Amino Acids
Numerous amino acids are found in nature, we are concerned primarily with those that make up the structures known as peptides and proteins. Peptides and proteins are both polyamides composed predominantly of α- amino acids linked through their carboxyl and α- amino functions.
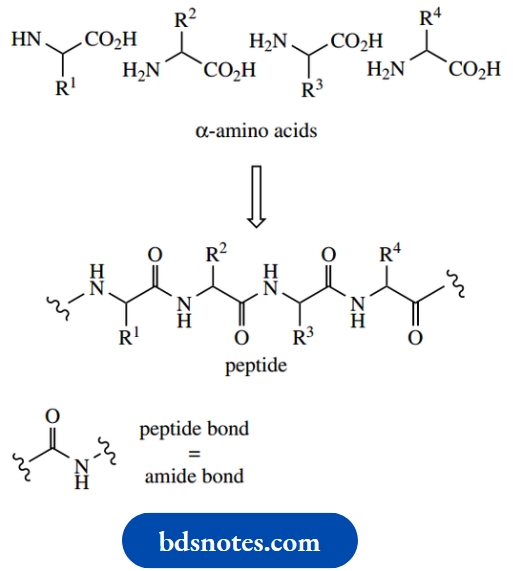
In biochemistry, the amide linkage is traditionally referred to as a peptide bond. Whether the resultant polymer is classified as a peptide or a protein is not clearly defined; generally, a chain length of more than 40 residues confers protein status, whereas the term polypeptide can be used to cover all chain lengths.
Proteins in all organisms are made up of the same set of 20 α-amino acids, though the organism is not necessarily capable of synthesizing all of these.
Some amino acids are obtained from the diet.
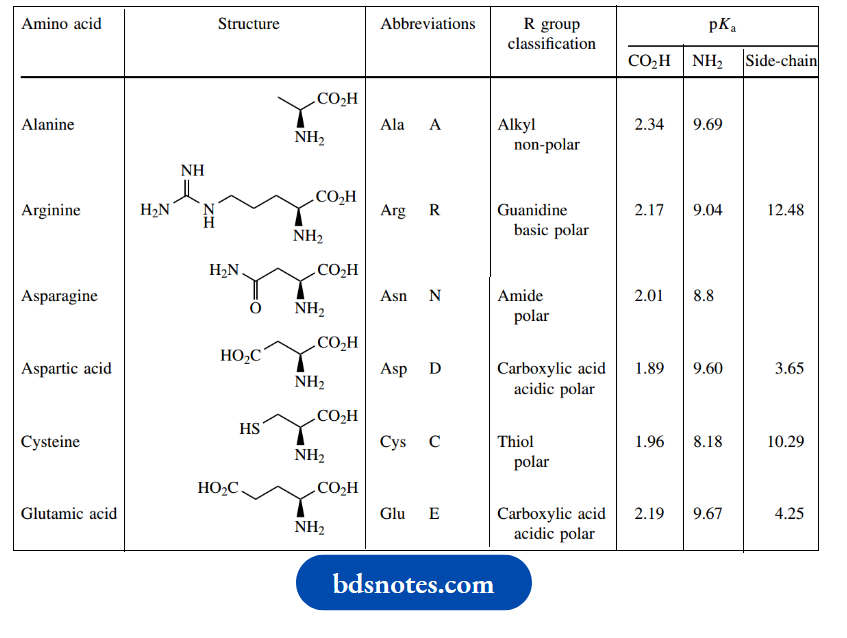
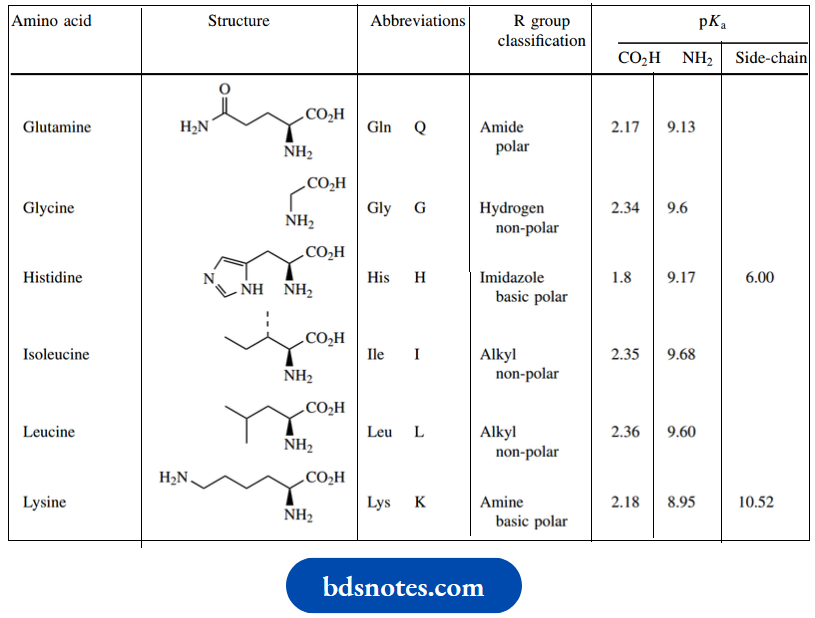
- The amino acids are combined in a sequence that is defined by the genetic code, the sequence of bases in DNA. gives the structures of these 20 amino acids together with the standard three-letter and one-letter abbreviations used to represent them.
- Proline is strictly an imino acid rather than an amino acid, but it is normally included as one of the 20 amino acids.
- The amino acids are also subclassified according to the chemical and physical characteristics of their R substituent.
- Since the polypeptide structure combines both the amino and carboxylic acid functions of an amino acid into amide linkages, the overall properties of the polypeptide are going to be defined predominantly by the characteristics of these R substituents.
The amino acid components of proteins have the L configuration, but many peptides are known that contain one or more D-amino acids in their structures.
D-Amino acids are not encoded by DNA, and peptides containing them are produced by what is termed ‘non-ribosomal peptide biosynthesis’. D-Amino acids generally arise by epimerization of L-amino acids. All the protein L-amino acids have the S configuration, except for glycine, which is not chiral, and L-methionine which is R, a consequence of the priority rules for systematic descriptors of configuration.
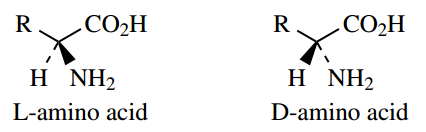
Amino acids structure and standard abbreviations:
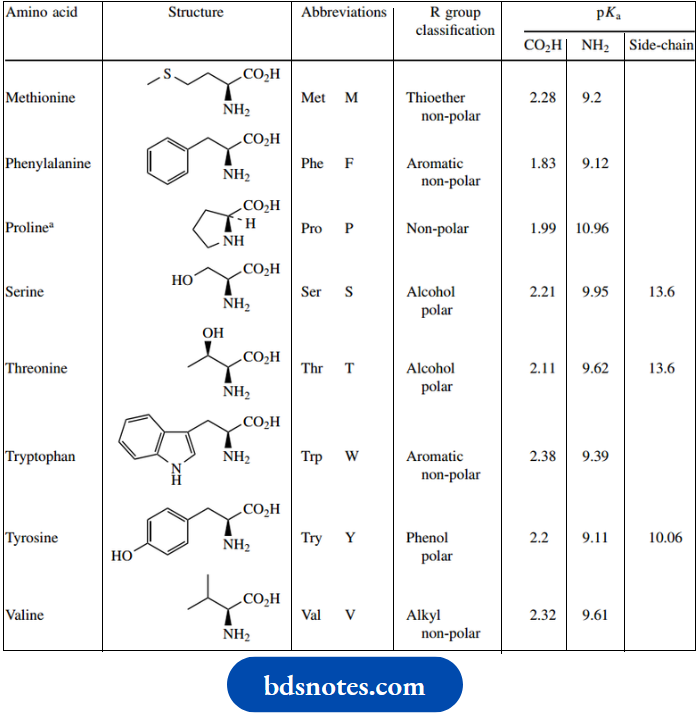
Two of the protein amino acids, threonine, and isoleucine, have two chiral centers; therefore, diastereoisomeric forms are possible. In proteins, each of these amino acids exists in a single diastereoisomeric form.
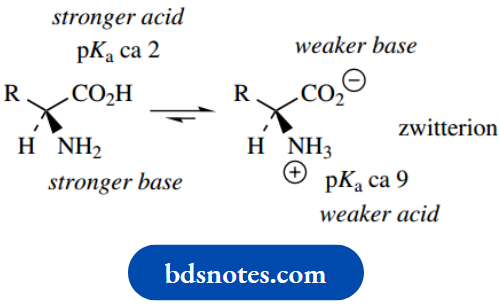
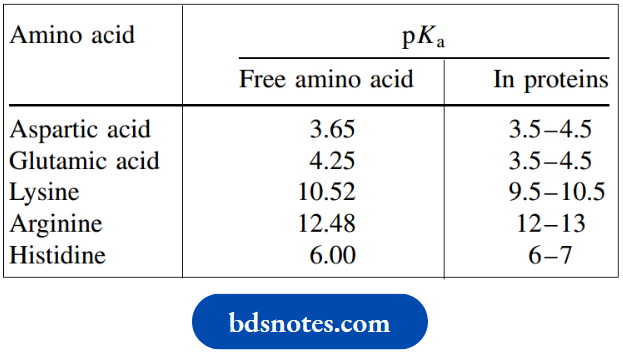
The pKa of the carboxylic acid group of amino acids is around 2, and that of the amino group (as conjugate acid) is around 9. As we saw this means that the carboxylic acid group (a stronger acid than the ammonium cation) will protonate the amino group (a stronger base than the carboxylate anion). At pH 7, therefore, amino acids with neutral R groups will exist mainly as the overall neutral, but doubly charged zwitterionic form (the weaker acid and weaker base).
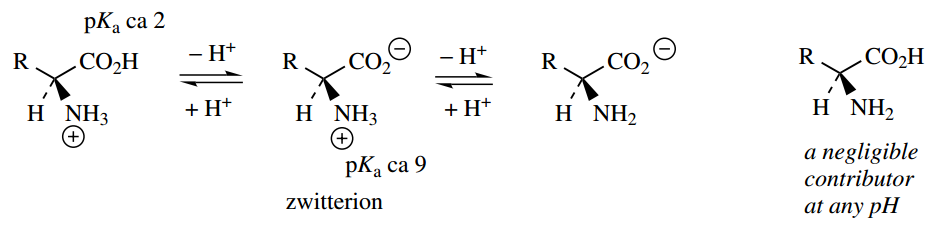
The carboxylate group becomes protonated as the pH decreases, whereas at higher pH the ammonium ion becomes deprotonated, in both cases yielding a singly charged species. The uncharged amino acid (as we almost always draw it!) is a negligible contributor at any pH
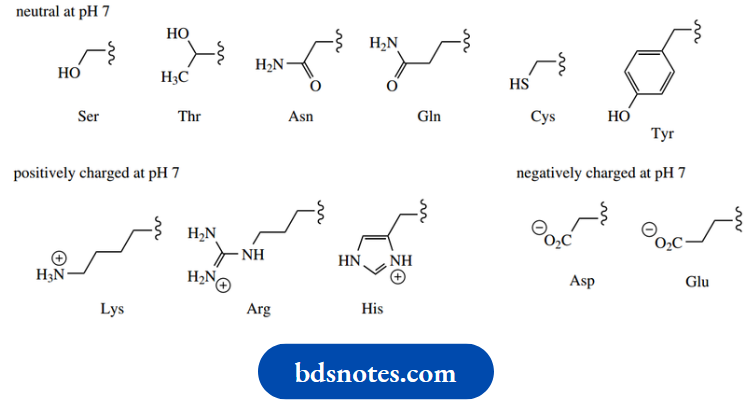
When the R group contains another ionizable group, the amino acid will have more than two dissociation constants. The carboxylic acid groups of aspartic acid and glutamic acid, the amine of lysine, and the guanidino group of arginine will all be ionized at pH 7, and the imidazole nitrogen of histidine will be partially protonated. However, neither the phenolic group of tyrosine nor the thiol group of cysteine will be ionized at this pH
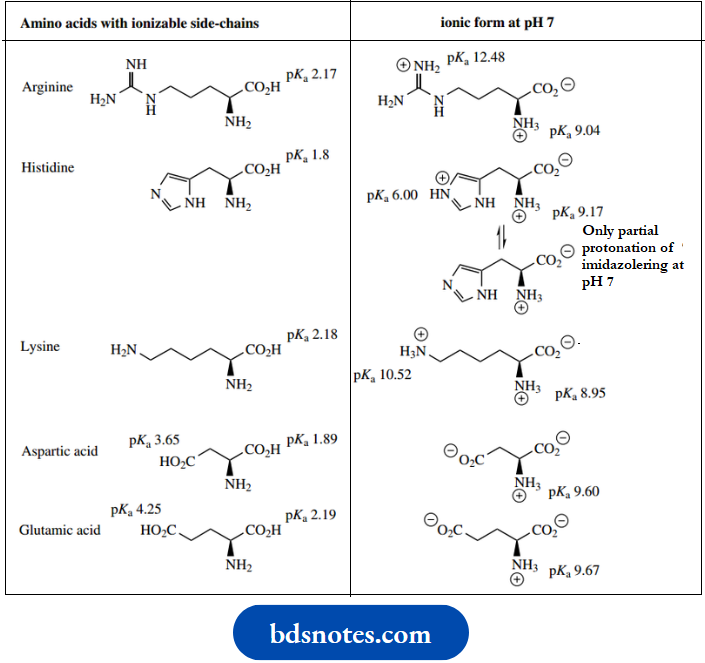
The ionization properties of side-chain substituents will usually carry through into the peptide or protein and influence the behavior of the polymer. However, the actual pKa values of the amino acid side-chains in the protein are modified somewhat by the position of the amino acid in the chain, and the environment created by other substituents. Typical pKa values
Note that the side chains of glutamine and asparagine are not basic; these side chains contain amide functions, which do not have basic properties. The heterocyclic ring in tryptophan can also be considered as non-basic since the nitrogen lone pair electrons form part of the aromatic π electrons and are unavailable for bonding to a proton
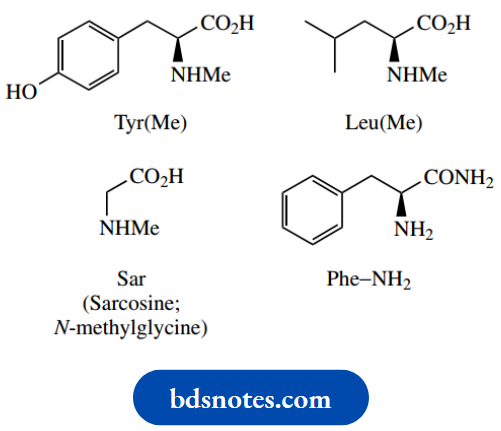
In addition to the 20 amino acids described, there are also a few amino acids quite frequently encountered that are not encoded by DNA. These are mainly found in peptides and are typically slightly modified versions of the common amino acids, such as N-methyl amino acids. These components are represented by an appropriate variation of the normal abbreviation,
Example:
N-methyl amino acids such as Tyr(Me) or Leu(Me), though N-dimethylglycine is often referred to as sarcosine (Sar)
pKa values for free and protein-bound amino acids:
A frequently encountered modification is the conversion of the C-terminal carboxyl into an amide. This is represented as Phe–NH2, for example, which must be considered carefully, and not be interpreted as an indication of the N-terminus.
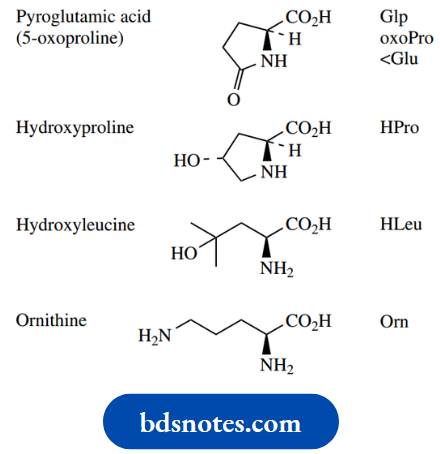
Some other variants are shown below, with their abbreviations. Pyroglutamic acid may be found where a terminal glutamic acid residue, linked to the chain through its carboxyl, forms a cyclic amide (lactam)
Some common amino acids not encoded by DNA
Peptides And Proteins
Although superficially similar, peptides and proteins display a wide variety of biological functions, and many have marked physiological properties. For example, they may function as structural molecules in tissues, as enzymes, as antibodies, or as neurotransmitters. Acting as hormones, they can control many physiological processes, ranging from gastric acid secretion and carbohydrate metabolism to growth itself. The toxic components of snake and spider venoms are usually peptides in nature, as are some plant toxins.
These different activities arise as a consequence of the sequence of amino acids in the peptide or protein (the primary structure), the three-dimensional structure that the molecule then adopts as a result of this sequence (the secondary and tertiary structures), and the specific nature of individual side-chains in the molecule. Many structures have additional modifications to the basic polyamide system shown, and these features may also contribute significantly to their biological activity.
The tripeptide formed from L-alanine, L-phenylalanine and L-serine by two condensation reactions is alanyl–phenylalanyl–serine, considering each additional amino acid residue as a substituent on the previous. This would be more commonly represented as Ala–Phe–Ser, using the standard three-letter abbreviations for amino acids
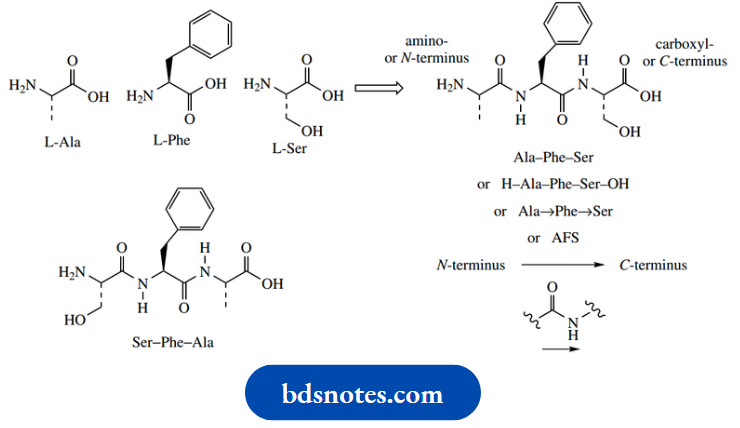
By convention, the left-hand amino acid in this sequence is the one with a free amino group, the N-terminus, and the right-hand amino acid has the free carboxyl, the C-terminus. Thus, Ser–Phe–Ala is different from Ala–Phe–Ser, and represents a quite different molecule. Sometimes, the termini identities are emphasized by showing H– and –OH; H– represents the amino group and –OH the carboxyl group. Some peptides are cyclic, and this convention can have no significance, so arrows are incorporated into the sequence to indicate peptide bonds in the direction CO→NH. As sequences become longer, one-letter abbreviations for amino acids are commonly used instead of the three-letter abbreviations, thus Ala–Phe–Ser becomes AFS.
