The Organic Chemistry Of Intermediary Metabolism
Intermediary Metabolism
Intermediary metabolism is the all-encompassing name given to the highly integrated network of chemical reactions by which organisms obtain energy from their environment and synthesize those molecules necessary for their continued well-being and existence.
These are the reactions that we consider to comprise ‘biochemistry’, and they are usually studied as part of a biochemistry course. However, if we choose to study these reactions as part of organic chemistry, we shall see some interesting, elaborate, and often quite complex transformations taking place.
Two very important characteristics differentiate these reactions from those we have already encountered. First, they are almost always enzyme-mediated reactions and thus take place readily at near-neutral pH and at ambient temperatures.
Second, they are also highly regulated, and their participation can be switched on or off, or otherwise finely controlled by the organism according to its needs.
As we look at some of the reactions of intermediary metabolism, we shall rationalize them in terms of the chemistry that is taking place.
In general, we shall not consider here the involvement of the enzyme itself, the binding of substrates to the enzyme, or the role played by the enzyme’s amino acid side chains.
We looked at specific examples where we know just how an enzyme is able to catalyze a reaction. Examples such as aldolase and triose phosphate isomerase, enzymes of the glycolytic pathway, and citrate synthase from the Krebs cycle were considered in some detail.
It may be advantageous to look back at these examples in order to underline the participation of an enzyme.
A large proportion of the substrates used in intermediary metabolism are in the form of phosphates. Phosphates are favored in nature since they usually confer water solubility on the compound, and provide a functional group that is able to bind to enzymes through simple electrostatic bonding.
In many cases, the phosphate group may also feature as a chemically reactive functional group – phosphates are good leaving groups. In many structures, the abbreviation P is used to represent the phosphate group and PP the diphosphate (or pyrophosphate) group:
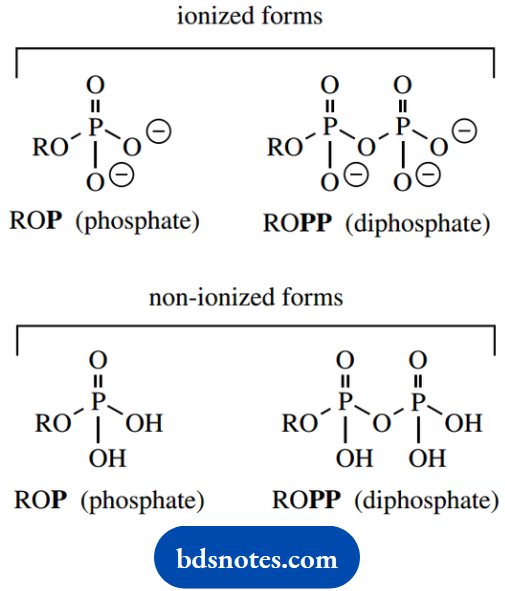
At physiological pH values, these groups will be ionized as shown, but in schemes where structures are given in full, the non-ionized acids are usually depicted.
This is done primarily to simplify structures, eliminate the need for counterions, and avoid mechanistic confusion. Likewise, amino acids are usually shown in non-ionized form, although they will typically exist as zwitterions:

Ionized and non-ionized forms of many com-pounds are regarded as synonymous in the text, thus citrate/citric acid, acetate/acetic acid or others may be used according to the author’s whim and context, and should not be considered as having any special relevance.
Oxidation Reactions And ATP
The currency unit for energy in biochemical reactions is the nucleotide derivative ATP, adenosine triphosphate. We have already discussed this molecule, where we rationalized many of the reactions of phosphates in terms of them being analogs of carbonyl compounds.
Only the triphosphate portion of ATP is involved chemically in the energy processes; the remaining complex part of the molecule is a recognizable feature that allows binding to the enzyme.
