nitrogen, only to carbons in the aromatic ring.
Hence, 3-methylpyridine is a weaker acid than either 2- or 4-methylpyridine. We predict, then, that a similar treatment of 2,3-dimethylpyridine will produce the methylstilbazole derivative shown.
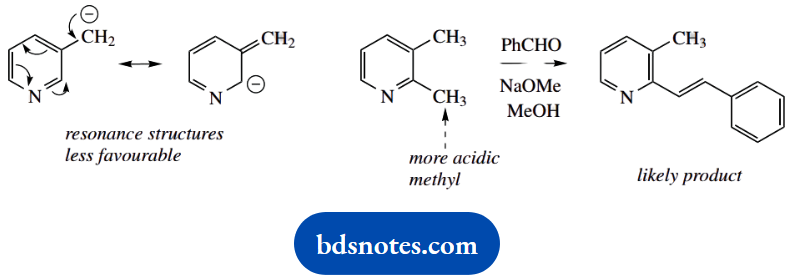
2. The reaction of 2-chloropyridine with sodium methoxide is a nucleophilic substitution. This is represented as an addition to the imine function, followed by loss of the chloride leaving group. The reaction is facilitated by the resonance stabilization in the intermediate anion.
Similar resonance structures may be drawn in the case of 4-chloropyridine, and this will also undergo nucleophilic substitution. However, 3-chloropyridine, despite having the same favourable leaving group, does not undergo substitution.
This may be deduced from consideration of the intermediate anion; resonance structures involve only aromatic carbons, with no stabilization contributed by the nitrogen.
It follows, therefore, that the product from the treatment of 3,4-dichoropyridine with sodium methoxide will be 3-chloro-4- methoxypyridine. The substitution will occur only at position 4.
3. The first reaction follows from part (a), in that we can generate the conjugate base of the alkylated pyridine derivative, and use this anion as a nucleophile in an SN2 reaction.
Note that we remove a proton from the position adjacent to the pyridine ring to obtain the resonance-stabilized anion.
In the second reaction, we also have a base-generated anion, this time an enolate anion from diethyl malonate. This reagent is more acidic than an alkylpyridine; and in any case, the unsaturated side-chain does not allow proton removal in the way we have just seen.
It can be seen that the enolate anion joins up with the heterocyclic reagent, but now the heterocycle must be responsible for providing the electrophile.
The double bond in the side chain gives a clue, and we note this is conjugated with the pyridine ring; the reaction is a nucleophilic attack on a conjugated imine, a Michael-like reaction.
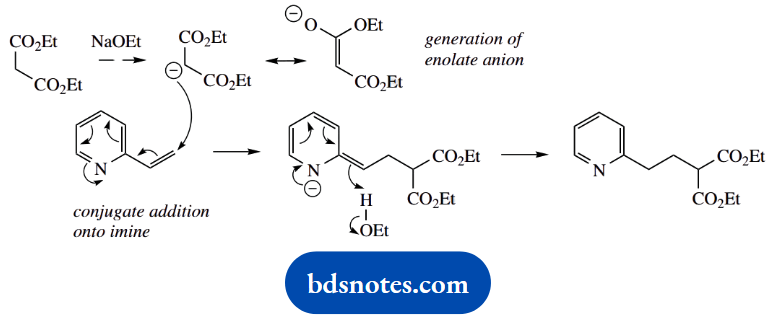
4. This is a two-step reaction, where the intermediate is not isolated; it is presented to give you a clue. The clue is that we have a Michael-like conjugate addition to the unsaturated ketone, and the heterocycle must provide the nucleophile.
Since the heterocycle is indole, this is quite reasonable; indoles are very reactive towards electrophiles with attack occurring at C-3.
We can now proceed. Indole attacks the unsaturated ketone in Michael fashion; because the conditions are acidic, we protonate the ketone. The product is an enol, so that is tautomerized to the ketone. This gives the ‘not isolated’ intermediate.
The next step is ring closure, and we have the iminium system of the heterocycle as an electrophile. The nucleophile should be distinguishable as the a-position of the ketone, an enolate anion equivalent. Under acidic conditions, we cannot have an enolate anion, so we must employ the enol.
This means a second tautomerization step. Ring closure follows; you may recognize it as equivalent to a Mannich reaction: attack of an enol onto an iminium cation. Loss of a proton and the job is done.
Leave a Reply