Abbreviations assume the L-configuration applies throughout, and any D-amino acids would be specifically noted,
Example: Ala–D-Phe–Ser.
Glutathione:
Glutathione is an important tripeptide; but it is a slightly unusual one, in that it has an amide linkage that involves the γ- carboxyl of glutamic acid rather than a normal amide bond utilizing the C-1 carboxyl group. To specify this bonding, the glutathione structure is written as γ-Glu–Cys–Gly
Peptides and proteins may be hydrolyzed to their constituent amino acids by either acid or base hydrolysis . However, because of its nature, the amide bond is quite resistant to hydrolytic conditions, a very important feature for natural proteins. Hydrolysis of peptides and proteins, therefore, requires heating with quite concentrated strong acid or strong base. Neither acid nor base hydrolysis provides the ideal hydrolytic conditions, however, since some of the constituent amino acids are found to be sensitive to the reagents.
Acid hydrolysis is preferred, but the indole system of tryptophan is largely degraded in strong acid, and the sulfur-containing amino acid cysteine is also unstable. Serine, threonine, and tyrosine may also suffer partial degradation. Those amino acids containing amide side-chains,
Example:
Asparagine and glutamine, will be hydrolyzed further, giving the corresponding structures with acidic side-chains, namely aspartic acid and glutamic acid
The Molecular Shape Of Proteins: Primary Secondary And Tertiary Structures
Peptides and proteins are composed of amino acids linked together via amide (peptide) bonds, the amino group of one condensing with the carboxylic acid of another. The sequence of amino acids in a peptide or protein is closely controlled by genetic factors.
Some peptides are synthesized via a multi-functional enzyme complex (non-ribosomal peptide synthesis), whereas others, including the larger proteins, are produced on the ribosome, and the sequence can be related directly to the nucleotide sequence of DNA.
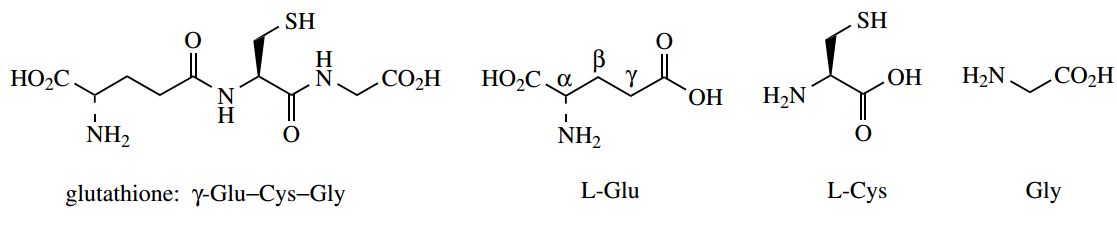
This amino acid sequence provides what we term the primary structure of the protein, although this term also includes the position of disulfide bridges, the result of covalent bonding between pairs of cysteine residues. Disulfide bridges produce cross-linking in the polypeptide chain.
This covalent bonding arises as a result of biochemical oxidation of the thiol groups in two cysteine residues, and it may also be achieved chemically with the use of mild oxidizing agents. This modification of thiol groups may thus loop a polypeptide chain or cross-link two separate chains.
It also significantly modifies the properties of a protein by removing two polar and potentially acidic (pKa 10.3) groups, replacing them with a nonpolar disulfide function. Under suitable hydrolytic conditions, a protein containing one or more disulfide bridges will yield cysteine residues still joined by this type of bonding. This amino acid ‘dimer’ is called cystine (Cys–Cys).
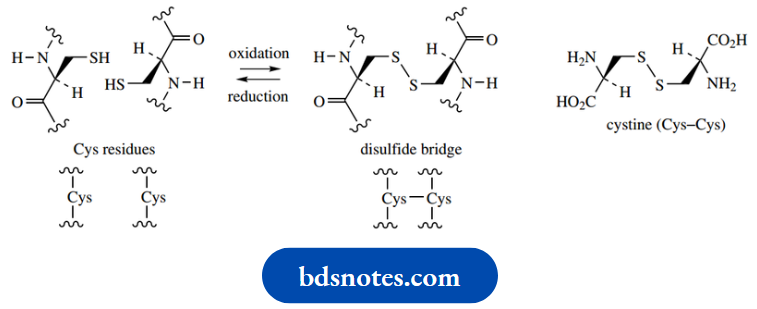
Because of the similarity in names, it is usual practice to differentiate them in speech by pronouncing cysteine as sis-tay-een, whereas cysteine is pronounced sis-teen. The disulfide bridge is easily formed and is just as easily broken. It may be cleaved to thiol groups by reduction with reagents such as sodium borohydride or by the use of other thiol reagents.
For example:
Mercaptoethanol (HSCH2CH2OH) is routinely used in protein analysis to help locate disulfide bridges through an equilibration reaction.
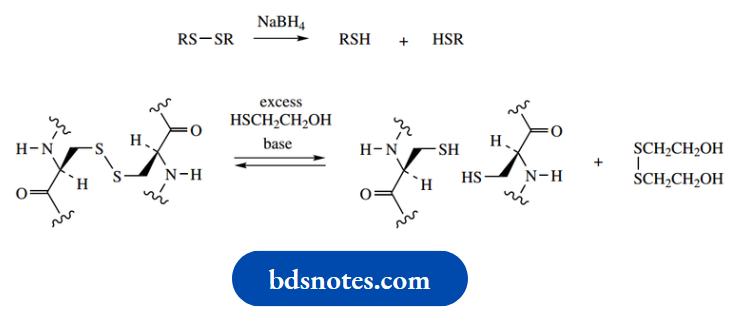
The mechanism for this reaction is shown below:
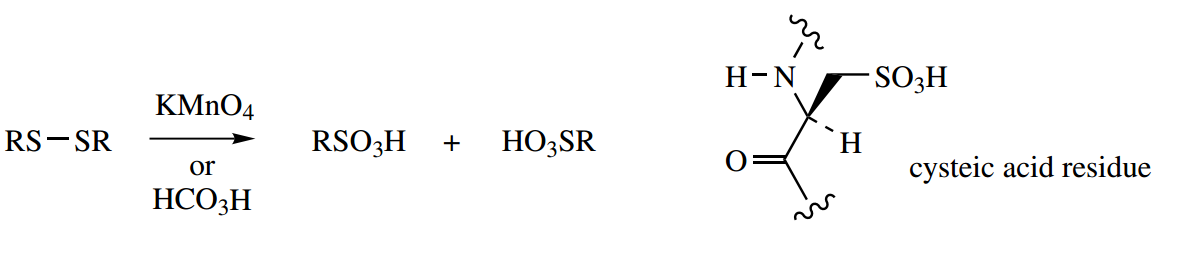
Oxidation with stronger oxidizing agents,
Example:
Potassium permanganate or performic acid, converts the disulfide to two molecules of a sulfonic acid, namely cysteic acid.
This reaction may be of value in sequence analysis, to determine the position of disulfide bridges (as opposed to unmodified cysteine residues) in the primary structure.
Disulfide bridges in insulin
The peptide hormone insulin is produced by the pancreas and plays a key role in the regulation of carbohydrate, fat, and protein metabolism.
- In particular, it has a hypoglycaemic effect, lowering the levels of glucose in the blood. A malfunctioning pancreas leads to a deficiency in insulin synthesis and the condition known as diabetes.
- This results in increased amounts of glucose in the blood and urine, diuresis, depletion of carbohydrate stores, and subsequent breakdown of fat and protein. Incomplete breakdown of fat leads to the accumulation of ketones in the blood, severe acidosis, coma, and death.
- Diabetes treatment requires daily injections of insulin; since insulin is a peptide, it would be degraded by stomach acid if taken orally. Insulin does not cure the disease, so treatment is lifelong.
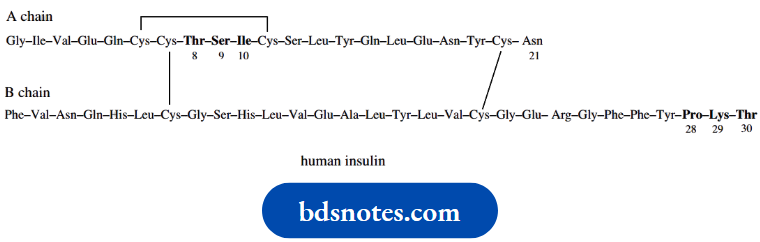
Human insulin:
Human insulin is composed of two straight-chain polypeptides joined by disulfide bridges.
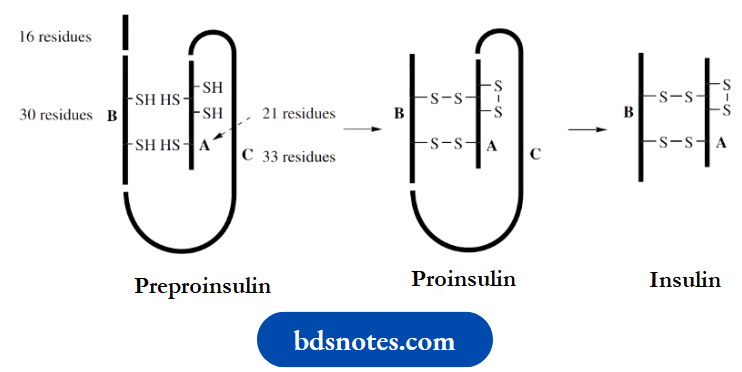
This structure is known to arise from a single straight-chain polypeptide, preproinsulin, containing 100 amino acid residues. This loses a 16-residue portion of its chain and forms proinsulin, in which disulfide bridges connect the terminal portions of the chain in a loop.
A central portion of the loop (the C chain) is then cleaved out, leaving the A chain (21 residues) bonded to the B chain (30 residues) by two disulfide bridges. There is also a third disulfide bridge interconnecting two cysteine residues in the A chain. This is the resultant insulin.
Mammalian insulins from different sources are very similar and may be used to treat diabetes. The compounds show variations in the sequence of amino acid residues 8–10 in chain A, and at amino acid 30 in chain B.
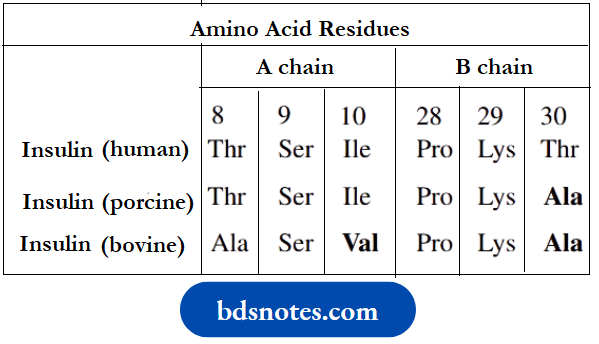
Porcine insulin and Bovine insulin for drug use are extracted from the pancreas of pigs and cattle respectively. More frequently, human insulin is now employed. This is produced by the use of recombinant DNA technology to obtain the two polypeptide chains, and then linking these chemically to form the disulfide bridges
Peroxides, including hydrogen peroxide (H2O2), can damage cells by causing unwanted oxidation reactions. The tripeptide glutathione (GSH) is able to participate in a cellular protection mechanism via its ability to form disulfide bridges.
In an enzymic reaction catalysed by glutathione peroxidase, GSH reacts with peroxides and becomes oxidized to form a dimer (GSSG) linked by a disulfide bridge.
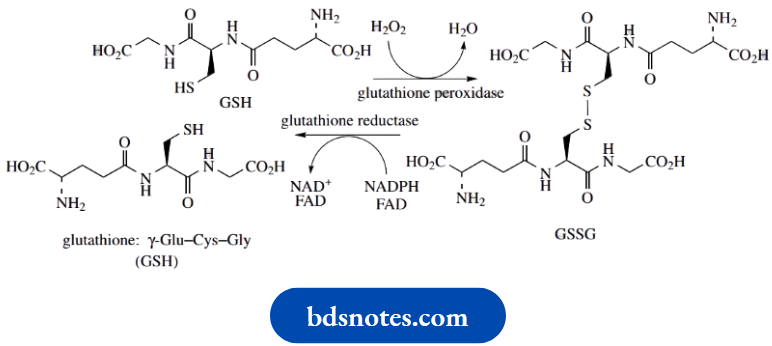
In so doing, it reduces the peroxide. In the case of H2O2, this generates water, whereas an organic peroxide would yield water and an alcohol
H2O2 + 2GSH → GSSG + 2H2O
ROOH + 2GSH → GSSG + ROH + H2O
In order maintain adequate levels of GSH, the oxidized dimer is then reduced back to the original thiol components. This is achieved using the enzyme GSH reductase in a reaction involving NADPH and FAD cofactors
Protein chains are not the sprawling, ill-defined structures that might be expected from a single polypeptide chain. Most proteins are compact molecules, and the relative positions of atoms in the molecule contribute significantly to its biological role. A particularly important contributor to the shape of proteins is provided by the peptide bond itself. Drawn in its simplest form, one might expect free rotation about single bonds, with a variety of conformations possible.
However there is resonance stabilization in an amide, via electron movement from the lone pair on the nitrogen to the carbonyl oxygen. We have already noted this type of resonance stabilization in amides , and also in esters. It was invoked in explaining reactivity and pKa values compared with other types of carbonyl compounds, and the non-basic behavior of the nitrogen atom in amides

To achieve this stabilization, the p orbital on nitrogen needs to be lined up with the carbonyl π bond. The immediate consequence of this are that five bonds in the peptide linkage must be coplanar. There is no free rotation about the N–C bond, because it is involved with a partial double-bond system. Of course, there are potentially two configurations with respect to this N–C bond, corresponding to cis and trans versions if it were a true double bond. It is not surprising that the transform is energetically favored, where we have the large groups, i.e. the rest of the chain, arranged to give minimum interaction.
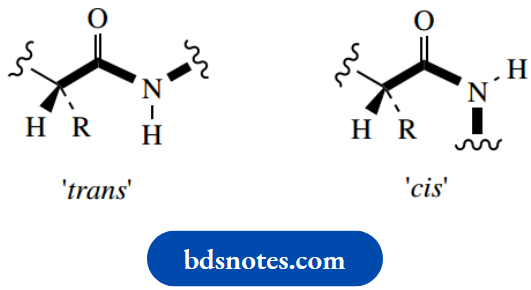
Zig-Zag conformation with main chain trans oriented
Rotational freedom about single bonds
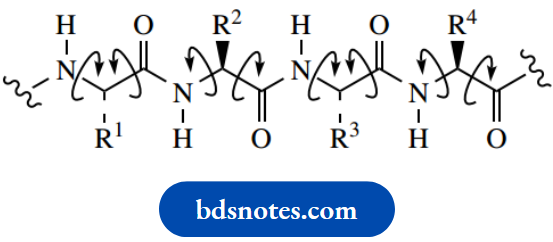
Rotation about the C–N bond is restricted
We now see good reasons for drawing a polypeptide chain in the accepted zigzag form. Note, however, that the remaining single bonds in the chain do allow rotation, and this is why we see a wide variety of different shapes in proteins. We can also appreciate that, in general, the carbonyl groups and N–H groups are all going to be coplanar. This leads to the secondary structure of proteins, a consequence of hydrogen bonding possible because of the regular array of carbonyl and N–H groups.
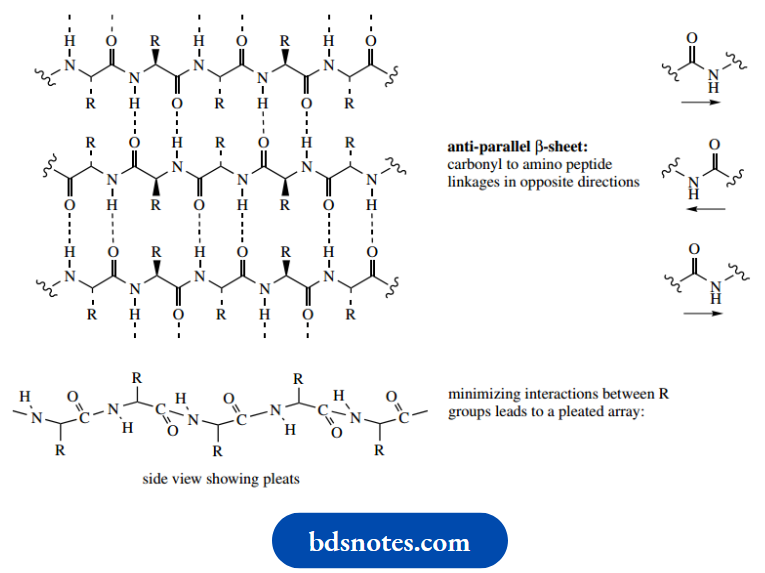
The most easily appreciated example of this is the β-pleated sheet, one of the ways in which a polypeptide chain can be arranged in an ordered fashion. Polypeptide chains align themselves side-by-side, stabilized by multiple hydrogen bonding, allowed by the regular array of carbonyl and N–H bonds.
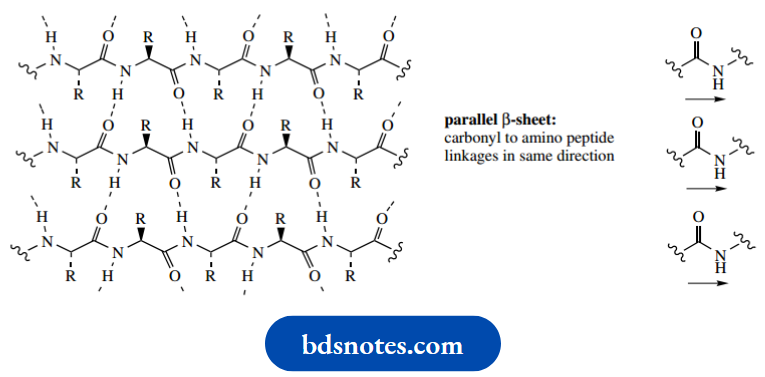
The alignment may be parallel, such that all the carbonyl to amino peptide linkages are in the same direction, or antiparallel where carbonyl to amino peptide linkages run in opposite directions. Although there are going to be groups of atoms that are planar, the whole chain is not planar. Instead, these arrangements take up a pleated array, which helps to minimize interaction between the large R groups.