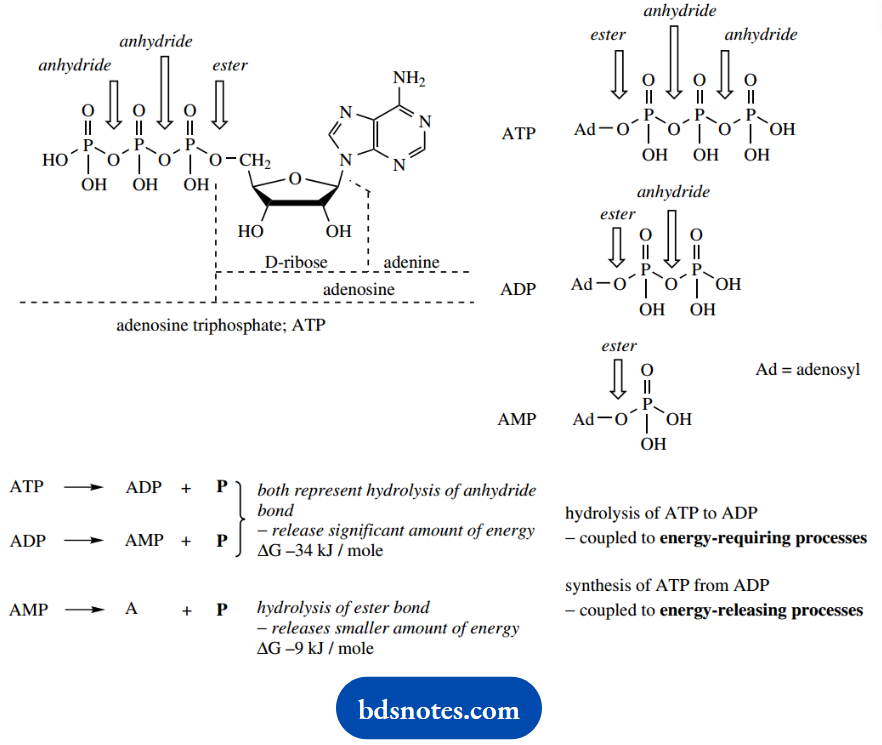
The triphosphate portion can be visualized as containing two anhydride functions and one ester function. We have seen that hydrolysis of anhydrides is achieved much more easily than hydrolysis of esters, an observation that can be related to the nature of the leaving group.
Accordingly, hydrolysis of the anhydride bond liberates considerably more energy than does hydrolysis of the ester bond, and it is anhydride hydrolysis that is crucial to ATP’s role in biochemistry. Hydrolysis of ATP to ADP liberates energy, which can be coupled to energy-requiring processes.
Alternatively, energy-releasing processes can be coupled to the synthesis of ATP from ADP.
Hydrolysis of ATP to ADP is rationalized simply as a nucleophilic attack of water onto the terminal P=O double bond, followed by cleavage of the anhydride bond and expulsion of ADP as the leaving group.
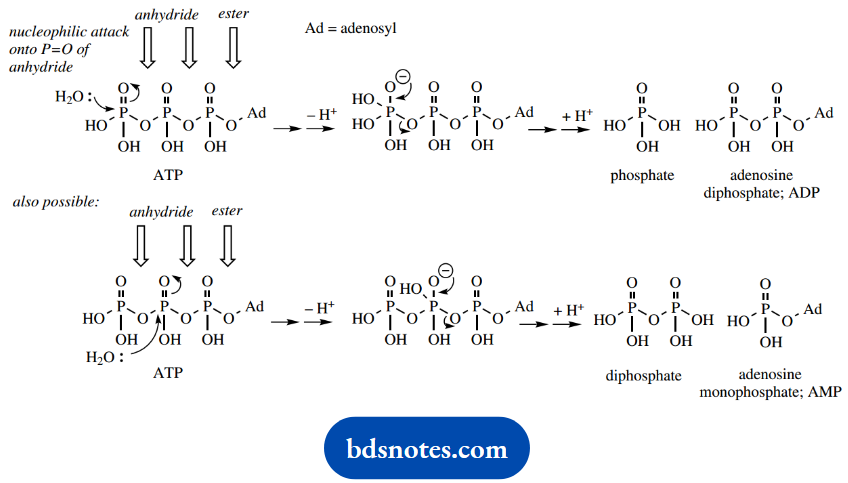
There are two anhydride linkages in ATP, but nucleophilic attack in the enzyme-controlled reaction usually occurs on the terminal P=O (hydrolysis of ATP to ADP), and only occasionally do we encounter attack on the central P=O (hydrolysis of ATP to adenosine monophosphate, AMP).
Both reactions yield the same amount of energy, ΔG—34 kJmol-1. This is not surprising, since in each case the same type of bond is being hydrolyzed.
The further hydrolysis of AMP to adenosine breaks an ester linkage and liberates only a fraction of the energy, ΔG — 9 kJmol-1, and this reaction is not biochemically important.
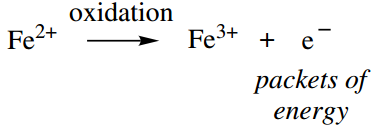
Oxidation reactions are the main providers of energy for ATP synthesis. Whilst oxidation usually involves the incorporation of one or more oxygen atoms, in its simplest form it can be thought of as a loss of electrons.