Parallel sheets may involve different polypeptide chains via intermolecular hydrogen bonds, or the same chain via intramolecular hydrogen bonds. For intramolecular interactions, the chain length needs to be substantial, i.e. proteins rather than peptides, and it will be necessary for the polypeptide chain to bend back upon itself. The commonest type of arrangement for bending back a chain is called the β-turn, resulting in hydrogen bonding between residues n and n + 3.
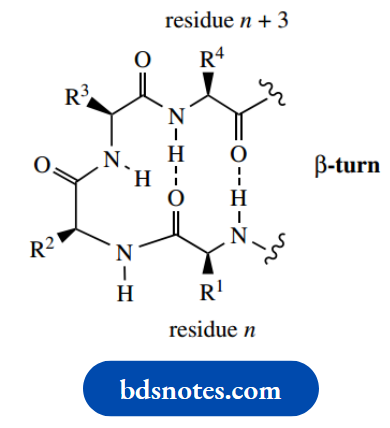
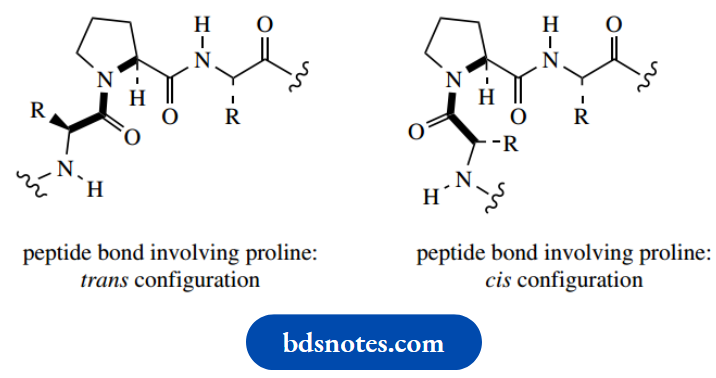
Note also that the imino acid proline must distort the regular zigzag array and introduce a bend into the chain; two configurations may be considered,
And both are possible since there is little difference in energy between them. Further, there is no N–H in proline, so hydrogen bonding involving this residue is not possible.
Alpha (α) Helix:
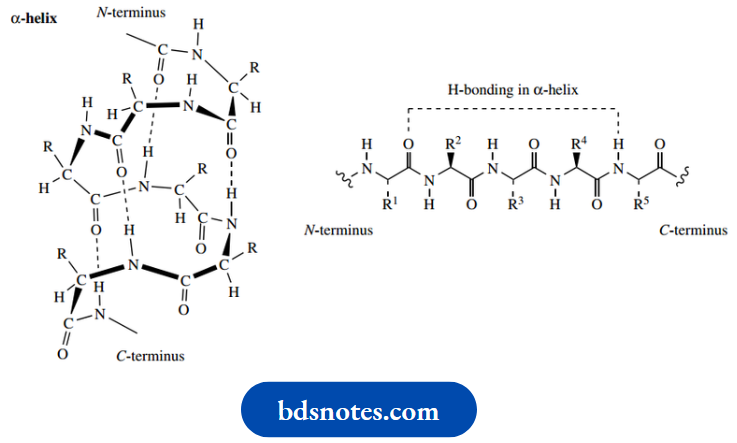
Whereas the β-pleated sheet provides a particularly nice and easily appreciated example of regular hydrogen bonding in polypeptide chains, the most common arrangement found in proteins is the α-helix. Do not worry about the α or β used in the nomenclature; this merely signifies that the helical structure ( α) was deduced earlier than that of the pleated sheet ( β).
- The α-helix is a right-handed helix, an ordered coil array stabilized by hydrogen bonding between carbonyl and N–H groups in the same chain. In a right-handed helix, movement along the chain involves a clockwise or right-handed twist, just like an ordinary screw – you turn the screwdriver clockwise.
- Hydrogen bonds link carbonyl and N–H bonds in amino acids that are separated by three other residues, and each turn of the helix is found to take up 3.6 amino acid residues. Note that all of the R groups, which in the majority of amino acids are quite bulky, are accommodated on the outside of the helix. Only the imino acid proline cannot fit into the regular array of the α-helix.
- We have just seen that proline must distort the regular array and introduce a bend into the chain, and that there is no N–H for hydrogen bonding. The secondary structure is responsible for some of the physical properties of proteins.
For example:
Structural proteins such as α-keratins in skin and hair are fibrous in nature and have good elastic properties. This elasticity can be traced back to the α-helix structure, in which weak hydrogen bonds are parallel to the direction of stretching, i.e. a spring-like structure.
On the other hand, proteins such as α-fibroin in silk are relatively inelastic since they contain the β-pleated sheet structure, where extension is resisted by the full strength of covalent bonds.
- In the β-pleated sheet, the weaker hydrogen bonds would be perpendicular to the direction of stretching.
- However, most proteins have a roughly spherical shape and are thus termed globular proteins. Globular proteins are likely to contain portions of the polypeptide chain that adopt both helical and sheet structures.
- In contrast to structural proteins like α-keratin or α-fibroin, the helical or sheet fragments in globular proteins are rather short and do not extend far without a change in direction. The overall folding of the polypeptide chain and the three-dimensional arrangement produced provide the tertiary structure of the protein.
The globular shape is facilitated, however, by some other non-covalent interactions.
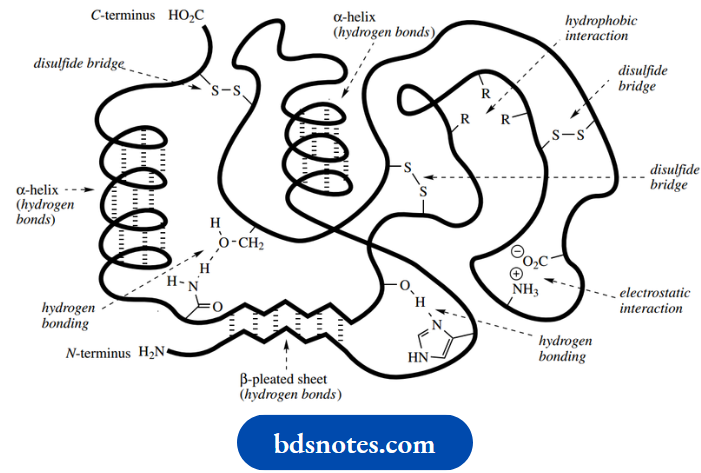
Terity structure intramolecular interaction
The conformation of a protein is determined and maintained by a range of intramolecular interactions that arise from some of the amino acid sidechain substituents. These are non-covalent interactions, although we must also remember that a disulfide bond formed between pairs of cysteine residues also contributes to the three-dimensional shape of the protein by providing cross-chain links.
Non-covalent interactions are relatively weak when compared with covalent bonds, but there are usually many such interactions in a protein, and, overall, a substantial degree of stabilization is attained.
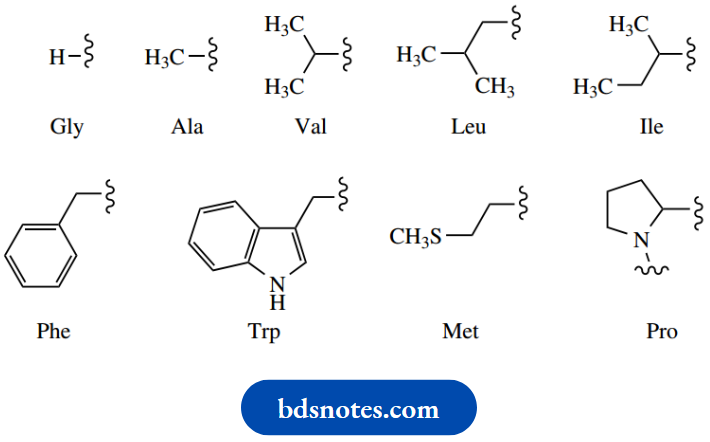
1. Hydrophobic interactions:
Hydrophobic interactions. Many of the amino acids contain side-chains that are hydrocarbon in nature, either aliphatic or aromatic
- In an aqueous environment, such groups are hydrophobic, and any folding in the protein that concentrates these hydrophobic areas together, and away from water, is going to be favored.
- This hydrophobic effect tends to encourage the burying of hydrophobic side chains in the interior of the protein and provides a significant part of the driving force for protein folding.
- Because so many amino acids have hydrocarbon side chains, not all can be accommodated in the interior, and hydrophobic groups tend to be about equally distributed between the interior and the surface of the molecule. On the other hand, hydrophilic sidechains are more likely to be found on the surface of a protein.
Non-polar, hydrophobic side-chains:
Polar, hydrophilic side-chains:
2. Hydrogen bonds:
Hydrogen bonds are responsible for the fundamental characteristics of the α-helix and β-pleated sheet; in addition, they contribute to the final shape of a globular protein. Hydrogen bonds can form in a variety of ways, involving the peptide backbone, polar amino acid side-chains, and also water molecules. Some of the hydrogen bonding situations are shown below; others can be deduced.
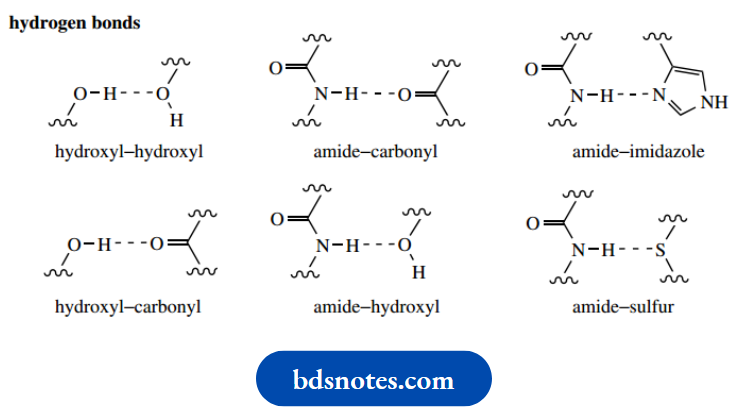
It should be appreciated that amino acids such as serine, threonine, tyrosine, and cysteine all contain side-chain alcohol or thiol groups that may participate in hydrogen bonding and stabilize a particular protein conformation.
3. Ionic bonds:
Ionic bonds. Carboxylic acid groups in amino acid side chains (aspartic acid, glutamic acid) will be ionized at pH 7, and nitrogen-containing groups (lysine, arginine) will similarly be protonated. Isolated hydrophilic groups such as these will never be found in the hydrophobic interior of a globular protein but will be positioned on the outer surface in proximity to water molecules.
- However, pairs of oppositely charged ions may be found in the interior since electrostatic interactions can provide the necessary attractive forces.
- Thus, non-covalent hydrophobic interactions, hydrogen bonds, and electrostatic bonds all contribute to the overall shape of a protein .
- As we shall see, many pertinent properties of a protein are then provided by the appropriate combination of the remaining amino acid side chains that reside on the surface, allowing specific binding to various molecules. This is the essence of enzymic activity and drug-receptor interactions.
- With some proteins, there is a further level of structure, i.e. quaternary structure, which may need to be considered. This arises because two or more protein chains aggregate to form the normal functional protein.
- Typically, the separate subunits, often the same, are held together by non-covalent interactions, as seen in the consideration of tertiary structure. Not all proteins have quaternary structure.
Protein binding sites
From our considerations above, we can see just how important the interactions of various amino acid side chains are to the structure and shape of proteins.
- These interactions tend to be located inside the protein molecule, stabilizing a particular conformation and generating the overall shape as in a globular protein.
- However, it is obvious that there are also going to be many amino acid side chains located on the surface of a protein, and these in turn will be capable of interacting with other molecules.
- These interactions will be intermolecular, rather than the intramolecular interactions that contribute to protein structure.
- Because there will also be several amino acid side-chains close, a combination of interactions may generate a site that has a specific shape, and a specific array of forces. The site will then be able to bind a particular molecule or part of a molecule.
- These amino acid side chains, therefore, allow strong binding to specific molecules, and the particular molecule can be regarded as being a perfect fit both in terms of the forces involved and in a geometric sense.
As a result, even a small change in the structure of the molecule could well spoil the fit and upset the interplay of forces – the binding is powerful and reasons by specific.
The sole function of many proteins is simply to bind other molecules. Thus, immunoproteins can bind to alien molecules (antigens) and destroy their activity by complexation. Some proteins act as hormones, influencing metabolic rates by binding to another molecule or structure. Binding to a protein may be the way an organism transports a molecule or even an ion to a different part of the organism
For example:
Hemoglobin transports molecular oxygen around the body, and cytochromes transport electrons within a cell.
- Drug–receptor interactions and enzymic activity are also a consequence of the binding of molecules to a protein.
- A substance that elicits a particular biological response by interaction at a receptor site is termed an agonist.
- An antagonist is a substance that inhibits the action of an agonist, often by competing for the same receptor site. The binding site on an enzyme is usually termed the active site.
Pain relief: morphine mimics natural peptides called endorphins
Although the pain-killing properties of the opium alkaloid morphine and related compounds have been known for a considerable time, the existence of endogenous peptide ligands for the receptors to which these compounds bind is a more recent discovery. It is now appreciated that the body produces a family of endogenous opioid peptides that bind to a series of receptors in different locations.
These peptides include enkephalins, endorphins, and dynorphins, and are produced primarily, but not exclusively, in the pituitary gland. The pentapeptides Metenkephalin and Leu-enkephalin were the first to be characterized. The largest peptide is β-endorphin (‘endogenous morphine’), which is several times more potent than morphine in relieving pain.

Although β-endorphin at its N-terminus contains the sequence for Met-enkephalin, the latter peptide and Leu-enkephalin are derived from a larger peptide, namely proenkephalin A, and β-endorphin itself is formed by cleavage of the peptide pro-opiomelanocortin. The proenkephalin. A structure contains four Met-enkephalin sequences and one of Leu-enkephalin. The dynorphins,
Example: Dynorphin A, is also produced by cleavage of a larger precursor, namely proenkephalin B (prodynorphin), and all contain the Leu-enkephalin sequence.
- Some 20 opioid ligands have now been characterized.
- When released, these endogenous opioids act upon specific receptors, inducing analgesia and depressing respiratory function and several other processes.
- The individual peptides have relatively high specificity towards different receptors.
- It is known that morphine, β-endorphin, and Met-enkephalin are agonists for the same site. The opioid peptides are implicated in analgesia brought about by acupuncture since opiate antagonists can reverse the effects.
- The hope of exploiting similar peptides as ideal, non-addictive analgesics has yet to be attained; repeated doses of endorphin or enkephalin produce addiction and withdrawal symptoms.
A common structural feature required for centrally acting analgesic activity in opioids is the combination of an aromatic ring and a piperidine ring that maintains the stereochemistry at the chiral center, as shown below.
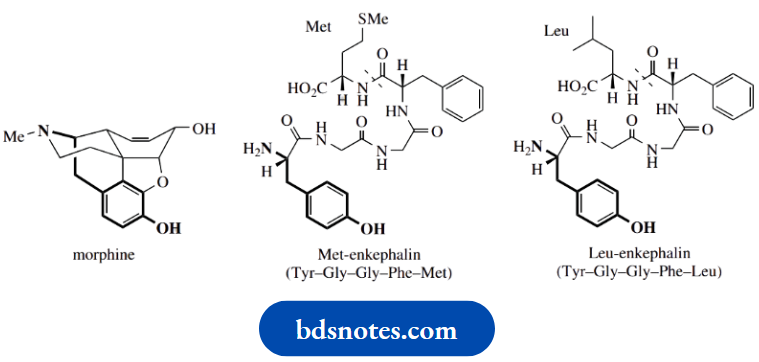
The three-dimensional disposition of the nitrogen function to the aromatic ring allows morphine and other analgesics to bind to a pain-reducing receptor in the brain. The terminal tyrosine residue in the natural agonist’s Met-enkephalin and Leu-enkephalin is mimicked by portions of the morphine structure
The Chemistry Of Enzyme Action
Enzymes are proteins that act as biological catalysts. They not only bind molecules but also provide a special environment in which the molecules are chemically modified.
- Enzymes cannot promote a reaction that is not energetically favorable, but by binding the reagents in the necessary orientation and nearby, they significantly reduce the activation energy for the transformation.
- By the amino acid side chains, enzymes can provide a highly specific binding site for their substrates, anchoring these reagents in an appropriate manner and suitable proximity so that a reaction can occur, as well as providing any necessary acid or base catalyst for the reaction. In some cases, a further reagent, a coenzyme, must also be bound for the reaction to occur.
- After the reaction is completed, the products are then released from the enzyme, so that the reaction can be repeated on further molecules of the substrates. These amino acid side chains confer immense catalytic power to the enzyme, giving it the ability to carry out reactions that organic chemists can only dream of.
Several types of bonding might be utilized to bind substrates to enzymes.
These are analogous to the bondings that contribute to the secondary and tertiary structures of a protein and include the following noncovalent interactions:
- Electrostatic bonding, via acids, bases, phosphates
- Hydrophobic interactions, via alkyl groups, aromatic rings
Hydrogen bonding, via NH, OH, SH, C=O. In addition, we can meet examples of covalent bonding that are responsible for binding a substrate, where a functional group in the substrate reacts chemically with a protein side-chain functional group.
Two important reactions are:
- Imine formation, via NH2 and C=O;
- Thioester formation, via SH and C=O.
A large proportion of the substrates used in intermediary metabolism are in the form of phosphates.
Phosphates are favored in nature since they usually confer water solubility on the compound, and they provide a functional group that can bind to enzymes through simple electrostatic bonding.
In many cases, the phosphate group may also feature as a chemically reactive functional group – phosphates are good leaving groups. We have considered the structures of the various amino acids in terms of polarity, basicity, acidity, etc.
Here is a useful reminder of the important functionalities that are pertinent to enzyme action
- Amino acids with hydroxyl groups: Ser, Thr, Tyr
- Amino acids containing thiol (sulfhydryl) groups: Cys
- Amino acids with acidic groups: Asp, Glu
- Amino acids with basic groups: Lys, Arg, His
It is not sensible to try to cover a wide range of enzymic reactions to demonstrate how amino acid side chains are responsible for the chemical changes the enzyme brings about. Instead, one or two suitable examples will suffice to illustrate the general principles.
Acid-base catalysis
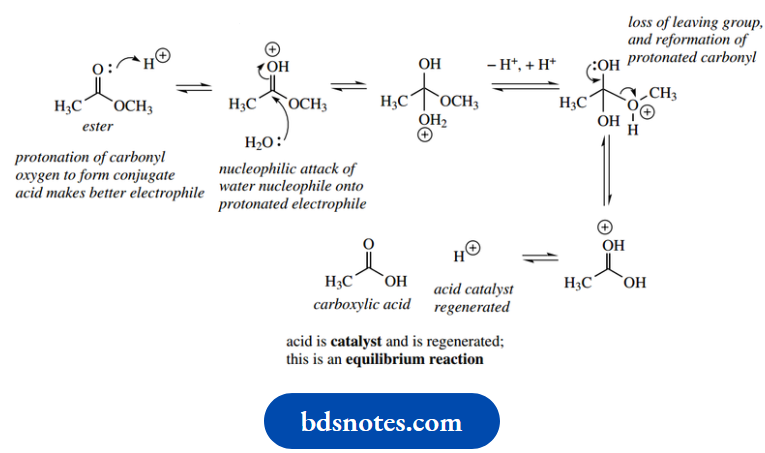
Let us first remind ourselves of the two mechanisms for hydrolysis of an ester, namely acid-catalysed hydrolysis and base-catalyzed hydrolysis
Acid-catalyzed hydrolysis of esters:
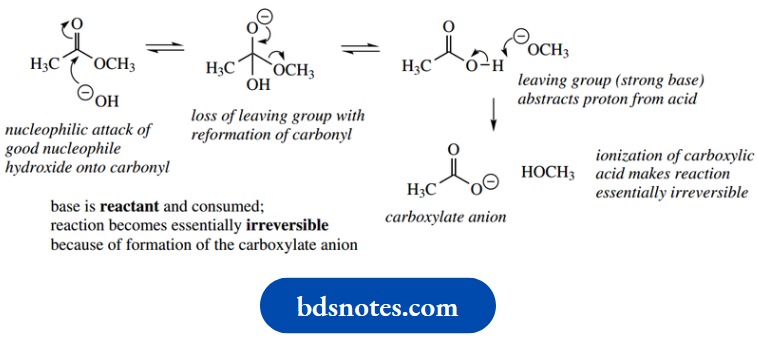
Base-catalyzed hydrolysis of esters:
Since enzymic reactions proceed in aqueous solution at pH 7 or thereabouts, where the concentrations of hydronium and hydroxide ions are both approximately 10−7 M, we need alternatives to strong acids and strong bases to formulate comparable enzymic mechanisms.
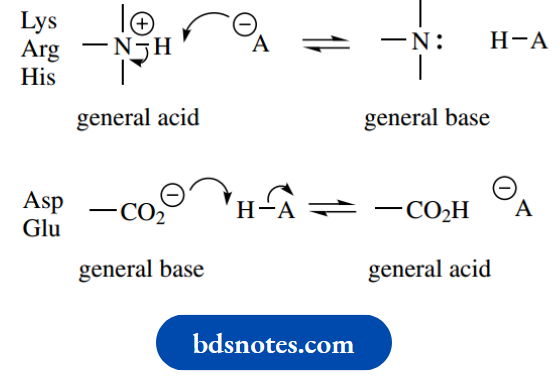
- Such reagents are provided by those amino acid side chains that are ionized at pH 7. Thus, the capacity for acid or base catalysis is built into the active site of many enzymes.
- Furthermore, the effective concentration of these groups at the active site is high, making them very effective acid and base catalysts. These donors and acceptors of protons are called general acid catalysts and general base catalysts respectively.
- The amino acids in question are the basic amino acids lysine, arginine, and histidine, and the acidic amino acids aspartic acid and glutamic acid.
- The side-chain functions of these amino acids, ionized at pH 7, act as acids or bases.
In a reverse sequence, protons may be acquired or donated to regenerate the conjugate acids and conjugate bases.
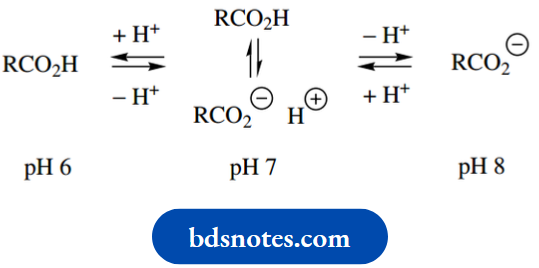
The most effective acid-base catalyst is one whose pKa is 7.0, since at pH 7.0 the concentrations of acid and conjugate base are equal. With just a slight decrease in pH it would become protonated and function as a general acid catalyst, whereas with a slight increase in pH, it would become unprotonated and, therefore, a general base catalyst.
Enzymes are active over a limited pH range; the pH value of maximum activity is known as the pH optimum, and this is characteristic of the enzyme. It typically reflects the pH necessary to achieve the appropriate ionization of amino acid side chains at the active site.
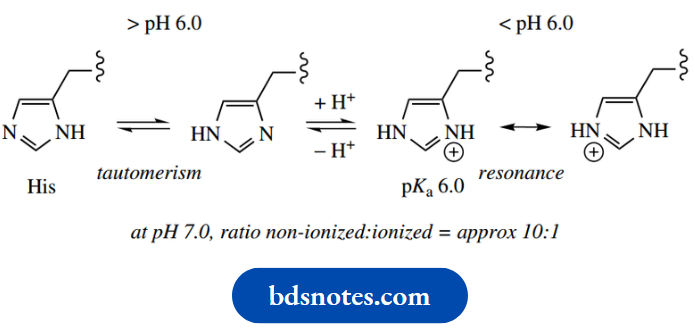
- The side-chain of histidine has a pKa value of 6.0; above pH 6.0, the imidazole ring acts as a proton acceptor or general base catalyst, whereas below pH 6.0 it is protonated and can act as a proton donor or general acid catalyst.
- At pH 7.0, the imidazole ring can be considered as partially protonated, since both ionized and non-ionized forms are present in the ratio of about 1: 10. Consequently, we find that the imidazole ring of histidine participates in acid-base catalysis in many enzymes.
- As we mentioned earlier, the pKa values of histidine side chains in a protein are modified by the neighboring amino acid residues and are typically in the range 6–7. These values provide a level of ionization somewhere between 9 and 50%, depending upon the protein.
- Remember that tautomerism can occur in imidazole rings When we meet structures for the amino acid histidine, we may encounter either of the tautomeric forms shown. Do not think there is a discrepancy in structures.
On the other hand, we can write resonance structures for the protonated ring, the imidazolium cation
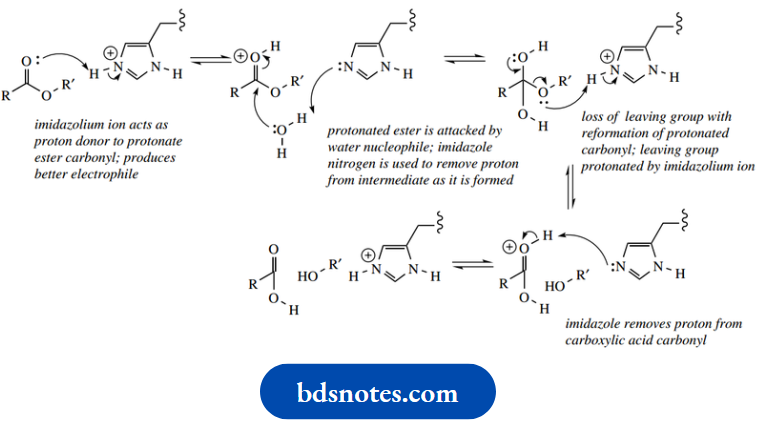
Now let us see how the imidazole grouping of histidine can be involved in the general acid-catalyzed and general base-catalyzed hydrolysis of esters by enzymes. In essence, the chemical and enzymic reactions are very similar.This is to be expected since enzymes can only catalyze an energetically favorable reaction. The role of acid or base is largely achieved in the enzyme reactions by implicating the imidazolium/imidazole system
General Acid-catalyzed ester hydrolysis:
General Base-catalyzed ester hydrolysis:
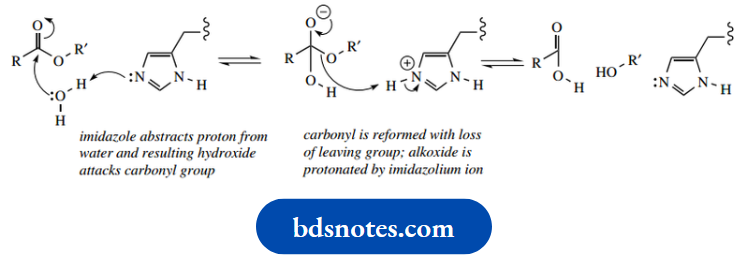
In the general acid-catalyzed mechanism, the imidazolium ion acts as a proton donor to protonate the carbonyl oxygen, thus producing a better electrophile. The protonated ester is then attacked by a water nucleophile, after which the imidazole nitrogen removes the now unwanted proton from the nucleophile; the imidazole nitrogen consequently becomes reprotonated.
The carboxylic acid is then formed via the loss of the leaving group, and this is facilitated by protonation so that the leaving group is an alcohol rather than an alkoxide.
The imidazolium proton is again a participant in this process.
- Finally, abstracting the proton from the protonated carbonyl regenerates the imidazolium ion. As in the chemical reactions, the general base-catalyzed process is mechanistically rather simpler. The imidazole nitrogen acts as a base to remove a proton from water, generating hydroxide that attacks the carbonyl.
- Subsequently, the alkoxide leaving group is reprotonated by the imidazolium ion
- However, not included in the above mechanisms are other amino acid side chains at the active site, whose special role will be to help bind the reagents in the required conformation for the reaction to occur.
- Examples of such interactions are found with acetylcholinesterase and chymotrypsin, representatives of a group of hydrolytic enzymes termed serine hydrolases, in that a specific serine amino acid residue is crucial for the mechanism of action.
- The proposed enzyme mechanisms just described, and those that follow, are depicted to show how certain amino acid residues become involved. Remember that the enzyme active site is three-dimensional, whereas our representation is only two-dimensional.
This means that bond angles and bond lengths sometimes look a little odd and distorted. However, such imperfect representations are easier to follow than if we had provided pictures that tried to emulate three-dimensional views
Acetylcholinesterase, a serine esterase
Acetylcholine:
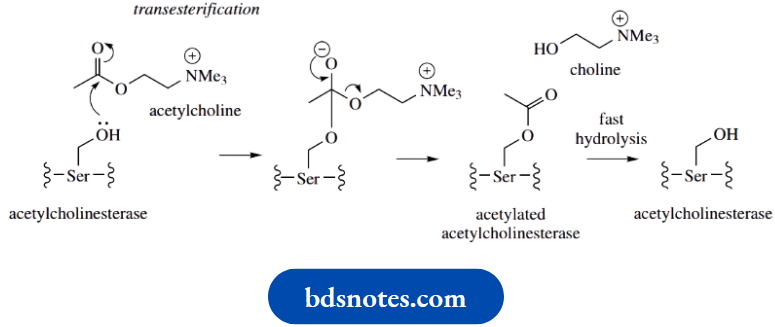
Acetylcholine is a relatively small molecule that is responsible for nerve-impulse transmission in animals. As soon as it has interacted with its receptor and triggered the nerve response, it must be degraded and released before any further interaction at the receptor is possible. Degradation is achieved by hydrolysis of acetate and choline by the action of the enzyme acetylcholinesterase, which is located in the synaptic cleft. Acetylcholinesterase is a serine esterase that has a mechanism similar to that of chymotrypsin
Hydrolysis involves nucleophilic attack by the serine hydroxyl onto the ester carbonyl. This leads to the transfer of the acetyl group from acetylcholine to the enzyme’s serine hydroxyl, i.e. formation of a transient acetylated enzyme, and the release of choline. . Hydrolysis of the acetylated enzyme then occurs rapidly, releasing acetate and regenerating the free enzyme.
The active site of the enzyme contains two distinct regions: an anionic region that contains a glutamic acid residue, and a region in which a histidine imidazole ring and a serine hydroxyl group are particularly important.
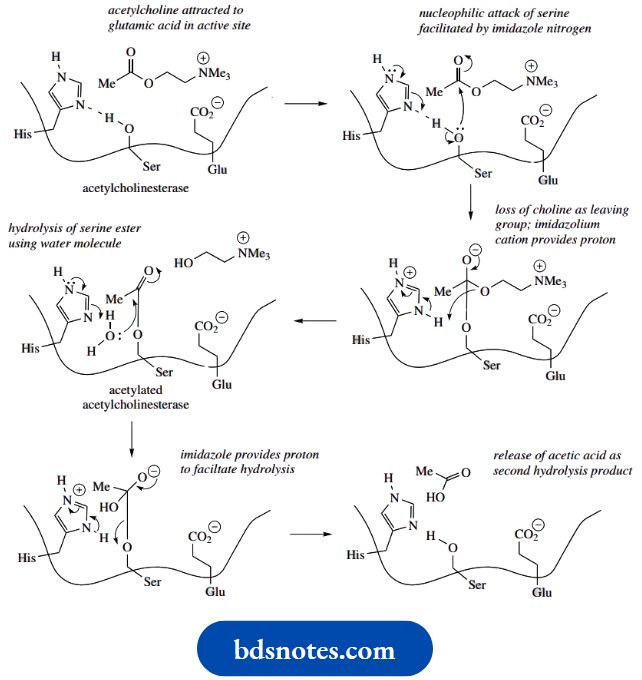
It is easy to see that the glutamic acid side-chain, ionized at pH 7, can attract the positively charged acetylcholine using ionic interactions. This allows binding and locates the ester function close to the serine side chain and the imidazole ring.
- Serine itself would be insufficiently nucleophilic to attack the ester carbonyl, so the reaction is facilitated by the participation of the imidazole ring of histidine. The basic nitrogen in this residue is oriented so that it can remove a proton from the serine hydroxyl, increasing nucleophilicity and allowing an attack on the ester carbonyl.
- This leads to the formation of the transient acetylated enzyme and the release of choline.
- Hydrolysis of the acetylated enzyme utilizes water as a nucleophile, but again involves the imidazole ring, and regenerates the free enzyme.
- Not included in this simplified description of the active site is the important role played by another amino acid residue.
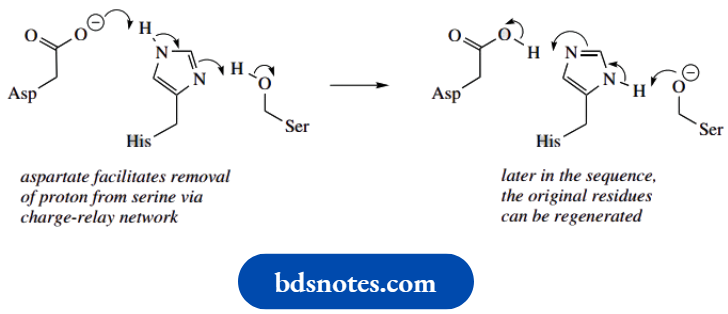
- The basicity of the histidine nitrogen is increased because of the proximity of a neighboring aspartate residue. This facilitates the removal of a proton from the active site serine.
- The relationship of these three residues provides a charge-relay network, in which a charge, here a proton, is effectively passed from one molecule to another.
- In due course, the groups can be restored to their original nature by a reversal of the sequence. We shall see this feature again with chymotrypsin below.
Aspartate−histidine−serine charge relay network
Acetylcholinesterase:
Acetylcholinesterase is a remarkably efficient enzyme; turnover has been estimated as over 10 000 molecules per second at a single active site. This also makes it a key target for drug action, and acetylcholinesterase inhibitors are of considerable importance. Some natural and synthetic toxins also function by inhibiting this enzyme
Chymotrypsin and other serine proteases
The serine proteases cleave amide (peptide) bonds in peptides and have a wide variety of functions, including food digestion, blood clotting, and hormone production. They feature as one of the best-understood groups of enzymes as far as the mechanism of action is concerned.
We can ascribe a function to many of the amino acid residues in the active site, and we also understand how they determine the specificity of the various enzymes in the group.
Example:
Chymotrypsin cleaves peptides on the C-terminal side of aromatic amino acid residues phenylalanine, tyrosine, and tryptophan,
And to a lesser extent some other residues with bulky side chains,
Example:
Leu, Met, Asn, Gln. On the other hand, trypsin cleaves peptides on the C-terminal side of the basic residues arginine and lysine.
Elastase usually catalyzes the hydrolysis of peptide bonds on the C-terminal side of neutral aliphatic amino acids, especially glycine or alanine. These three pancreatic enzymes are about 40% identical in their amino acid sequences, and their catalytic mechanisms are nearly identical.
Cleavage sites for serine proteases:
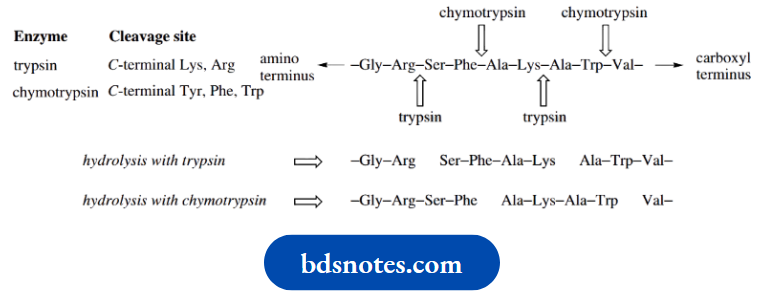
Because of their known specificity, these enzymes, especially trypsin, and chymotrypsin, have been widely utilized in helping to determine the amino acid sequences of peptides (see Hydrolysis using these enzymes generate smaller peptide fragments via hydrolysis at specific amino acid residues. The shortened chains can then be sequenced and, with a little logic and reasoning, the order in which they are attached in the larger peptide can be deduced.
The differences in specificity are known to be a consequence of the amino acid sequences at the binding sites of the enzymes; these sequences are almost identical.
- Thus, trypsin and chymotrypsin differ in only one residue at the binding site.
- This residue is located in a so-called ‘pocket’ in the binding site and allows the binding of substrates containing specific amino acids in their structure.
- The pocket in chymotrypsin contains a serine residue, and the pocket provides a hydrophobic environment allowing the binding of aromatic amino acid side-chains.
- On the other hand, the trypsin pocket has an aspartate residue and binds substrates with the positively charged amino acid residues lysine and arginine.
For simplicity, the additional binding resulting from the pocket residues has not been included in the mechanistic interpretation below.
The mechanism of action of chymotrypsin can be rationalized as follows.
- The enzyme-substrate complex forms, with the substrate being positioned correctly through hydrogen bonding and interaction with the ‘pocket’ as described above.
- The nucleophilicity of a serine residue is only modest, but here it is improved by
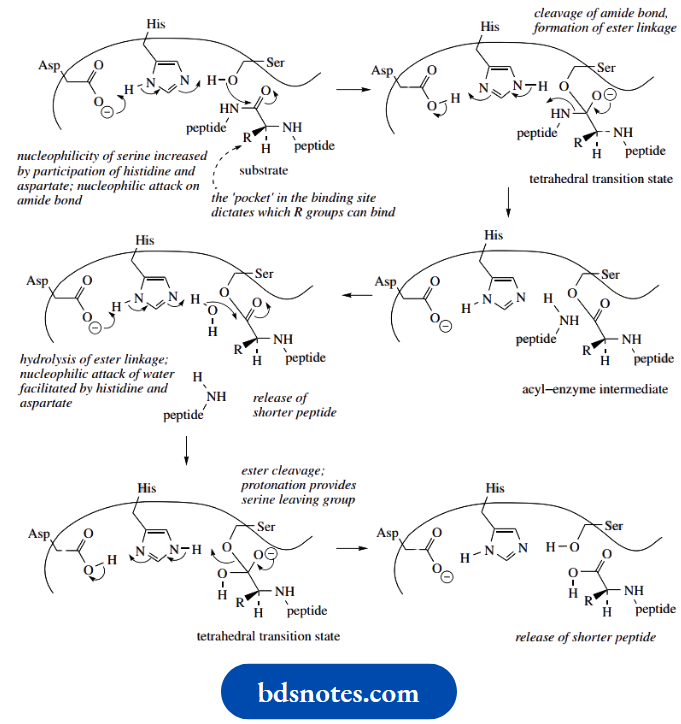
The participation of the histidine group, with the basicity of the histidine nitrogen also being increased because of the proximity of a neighboring aspartate residue – a charge-relay network, as seen with acetylcholinesterase
- This allows nucleophilic attack on the peptide carbonyl, giving an initial tetrahedral transition state.
- We also know that specific amino acid residues are positioned so that they help to stabilize this anionic transition state.
- The formation of the carbonyl group is followed by cleavage of the peptide bond.
- The proton required to form the amino group is acquired from the imidazole. The product is now an acyl–enzyme intermediate, actually an ester involving the serine hydroxyl.
- This ester is hydrolysed by a water nucleophile, and deprotonation is achieved via the aspartate–histidine system once again.
- This generates another tetrahedral transition state, which collapses and allows the release of the carboxylic acid and regeneration of the serine hydroxyl by protonation from the imidazole system.
Note that penicillins and structurally related antibiotics are frequently deactivated by the action of bacterial β-lactamase enzymes.
- These enzymes also contain a serine residue in the active site, and this is the nucleophile that attacks and cleaves the β-lactam ring.
- The β-lactam (amide) linkage is hydrolyzed, and then the inactivated penicillin derivative is released from the enzyme by further hydrolysis of the ester linkage, restoring the functional enzyme.
- The mode of action of these enzymes thus closely resembles that of the serine proteases; . Whilst chymotrypsin and trypsin are especially useful in peptide sequence analysis, they also have medicinal applications.
- Their ability to hydrolyze proteins makes them valuable for wound and ulcer cleansing (trypsin) or during cataract removal (chymotrypsin).
Enoliztionand enolate anion biochemistry
Let us now look at an example of how nature exploits the equivalent of enol and enolate anion chemistry. Enolization provides another application of acid-base catalysis. We saw that the chemical process for enolization could be either acid- or base-catalyzed, and the following scheme should remind us of the mechanism for base-catalyzed enolization of acetone.
Base-catalysed enolization:
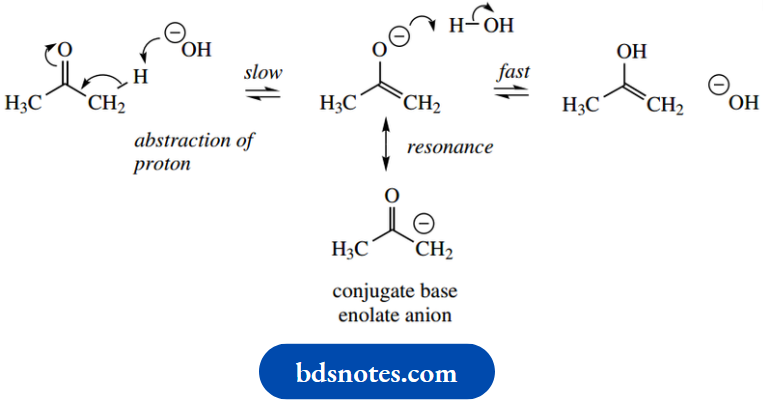
The enzymic processes appear exactly equivalent, except that protons are removed and supplied through the involvement of peptide side chains. It is unlikely that a distinct enolate anion is formed; instead, we should consider the process as concerted with a smooth flow of electrons. Thus, as a basic group removes a proton from one part of the molecule, an acidic group supplies a proton at another.
The example of triose phosphate isomerase in provides us with an easily understood analogy.
Enzyme-catalysed enolization:
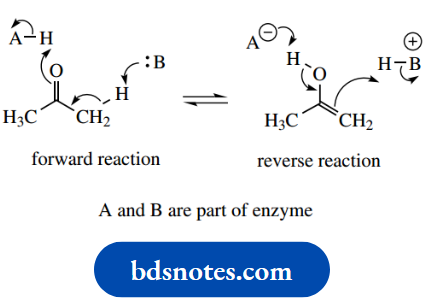
Triose phosphate isomerase Enolization via acid-base catalysis
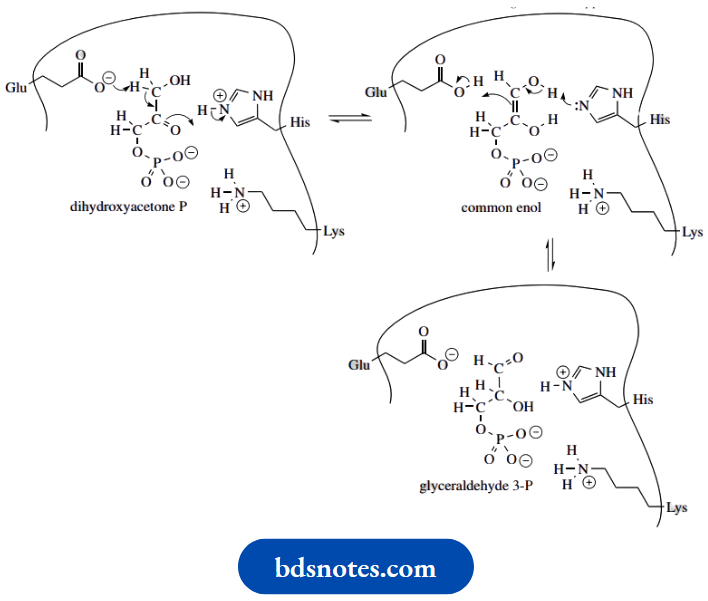
Triose phosphate isomerase is one of the enzymes of glycolysis and is responsible for converting dihydroxyacetone phosphate into glyceraldehyde 3-phosphate by a two-stage enolization process. An intermediate enediol is involved – this common enol can revert to a keto form in two ways, thus providing the means of isomerization
The active site of the enzyme contains a glutamic acid residue that is ionized at pH 7 and supplies the base. A histidine residue, partially protonated at pH 7, in turn, supplies the proton necessary to form the common enol
The process continues, in that the now uncharged histidine is suitably placed to remove a proton from the second of the two hydroxyls, and tautomerization is achieved by abstraction of a proton from the now non-ionized
We have seen many examples of chemical reactions involving enolate anions, and should now realize just how versatile they are in chemical synthesis.
We have also seen several examples of how equivalent reactions are utilized in nature.
- For the triose phosphate isomerase mechanism above, we did not invoke a distinct enolate anion intermediate in the enolization process but proposed that there was a smooth flow of electrons.
- For other reactions, we shall also need to consider whether enolate anions are actually involved, or whether a more favorable alternative exists. The aldol-type reaction catalysed by the enzyme aldolase is an excellent illustration of nature’s approach to enolate anion chemistry.
- Aldolase catalyzes both aldol and reverse aldol reactions according to an organism’s needs. In glycolysis, the substrate fructose 1,6-diphosphate is cleaved by a reverse aldol reaction to provide one molecule of glyceraldehyde 3-phosphate and one molecule of dihydroxyacetone phosphate.
In carbohydrate synthesis, these two compounds can be coupled in an aldol reaction to produce fructose 1,6- diphosphate
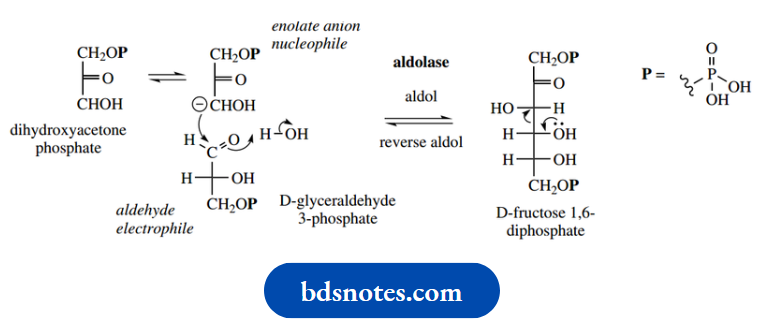
It is conceptually easier to consider initially the aldol reaction rather than the reverse aldol reaction. This involves generating an enolate anion from the dihydroxyacetone phosphate by removing a proton from the position α to the ketone group.
- This enolate anion then behaves as a nucleophile towards the aldehyde group of glyceraldehyde 3-phosphate, and an addition reaction occurs, which is completed by abstraction of a proton, typically from solvent.
- In the reverse reaction, the leaving group would be the enolate anion of dihydroxyacetone phosphate.
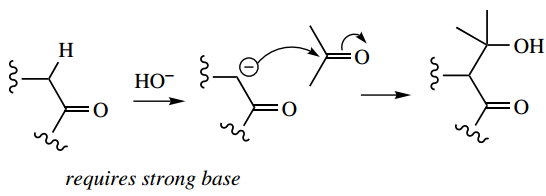
- Now let us consider the difficulties associated with this reaction, should we attempt it using chemical reagents. In contrast to the chemical aldol reaction,
The enzymic reaction has several remarkable advantages:
- The reaction is conducted at room temperature;
- It is conducted at pH 7 without the need for a strong base to generate the enolate anion
- Although it is a mixed aldol reaction, it is quite specific, giving a single product
- Both substrates have the potential to form an enolate anion
- Both substrates have the potential to act as an electrophile
- Dihydroxyacetone phosphate has the potential to form two enolate anions
- Other functional groups in the substrates remain unchanged;
The reaction is reversible and can be employed in either direction under similar conditions. How this is achieved with the enzyme and the role played by some of the amino acid side-chains can now be considered
Chemical aldol reaction:
Enzymic aldol reaction:
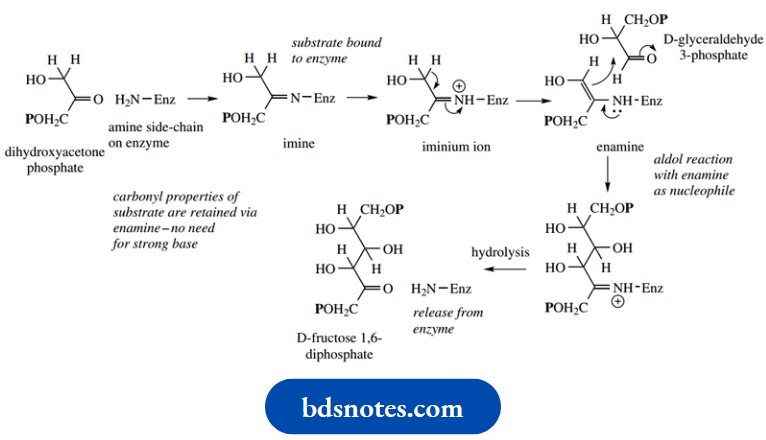
A particularly important interaction with the enzyme is that dihydroxyacetone phosphate is bound to the protein using an imine linkage between the ketone group and an amino group on the enzyme.
This produces a two fold advantage:
- First, it anchors the substrate to the enzyme through a covalent linkage; second, it allows the formation of an enamine by removal of the proton originally α to the ketone.
- An enamine is the equivalent of an enolate anion, but enamine formation is much easier than enolate anion formation and can occur without the need for a strong base.
- Proton removal is achieved by the participation of one of the basic groups on the enzyme.
- With the second substrate glyceraldehyde 3- phosphate appropriately positioned, the aldol addition can then take place, the completion of which requires a supply of a proton from the enzyme.
- The product can then be released from the enzyme by hydrolysis of the imine bond, restoring the original ketone of the substrate and the amino group on the enzyme. The reverse aldol reaction can be rationalized similarly.
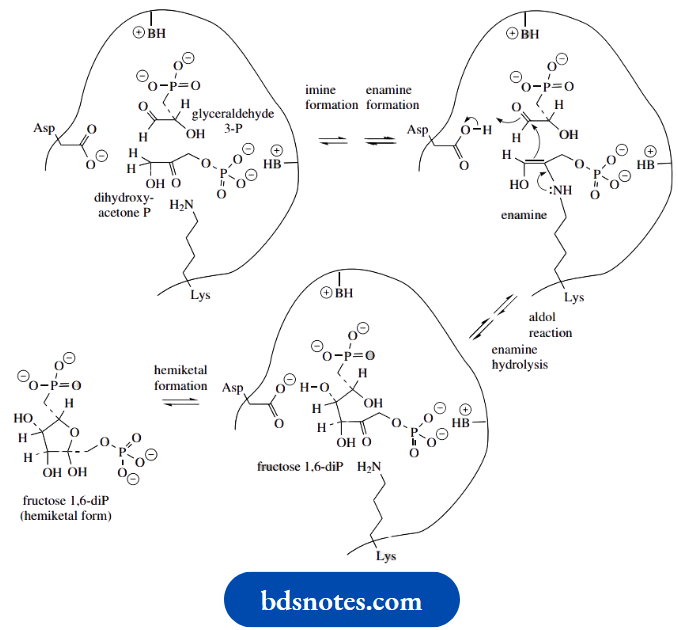
The active site of an aldolase
The active site of the aldolase enzyme is believed to be as shown. Although several amino acid residues are involved with bonding the substrates at the active site, the critical amino acid residues are a lysine and an aspartic acid residue.
- The lysine forms a substrate–enzyme bond via an imine linkage, and the aspartic acid residue functions as a general acid-base.
- Basic amino acid residues are involved in binding the phosphate substrates; to simplify the overall picture, these are not specified here.
- A lysine residue reacts with the carbonyl of dihydroxyacetone phosphate, forming first an addition product that dehydrates to give an imine linkage.
- An aspartate residue is suitably positioned to function as the active site base that removes a proton from the imine and generates the enamine.
- The resultant aspartic acid residue is then involved again in providing a proton to complete the aldol addition.
- The active site also facilitates the ketone–hemiketal interconversion, so that the product liberated is the hemiketal form of fructose 1,6-diphosphate.
- In the reverse reaction, aspartate removes a proton from the alcohol, which allows the formation of a transient carbanion or enamine.
The carbanion/enamine is subsequently protonated via aspartic acid
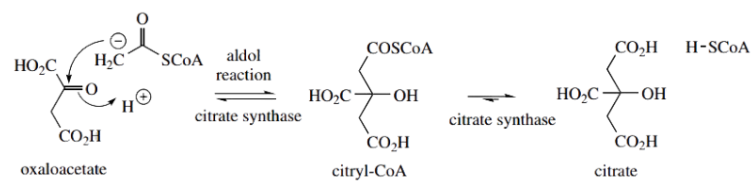
Citrate synthase catalyzes an aldol reaction rather than a Clasien reaction
The reaction is catalyzed by citrate synthase in the Krebs cycle. is primarily an aldol reaction, but the subsequent step, hydrolysis of a thioester linkage, is also catalyzed by the same enzyme. This is shown below
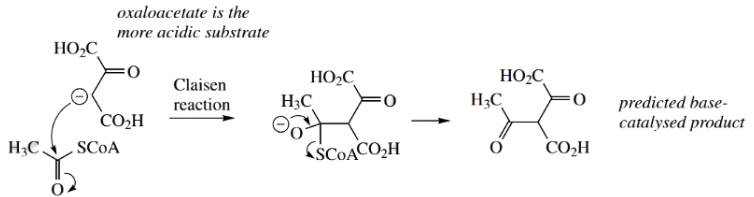
Mechanistically, we can consider it as an attack of an enolate anion equivalent from acetyl-CoA onto the ketone group of oxaloacetate. However, if we think carefully, we conclude that this is not what we would predict Of the two substrates, oxaloacetate is the more acidic reagent, in that two carbonyl groups flank a methylene.
According to the enolate anion chemistry, we would predict that oxaloacetate should provide the enolate anion and that this might then attack acetyl-CoA in a Claisen reaction The product expected in a typical base-catalyzed reaction would, therefore, be an acetyl derivative of oxaloacetate
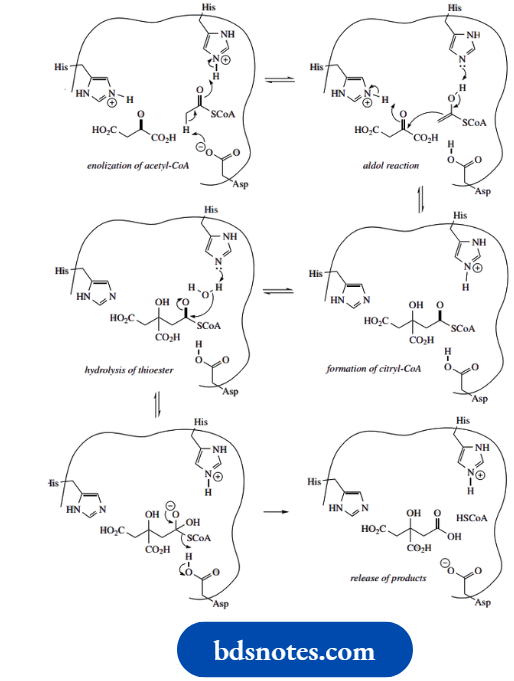
That this is not the case for the enzyme citrate synthase suggests we must look at the enzyme binding site to rationalize the different reaction sequence. It becomes clear that the enzyme binding site positions the substrates so that there are acidic and basic amino acid residues available to produce the enolate anion equivalent of acetyl-CoA (shown here as the enol), but not for the oxaloacetate
Imidazole rings of histidine residues are suitably oriented to participate in the aldol reaction. A histidine residue is also involved in the next step, the hydrolysis of citryl-CoA, and release of citric acid as the final product. It is the hydrolysis of the thioester that disturbs the equilibrium and drives the reaction to completion.
As with other examples of enzyme mechanisms, we can see that the exact array of amino acid residues in the binding site dictates binding of substrates and their chemical interaction to yield products.
Thioesters as intermediates
The reaction of an amino group with an aldehyde or ketone leads to an imine, which, as we have just seen with aldolase, provides a splendid example of how to bond a carbonyl substrate to an enzyme, and yet maintain its chemical reactivity in terms of enolate anion chemistry.
- Another type of covalent interaction is quite commonly encountered, and this exploits the thiol group of cysteine.
- Thiols are more acidic than oxygen alcohols, sulfur is a better nucleophile than oxygen, and sulfur derivatives provide better-leaving groups than the corresponding oxygen ones (see
- It is not surprising that nature makes very good use of these properties.
We shall meet several examples of this type of process, and so only the general mechanism will be considered at this stage.
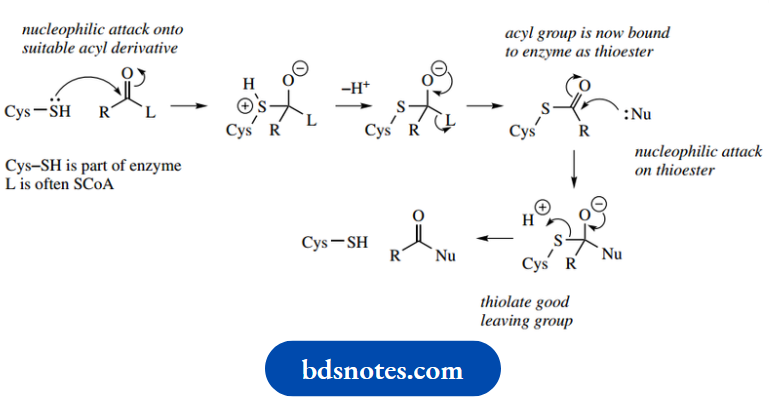
The thiol group of a cysteine residue acts as a nucleophile towards a suitable carbonyl system, which may frequently be a coenzyme A ester.
- Normal addition–elimination occurs, and the leaving group is expelled. This process effectively anchors the acyl residue to the enzyme through a thioester linkage.
- This now allows a nucleophilic substrate to approach the enzyme active site, and become acylated by reacting with the bound acyl group.
- This results in regeneration of the cysteine thiol group. Protons are likely removed and supplied as necessary by the participation of general acids or bases at the active site.
- We have shown the cysteine thiol group as uncharged.
The pKa for this group in cysteine is an application of the Henderson–Hasselbach equation indicating there will be negligible ionization at pH 7. Nevertheless, under the influence of a suitable basic group,
Example:
Arginine pKa 12.5, ionization to thiolate may be possible. In such an environment, thiolate may act as the nucleophile in the mechanism
Enzyme inhibitors
Nature has designed enzymes to carry out modest chemical modifications on a specific substrate. In certain cases, a small number of related substrates may be modified similarly, though not always with the same efficiency, i.e. the enzyme shows broad substrate specificity.
- The chemical change catalyzed is usually small, and several enzymes will be required to change the structure of the substrate significantly. This is made clear when we consider the pathways of intermediary metabolism.
- In a few of these pathways, we shall meet examples of where several enzyme activities are combined, either as a multi-functional enzyme or as an enzyme complex where the individual components may be separated.
- This allows a significant chemical change to be catalyzed by a single protein system. Whatever the arrangement of enzymes, it is clear to see that a single enzyme activity functions as a link in a chain and, therefore, can be used to control whether or not a sequence of reactions proceeds. We can thus exploit a chain’s weakest link
- Enzyme inhibitors are chemicals that may serve as a natural means of controlling metabolic activity by reducing the number of enzyme molecules available for catalysis. In many cases, natural or synthetic inhibitors have allowed us to unravel the pathways and mechanisms of intermediary metabolism.
Enzyme inhibitors may also be used as pesticides or drugs. Such materials are designed so that they inhibit a specific enzyme that is peculiar to an organism or a disease state. For example, a good antibiotic may inhibit a bacterial enzyme, but it should not affect the host person or animal.
We may consider enzyme inhibitors as either irreversible or reversible inhibitors. Some inhibitors become covalently linked to the enzyme and are bound so strongly that they cannot be removed. As a result, the enzyme activity decreases and eventually becomes zero.
- Irreversible inhibitor: E + → EI
- Reversible inhibitor: E + → EI
Irreversible inhibition
Irreversible inhibition in an organism usually results in a toxic effect.
Examples of this type of inhibitor are the organophosphorus compounds that interfere with acetylcholinesterase.
- The organophosphorus derivative reacts with the enzyme in the normal way, but the phosphorylated intermediate produced is resistant to normal hydrolysis and is not released from the enzyme
- The enzyme becomes inactivated, and a toxic level of acetylcholine builds up. Organophosphorus compounds provide a range of insecticides and nerve gases.
Reversible inhibitors:
Reversible inhibitors are potentially less damaging. In the presence of a reversible inhibitor, the enzyme activity decreases, but to a constant level as equilibrium is reached.
- The enzyme activity reflects the lower level of enzyme available for catalysis. We can subdivide the reversible inhibition into three types, i.e. competitive, non-competitive, and allosteric inhibition.
Competitive inhibitors
Competitive inhibitors bind to specific groups in the enzyme active site to form an enzyme–inhibitor complex. The inhibitor and substrate compete for the same site, so that the substrate is prevented from binding.
- This is usually because the substrate and inhibitor share considerable structural similarity. Catalysis is diminished because a lower proportion of molecules have a bound substrate.
- Inhibition can be relieved by increasing the concentration of substrate. Some simple examples are shown below.
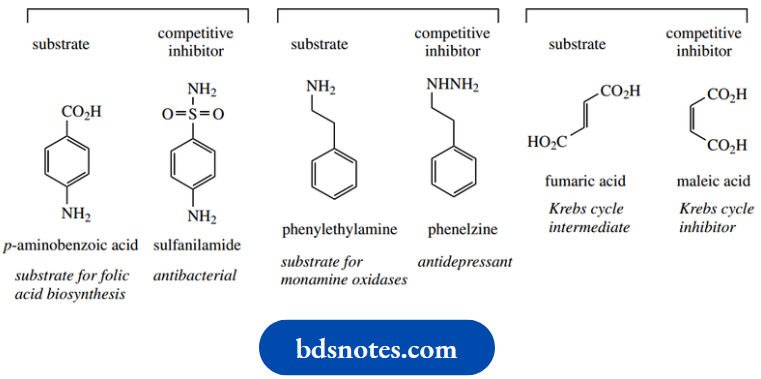
- Thus, sulfanilamide is an inhibitor of the enzyme that incorporates p-aminobenzoic acid into folic acid, and has antibacterial properties by restricting folic acid biosynthesis in the bacterium.
- Some phenylethylamine derivatives,
Example:
Phenelzine provides useful antidepressant drugs by inhibiting the enzyme monoamine oxidase.
The cis-isomer maleic acid is a powerful inhibitor of the enzyme that utilizes the trans-isomer fumaric acid in the Krebs cycle.
Non-competitive inhibitors do not bind to the active site but bind at another site on the enzyme and distort the shape of the protein, resulting in a lowering of activity. Both inhibitor and substrate can bind simultaneously to the enzyme.
A non-competitive inhibitor decreases the activity of the enzyme rather than lowering the proportion of molecules with a bound substrate. In contrast to competitive inhibition, increasing the concentration of substrate has no effect on the level of inhibition. The chemical structures of non-competitive inhibitors frequently bear no similarity to the natural substrate structures
For example:
Heavy metal ions, such as Pb2+ and Hg2+, inhibit the activity of some enzymes by binding to thiol groups, and cyanide reacts with and inhibits iron–porphyrin enzymes
- The third type of inhibition is called allosteric inhibition and is particularly important in the control of intermediary metabolism.
- This refers to the ability of enzymes to change their shape (tertiary and quaternary structure when exposed to certain molecules.
- This sometimes leads to inhibition, whereas in other cases it may activate the enzyme.
- The process allows subtle control of enzyme activity according to an organism’s demands. Further consideration of this complex phenomenon is outside our immediate needs.
Angiotensin-converting enzyme (ACE) inhibitors: captopril
Captopril was the first of a range of orally active drugs to counter high blood pressure, a group known collectively as ACE inhibitors. ACE is the abbreviation for angiotensin-converting enzyme, a protein that converts the decapeptide angiotensin I into the octapeptide angiotensin 2 by hydrolytic removal of a pair of amino acids.
- Angiotensin 2 has a powerful vasoconstrictor effect, so increases blood pressure. By inhibiting the action of ACE, angiotensin 2 levels are limited, blood vessels dilate, and blood pressure is reduced.
- This is of particular value in reducing the risk of heart attacks in patients prone to high blood pressure.
ACE is a carboxypeptidase enzyme that splits off a pair of amino acids from the C-terminal end; its active site is known to contain a zinc atom.
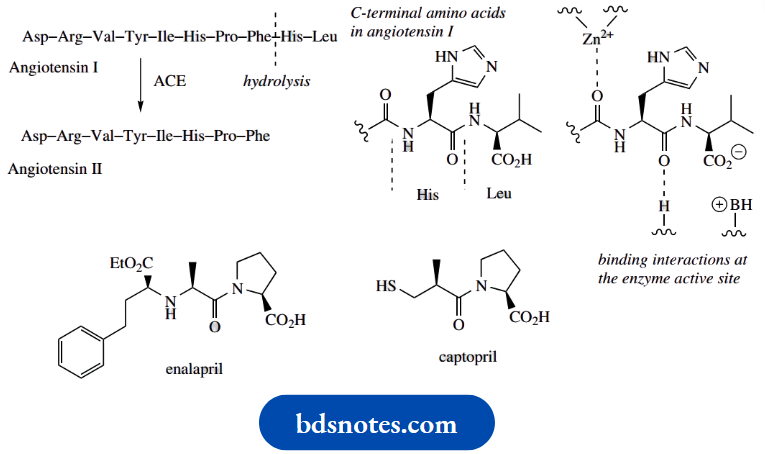
The development of captopril was one of the first examples of successful drug design based upon knowledge of the active site of the target enzyme. It was designed to fit the known active site of carboxypeptidase A, an enzyme very similar to ACE.
Captopril resembles the terminal dipeptide cleaved from angiotensin I, in that the proline carboxylate can bind to a positive center, the amide carbonyl can hydrogen bond, and the thiol group is a good ligand for the Zn2+ component. Captopril is thus a competitive inhibitor of the enzyme; it can bind to the enzyme, but, in so doing, inhibits its hydrolytic action. Several captopril-like drugs are now available, their main advantage over captopril being their increased duration of action, Example: Enalapril
Peptide Biosynthesis
Synthesis and biosynthesis of peptides and proteins requires the combination of amino acids via amide bonds. We have seen earlier that the chemical reaction of amines and acids to produce a simple amide is severely hindered by initial salt formation and that
a more efficient way of making amides is to employ a carboxylic derivative that is non-acidic and has a better-leaving group. Thus, acyl halides, anhydrides, or even esters provide better substrates. In nature, we find that esters or thioesters are the reactive species employed.
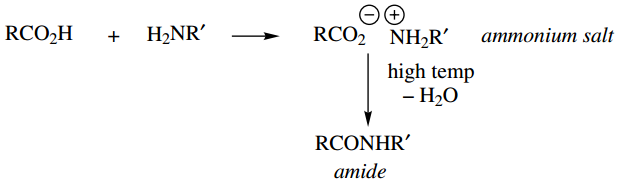
⇒\(\mathrm{RCOCl}+\mathrm{H}_2 \mathrm{NR}^{\prime} \longrightarrow \mathrm{RCONHR}^{\prime}+\mathrm{HCl}\)
⇒ \((\mathrm{RCO})_2 \mathrm{O}+\mathrm{H}_2 \mathrm{NR}^{\prime} \longrightarrow \mathrm{RCONHR}^{\prime}+\mathrm{RCO}_2 \mathrm{H}\)
⇒ \(\left.\begin{array}{l}
\mathrm{RCO}_2 \mathrm{R}+\mathrm{H}_2 \mathrm{NR}^{\prime} \longrightarrow \mathrm{RCONHR}^{\prime}+\mathrm{ROH} \\
\mathrm{RCOSR}+\mathrm{H}_2 \mathrm{NR}^{\prime} \longrightarrow \mathrm{RCONHR}^{\prime}+\mathrm{RSH}
\end{array}\right\} \begin{aligned}
& \begin{array}{l}
\text { esters and thioesters } \\
\text { are } \text { used in nature }
\end{array}
\end{aligned}\)
A further requirement for the chemical synthesis of peptides would be to take steps to avoid any sidechain functional groups reacting under the conditions used for amide bond formation.
- This can be accomplished by the use of appropriate protecting groups, though these will then have to be removed at a later stage in the synthesis.
- Nature employs enzymic reactions that position the functional groups in an appropriate orientation to react.
- Consequently, any side-chain functionalities are kept well away and do not interfere with the processes of amide bond formation.
- The final consideration is to assemble the amino acids in the correct order. In all cases, we need to choose the correct amino acid at each step, but we shall see that nature uses quite sophisticated techniques, and specificity is conferred by nucleic acids and enzymes. In the laboratory, we must pick up reagent bottles in the correct sequence.
Peptides are produced in nature by one of two methods, termed ribosomal peptide biosynthesis and non-ribosomal peptide biosynthesis.
In the former process, peptide biosynthesis takes place on the ribosomes, and the amino acid precursors are combined in a sequence that is defined by the genetic code, the sequence of bases in DNA. In nonribosomal peptide biosynthesis, peptides are synthesized by a more individualistic sequence of enzyme-controlled reactions. Despite the differences in programming the sequence, the chemical linkage of amino acid residues is achieved in a rather similar fashion.
Ribosomal peptide biosynthesis
A simplified representation of peptide biosynthesis, as characterized in the bacterium Escherichia coli. The major aspect to be considered here relates to the bond forming processes involved in linking the amino acids.
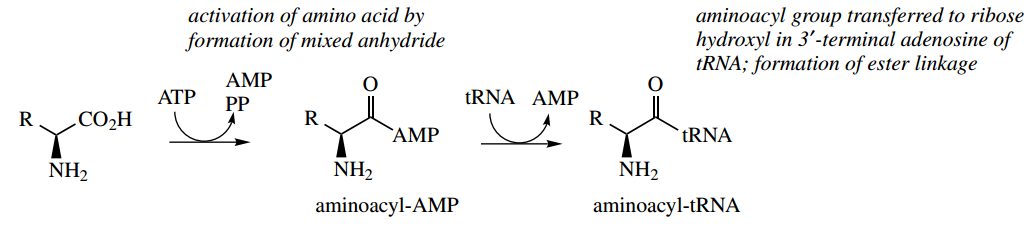
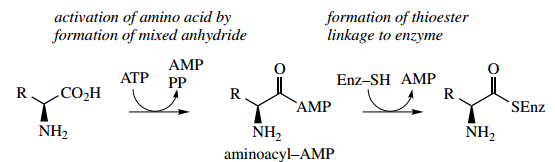
Initially, the amino acid is activated by an ATP-dependent process, producing an aminoacyl-AMP. This may be considered to be a nucleophilic attack of the amino acid carboxylate group onto the P=O system of ATP with the expulsion of diphosphate as the leaving group.
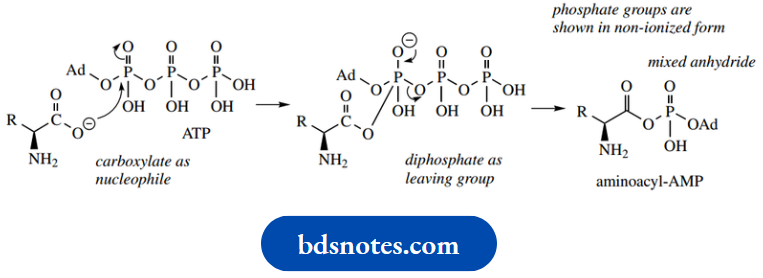
Carboxylate is not an especially good nucleophile, but we have seen it used in SN2 reactions to synthesize esters. Here, the attack is Carboxylate is not an especially good nucleophile, but we have seen it used in SN2 reactions to synthesize esters. Here, the attack is on a reactive anhydride; a similar type of reaction is seen in fatty acid degradation
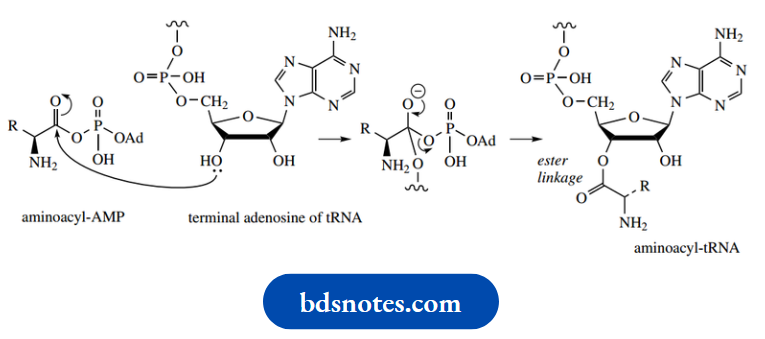
The intermediate aminoacyl-AMP can also be seen to be an anhydride but in this case a mixed anhydride of carboxylic and phosphoric acids.
This can react with a hydroxyl group in ribose, part of a terminal adenosine group of transfer-RNA (tRNA). This then binds the amino acid via an ester linkage, giving an aminoacyl-tRNA.
The tRNA involved will be specific for the particular amino acid.
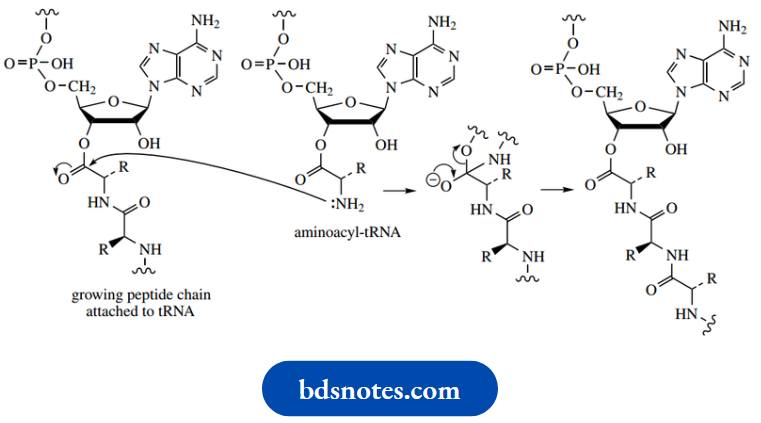
Peptide bond formation is the result of two such aminoacyl-tRNA systems interacting, the amino group in one behaving as a nucleophile and displacing the tRNA from the second, i.e. simply amide formation utilizing an ester substrate. The process is repeated as required. The sequence of amino acids is controlled by messenger RNA (mRNA), the message being stored as a series of three-base sequences (codons) in its nucleotides.
Elongation of the peptide continues until a termination codon is reached, and the peptide or protein is then hydrolyzed and released from the tRNA carrier.
Non-ribosomal peptide biosynthesis
In marked contrast to the ribosomal biosynthesis of peptides and proteins where a biological production line interprets the genetic code of mRNA, many natural peptides are known to be synthesized by a more individualistic sequence of enzyme-controlled processes, in which each amino acid is added as a result of the specificity of each enzyme involved.
- The many stages of the whole process appear to be carried out by a multi-functional enzyme nonribosomal peptide synthase (NRPS) comprised of a linear sequence of modules.
- Each module is responsible for inserting a particular amino acid to generate the sequence in the peptide product. The amino acids are first activated to aminoacyl-AMP derivatives as for ribosomal peptide biosynthesis.
- These are then converted into thioesters, by reaction with thiol functions in the enzyme.
The process is exactly analogous to forming aminoacyl-tRNA units but utilizes SH rather than OH as a nucleophile.
The residues are held so as to allow a sequential series of peptide bond formations gives a simplified representation), until the peptide is finally released from the enzyme A typical module consists of an adenylation (A) domain, a peptidyl carrier protein (PCP) domain, and a condensation (C) or elongation domain.
- The A domain activates a specific amino acid as an aminoacyl-AMP mixed anhydride, which is then transferred to the PCP domain to form an aminoacyl thioester.
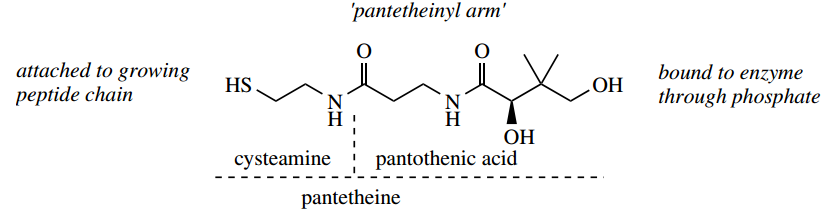
- The thioester linkage is not to a cysteine residue in the protein,
- Instead, it involves pantothenic acid (vitamin B5) bound to the enzyme as pantetheine, and this is used to carry the growing peptide chain via its thiol group.
The important significance of this is that the long ‘pantheinyl arm’ allows different active sites on the multi-functional enzyme to be reached in the chain assembly process
Nucleophilic attack by the amino group of the neighboring aminoacyl thioester is catalyzed by the C domain, and this results in amide (peptide) bond formation.
- Enzyme-controlled biosynthesis in this manner is a feature of many microbial peptides, especially those containing unusual amino acids not encoded by DNA and where post-translational modification is unlikely.
- As well as activating the amino acids and catalyzing the formation of the peptide linkages, the enzyme may possess other domains that are responsible for epimerizing L-amino acids to D-amino acids probably through enol-like tautomers in the peptide.
- A terminal thioesterase domain is also required. This is responsible for terminating the chain extension process by hydrolyzing the thioester and releasing the peptide from the enzyme.
Many medicinally useful peptides have cyclic structures. Cyclization may result if the amino acids at the two termini of a linear peptide link up to form another peptide bond. Alternatively, ring formation can very often be the result of ester or amide linkages that utilize side-chain functionalities (CO2H, NH2, OH) in the constituent amino acids, probably through enol-like tautomers in the peptide.
A terminal thioesterase domain is also required. This is responsible for terminating the chain extension process by hydrolyzing the thioester and releasing the peptide from the enzyme.
Many medicinally useful peptides have cyclic structures. Cyclization may result if the amino acids at the two termini of a linear peptide link up to form another peptide bond. Alternatively, ring formation can very often be the result of ester or amide linkages that utilize side-chain functionalities (CO2H, NH2, OH) in the constituent amino acids.
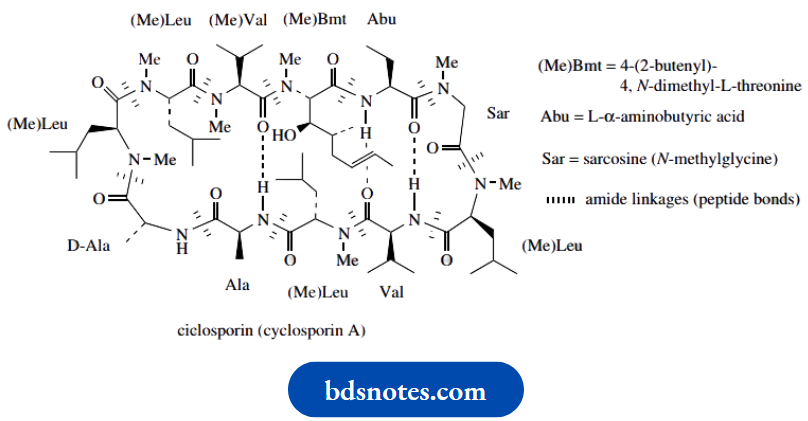
Ciclosporin, a cyclic peptide composed mainly of unusual amino acids
The cyclosporins are a group of cyclic peptides produced by fungi such as Cylindrocarpon lucidum and Tolypocladium inflatum. These agents show a rather narrow range of antifungal activity, but high levels of immunosuppressive and anti-inflammatory activities. The main component from the culture extracts is cyclosporin A, but some 25 naturally occurring cyclosporins have been characterized
Cyclosporin
Cyclosporin A contains 11 amino acids, joined in a cyclic structure by peptide bonds. The structure is also stabilized by intramolecular hydrogen bonds. Only two of the amino acids, i.e. alanine and valine, are typical of proteins.
- The compound contains several N-methylated amino acid residues, together with the even less common L- α-aminobutyric acid and an N-methylated butenyl methyl threonine. There is one D-amino acid, i.e. D-alanine, and the assembly of the polypeptide chain is known to start from this residue.
- Many of the other natural cyclosporin structures differ only concerning a single amino acid (the α-aminobutyric acid residue) or the number of amino acids that have the extra N-methyl group.
- Of all the natural analogs, and many synthetic ones produced, cyclosporin A is the most valuable for drug use, under the drug name ciclosporin.
It is now widely exploited in organ and tissue transplant surgery, to prevent rejection following bone marrow, kidney, liver, and heart transplants.
It has revolutionized organ transplant surgery, substantially increasing survival rates in transplant patients. It is believed to inhibit T-cell activation in the immunosuppressive mechanism by first binding to a receptor protein, giving a complex that then inhibits a phosphatase enzyme called calcineurin.
The resultant aberrant phosphorylation reactions prevent appropriate gene transcription and subsequent T-cell activation.
Penicillins and cephalosporins are modified tripeptides
Penicillin and cephalosporin antibiotics are usually classed as β-lactam antibiotics, since their common feature is a lactam function in a four-membered ring, typically fused to another ring system. This second ring takes in the β-lactam nitrogen atom and also contains sulfur.
In the case of penicillins,
Example: Benzylpenicillin, the second ring is a thiazolidine,
And in the cephalosporins,
Example: Cephalosporin C, this ring is a dihydrothiazine.
What is not readily apparent from these structures is that they are both modified tripeptides and their biosyntheses share a common tripeptide precursor.
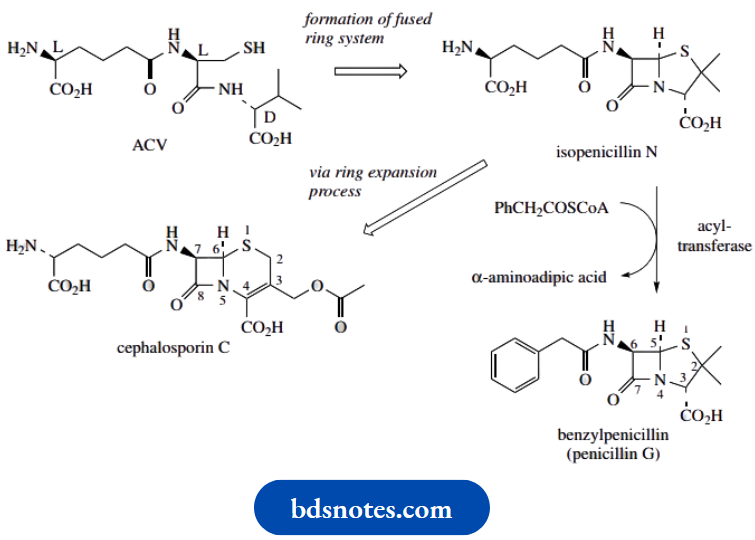
The tripeptide precursor is called ACV, an abbreviation for δ-(L- α-aminoacyl)-L-cysteinyl-D-valine. ACV is an acronym and does not refer to the systematic abbreviations for amino acids described in. ACV is the linear tripeptide that leads to isopenicillin N, the first intermediate with the fused-ring system found in the penicillins.
ACV is produced by the modular system for non-ribosomal peptide biosynthesis. The amino acid precursors are L- α-aminoadipic acid (an unusual amino acid derived by modification of L-lysine), L-cysteine, and L-valine; during tripeptide formation, the L-valine is epimerized to D-valine
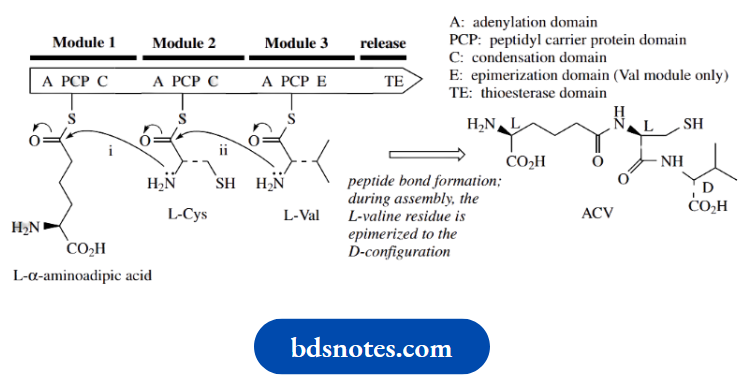
Medicinally useful penicillins are formed by replacing the acyl group of the side-chain amide in isopenicillin N with an alternative acyl group. This is sometimes achieved biochemically in the fungal culture, but more frequently it is accomplished through semi-synthetic procedures.
Isopenicillin N is also the precursor of the cephalosporins, the formation of which requires a ring expansion. The five-membered thiazolidine ring of the penicillin is expanded, taking in one of the methyl groups, to produce a six-membered heterocycle
Bacterial peptidoglycans D-amino acids and the antibacterial action of penicillins
Bacterial cell walls contain peptidoglycan structures in which carbohydrate chains (composed of alternating β1 → 4-linked N-acetylglucosamine and O-lactyl-N-acetylglucosamine residues) are cross-linked via peptide structures.
Part of the peptidoglycan of Staphylococcus aureus is shown here, illustrating the involvement of the lactyl group of the O-lactyl-N-acetylglucosamine (also called N-acetylmuramic acid) in linking the peptide with the carbohydrate via an amide/peptide bond. The peptide cross-links include some D-amino acids, namely D-alanine and D-glutamic acid.
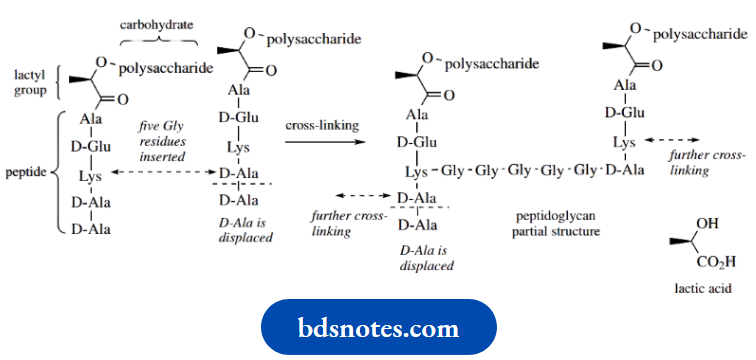
At the start of the cross-linking process, the peptide chains from the N-acetylmuramic acid residues have a terminal –Lys–D-Ala–D-Ala sequence. The lysine from one chain then becomes bonded to the penultimate Dalanine of another chain through five glycine residues, at the same time displacing the terminal D-alanine. The mechanism involves a serine residue at the active site of the enzyme. This residue is used to convert an amide linkage into an ester, and a reversal of this sequence provides the new peptide bond
Cross-linking in peptidoglycan biosynthesis:
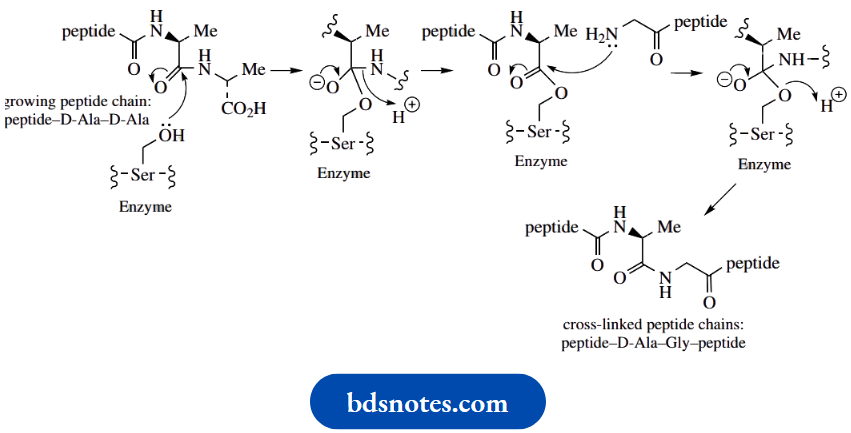
The biological activities of the β-lactam antibiotics,
Example: Penicillins and cephalosporins
- Stem from an inhibition of the cross-linking mechanism during the biosynthesis of the bacterial cell wall.
- The β-lactam drugs bind to enzymes (penicillin-binding proteins) that are involved in the late stages of the biosynthesis of the bacterial cell wall.
- During the cross-linking process, the peptide–D-Ala–D-Ala intermediate in its transition state conformation closely resembles the penicillin molecule.
Enzyme inhibition by β-lactams:
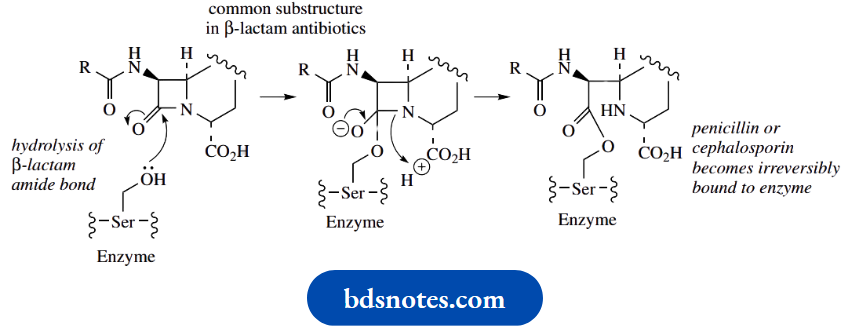
As a result, the penicillin occupies the active site of the enzyme and becomes bound via the active-site serine residue.
- This binding causes irreversible enzyme inhibition and stops cell-wall biosynthesis. Growing cells are killed due to rupture of the cell membrane and loss of cellular contents.
- The binding reaction between penicillinbinding proteins and penicillins is chemically analogous to the action of β-lactamases however, in the latter case, penicilloic acid is subsequently released from the β-lactamase, and the enzyme can continue to function.
- Inhibitors of acetylcholinesterase also bind irreversibly to the enzyme through a serine hydroxyl.
- The penicillins are very safe antibiotics for most individuals. The bacterial cell wall has no counterpart in mammalian cells, and the action is thus very specific.
- However, a significant proportion of patients can experience allergic responses, ranging from a mild rash to fatal anaphylactic shock.
- Cleavage of the β-lactam ring through nucleophilic attack of an amino group in a protein is believed to lead to the formation of antigenic substances that then cause the allergic response.
Peptide Synthesis
Many different approaches have been developed for peptide synthesis, and it is not the intention to cover more than the basic principles here, with a suitable example.
- The philosophy to convert two amino acids into a dipeptide is to transform each difunctional amino acid into a monofunctional compound, one of which has the amino group protected, whilst the other has the carboxyl group protected.
- This allows the remaining amino and carboxyl groups to react, provided the carboxyl group is suitably activated to make it more reactive, as discussed above.
- After coupling and formation of the new amide bond, the product can be deprotected to yield the dipeptide. Alternatively, one or other of the protecting groups can be removed, allowing the sequence to be repeated, leading to larger peptides.
This is shown in the following general scheme.
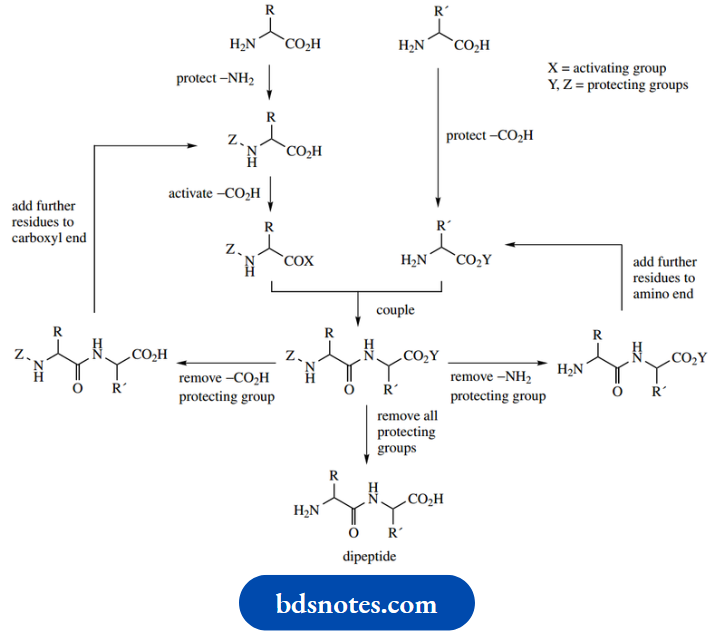
Protecting groups
What is not included here is the need also to protect any vulnerable functional groups in the amino acid side-chains.
- A range of methods is available to protect amino, carboxyl, thiol, and hydroxyl groups and prevent them from reacting during the amide bond synthesis.
- Such groups also have to be removed after their job is done, using conditions that do not destroy the new amide bonds.
- Where amino acid sidechains have carboxylic acid or amino groups, you will readily appreciate that manipulating protecting groups on these groups separately from those related to making the peptide linkage can turn out to be a highly delicate operation.
Let us consider one method to synthesize the dipeptide Ala–Leu. It is necessary to protect the amino group of Ala and the carboxyl group of Leu.
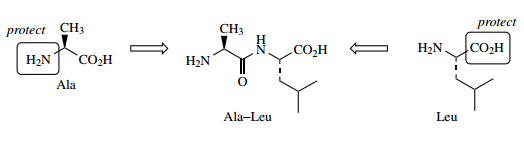
Amino group protection:
Amino group protection may be achieved by converting the amine into its N-tert-butyloxycarbonyl (tBOC or just BOC) derivative, by reaction with di-tert-butyl dicarbonate. This reagent should be considered as a variant of a carboxylic acid anhydride; it reacts in just the same way. The product is termed BOC-Ala and is strictly a carbamate, a half ester–half amide of carbonic acid.
Protection of amino group: tBOC
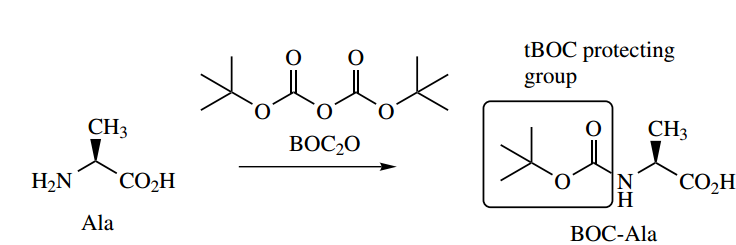
Carbamates behave like amides; the amino group is no longer basic or nucleophilic. The BOC-protecting group can thus be removed readily by treating it with dilute aqueous acid.
The process involves protonation, loss of the tert-butyl cation, and then decarboxylation. On the other hand, the carbonyl group is too hindered to be attacked by the base.
Removal of tBOC protecting group:
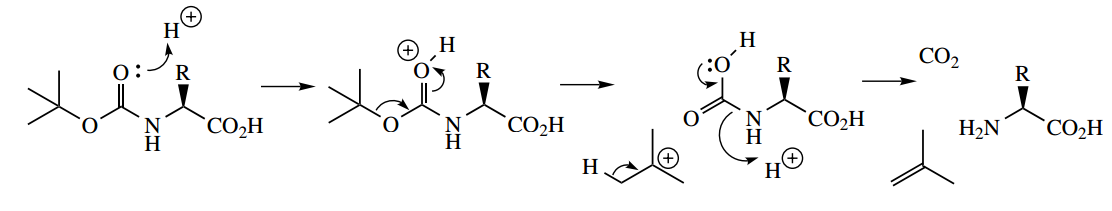
Carboxyl protection:
Carboxyl protection of the second amino acid is usually achieved by conversion to an ester using an appropriate alcohol and acidic catalyst Although methyl and ethyl esters work perfectly well, their removal typically requires alkaline hydrolysis, which may be undesirable. More acceptable are esters that can be removed via catalytic hydrogenolysis,
Example: Benzyl esters.
Protection of carboxyl group: benzyl ester:
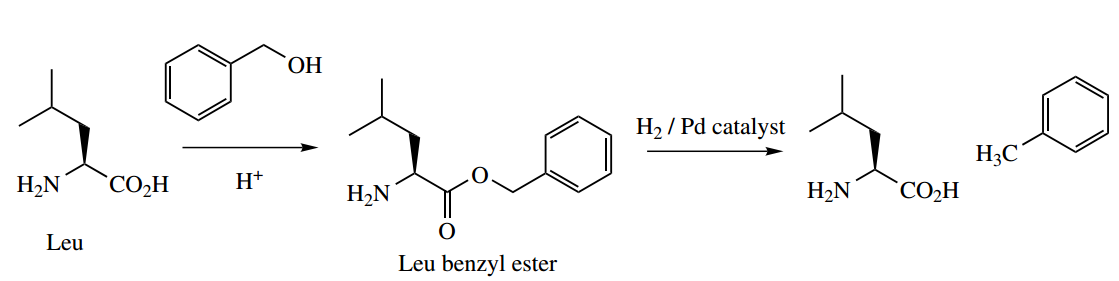
The dicyclohexyl carbodiinide coupling reaction:
Activation of the carboxyl and coupling may be achieved through the use of a single reagent, dicyclohexylcarbodiimide (DCC).
- This compound removes a proton from the carboxylic acid, producing a cation that is readily attacked by the carboxylate nucleophile across one of the C–N double bonds – the protonated imine behaves as a good electrophile.
- The product is now an activated ester (an O-acylisourea) that can be attacked by any available nucleophile. The amino group of the second amino acid derivative provides the nucleophile, resulting in the expulsion of a very stable urea as the leaving group, and production of the protected dipeptide.
DCC is a very attractive reagent, in that there is no need to generate the activated derivative separately. One merely mixes the two protected amino acid derivatives in an aprotic solvent such as CH2Cl2, adds DCC, and dicyclohexylurea is removed as an insoluble by-product. The desired dipeptide can then be obtained by removal of the protecting groups, as already outlined. Note that, in the example shown, we are extending the chain by adding new amino acid residues to the carboxyl terminus
Pepti de synthesis on polymeric supports
Synthesis of peptides in solution using the method outlined above, or alternative procedures, is laborious and often low-yielding since each intermediate needs isolating and purifying at each stage of the synthesis.
An alternative approach developed by Merrifield is to attach the growing peptide chain to a polymer, which renders it insoluble. This allows the use of excess reagents, and the removal of impurities merely by washing the polymer, which is usually in the form of beads. This approach is the basis of automated peptide synthesizers since the process can be fast, simple, and readily repeated.
In the initial step, the first BOC-protected amino acid is bound to the polymer, e.g. polystyrene in which a proportion of the phenyl rings have chloromethyl substitution. Attachment to these residues is through the carboxyl via an ester linkage.
This involves a simple nucleophilic substitution reaction, with the carboxylate as the nucleophile and chloride as the leaving group. After each stage, the insoluble polymer–product combination is washed free of impurities
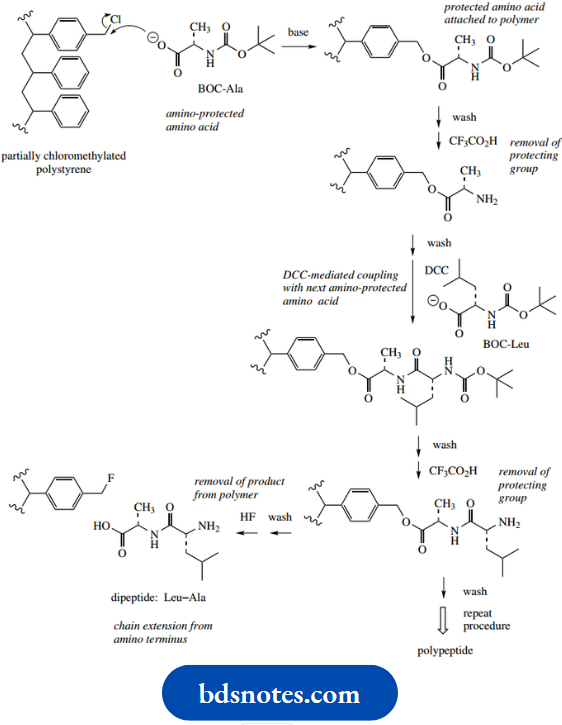
The BOC-protecting group is then removed from the amino acid, allowing the next protected amino acid to be bonded to the polymer-bound substrate via the DCC coupling reaction.
The processes of BOC removal and DCC coupling are then repeated with as many amino acid residues as required. This procedure extends the chain by adding new amino acid residues to the amino terminus.
Finally, the polypeptide is released from the polymer by treatment with HF. All the steps are carried out without isolating any intermediate. An early peptide synthesizer produced the 125 amino acid protein ribonuclease in an overall yield of 17%, a quite staggering achievement
Determination Of Peptide Sequence
Chemical methods for determining the amino acid sequence of a peptide or protein have been developed, and the normal approach is to exploit the properties of the amino group at the N-terminus. A long-established procedure for identifying the N-terminal amino acid is use of the Sanger reagent 2,4-dinitrofluorobenzene. This reacts with an amine by nucleophilic displacement of the fluorine.
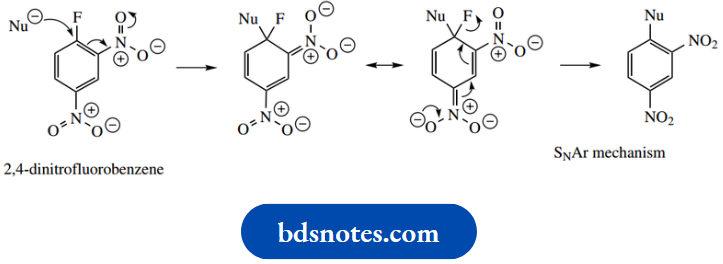
Normally, substitution on a benzene ring is achieved by electrophilic attack, with subsequent loss of a proton. With the Sanger reagent, the presence of three strongly electron-withdrawing substituents allows nucleophilic attack and then displacement of fluoride as a leaving group.
- The initial addition of a nucleophile to the aromatic system generates a transient carbanion, which is stabilized by the nitro groups. Charge is then lost by expelling fluoride as a leaving group, restoring the aromatic ring system
- We have already noted that fluoride is not normally a very effective leaving group. Here, the nucleophilic addition is the rate-determining step, though it is favored by the very large inductive effect from the fluorine and the stabilization from the nitro groups.
- This allows the formation of the additional carbanion, and, even though fluoride is a poor leaving group, it can be lost from the anion to restore aromaticity.
This type of reaction is strictly an addition–elimination mechanism but is referred to as an SNAr mechanism, or nucleophilic aromatic substitution.
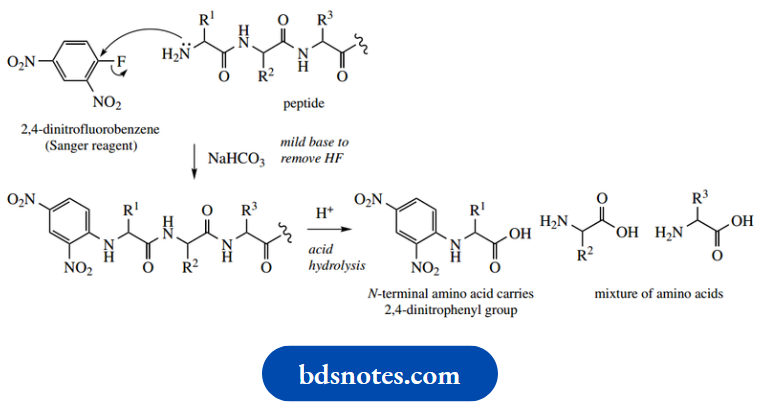
After treatment of the peptide with the Sanger reagent, all peptide bonds are then cleaved by hydrolysis, giving a mixture of amino acids, with the N-terminal one carrying a 2,4-dinitrophenyl group. Being yellow, this compound is readily detected and can be characterized easily by chromatographic comparison with standards.
Although 2,4-dinitrofluorobenzene will also react with any free amino group in an amino acid side-chain,
Example: That in lysine, only the N-terminal amino acid will carry the
2,4-dinitrophenyl residue in its α-amino group. A more useful procedure, in that it allows sequential determination of the N-terminal amino acids in a peptide, is the Edman degradation. This process removes the N-terminal amino acid, but leaves the rest of the chain intact, so allowing further reactions to be applied. The reagent used here is phenyl isothiocyanate
Edman degradation
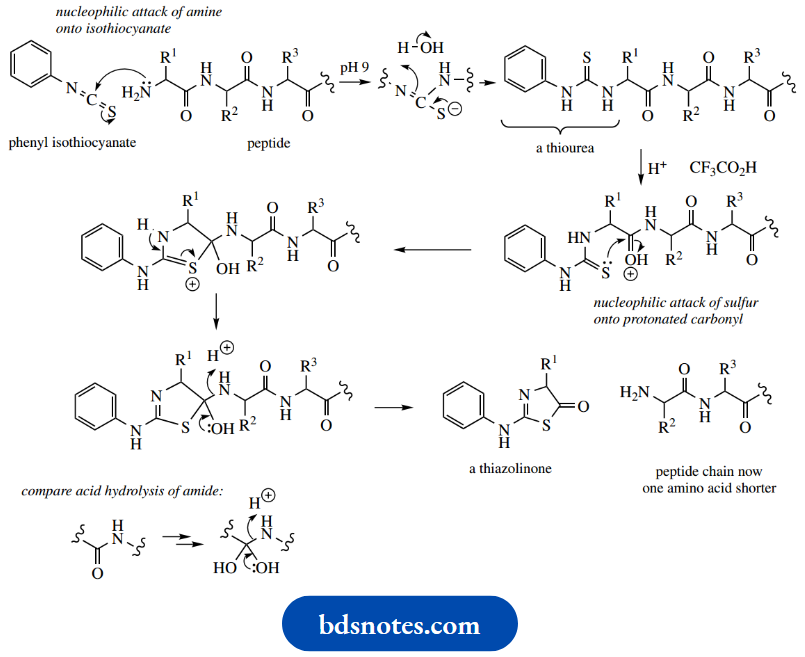
The carbon in the isothiocyanate grouping is highly susceptible to nucleophilic attack by the peptide’s free amino group. Overall addition to the C=N creates a thiourea derivative. Making the conditions strongly acidic then promotes nucleophilic attack by the sulfur of the thiourea onto the carbonyl of the first peptide bond, producing a five-membered thiazoline heterocycle.
Proton loss occurs from the nitrogen, and this creates an intermediate that is equivalent to the addition product in simple acid-catalyzed amide hydrolysis, though here we have employed a sulfur rather than an oxygen nucleophile. Bond cleavage follows, leaving the first amino acid as part of a thiazolinone system. The rest of the peptide chain is unaffected.
Thus, the N-terminal amino acid can be identified by analysis of the thiazolinone, and the process can be repeated on the one-unit-shortened polypeptide chain. Under the acidic conditions, the thiazolinone is actually unstable, and rearranges to a phenylthiohydantoin.
The reasons for the rearrangement need not concern us; a mechanism is shown merely to demonstrate that it can be rationalized. The phenylthiohydantoin derivative produced can be identified simply by chromatographic comparison with authentic standards
The repetitive cycle to identify a sequence of N-terminal amino acids has been automated. In practice, it is limited to about 20–30 amino acids, since impurities build up and the reaction mixture becomes too complex to yield unequivocal results. The usual approach is to break the polypeptide chain into smaller fragments by partial hydrolysis, preferably at positions relating to specific amino acid residues in the peptide chain.
There are ways of doing this chemically, and the enzymes chymotrypsin and trypsin are also routinely used for this purpose. The shortened chains can then be sequenced and, with a little logic and reasoning, the order in which they are attached can be deduced, leading us to the entire amino acid sequence. The process can be exemplified using a simple hypothetical example containing 12 amino acid residues, although, in practice, this is small enough to be achieved by an automatic amino acid sequencer.
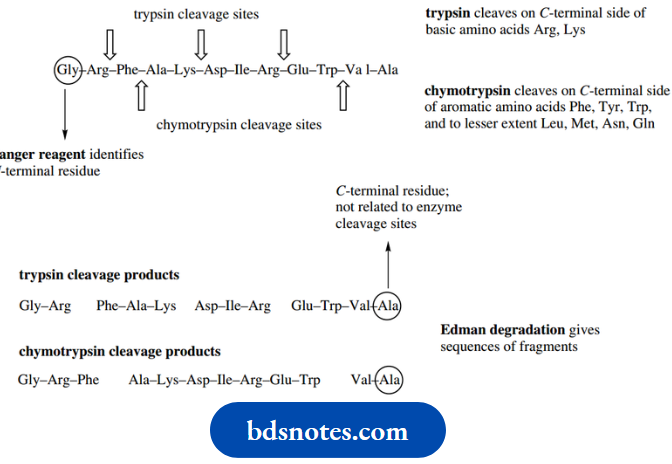
The N-terminal amino acid can be ascertained by the Sanger method. Enzymic cleavage using either chymotrypsin or trypsin will break the peptide into smaller fragments. The fragments obtained will be different, depending on the enzyme and its specificity. The smaller fragments are then each sequenced by the Edman technique.
C-terminal residues in the smaller peptides can be related to knowledge of the enzyme cleavage sites; this may point to the C-terminal residue of the full peptide if it does not correspond to an enzymic cleavage site. The full sequence can be deduced from these fragments by lining up matching sequences of overlapping portions.
Leave a Reply