Thus, the transformation of ferrous ion to ferric ion is an oxidation reaction and involves the loss of one electron. Such electrons can be considered as carrying the energy released from the oxidation reactions.
In biochemical reactions, these electrons are eventually passed to oxygen, which becomes reduced to water. Overall, the oxidation of a substrate AH2 could be represented by the equation
⇒ \(\mathrm{AH}_2+1 / 2 \mathrm{O}_2 \rightleftharpoons \mathrm{A}+\mathrm{H}_2 \mathrm{O} \text { large negative } \Delta \mathrm{G}\)
and this reaction has the potential to liberate energy, i.e. it has a large negative ΔG.
Now this reaction is not possible directly. We are not accustomed to seeing our food spontaneously reacting with atmospheric oxygen and igniting because of the energy released!
However, food such as carbohydrates, fat, and protein is oxidized after we have eaten it, and energy is released and utilized by our bodies. The secret is to react AH2 through the involvement of a suitable coenzyme, not directly with oxygen. This reaction can be considered as
⇒ \(\left.\mathrm{AH}_2+\mathrm{X}(\text { oxidized }) \rightleftharpoons \mathrm{A}+\mathrm{X} \text { (reduced }\right)\)
where X is the coenzyme. The reaction is catalyzed by an enzyme termed dehydrogenase, which removes two hydrogen atoms from the substrate. The coenzyme system involved can generally be related to the functional group being oxidized in the substrate.
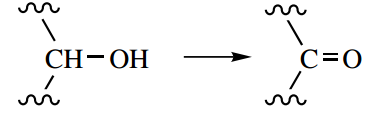
If the oxidation process is then a pyridine nucleotide, nicotinamide adenine dinucleotide (NAD+) or nicotinamide adenine dinucleotide phosphate (NADP+), tends to be utilized as hydrogen acceptor. One hydrogen from the substrate (that bonded to carbon) is transferred as hydride to the coenzyme, and the other, as a proton, is passed to the medium.
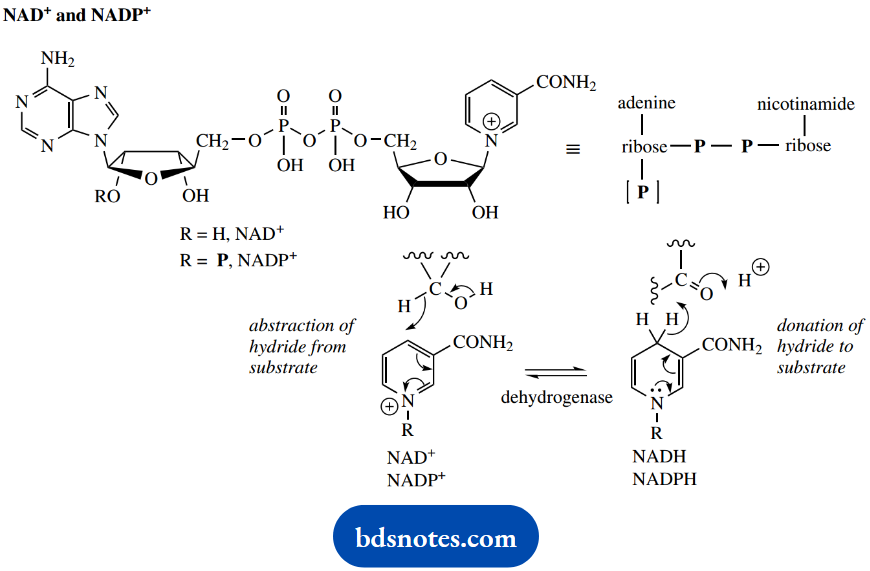
NAD+ and NADP+ may also be used in the oxidations
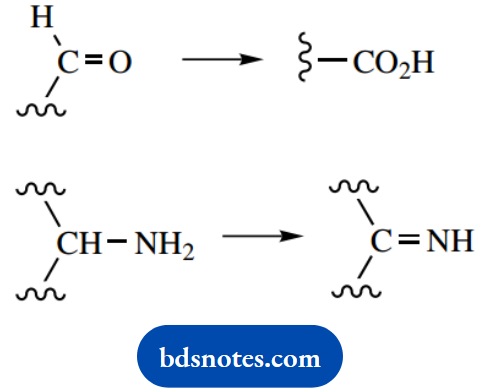
The reverse reaction, i.e. reduction, is also indicated in the scheme and may be compared with the chemical reduction process using complex metal hydrides, for example, LiAlH41or NaBH4, namely nucleophilic addition of hydride and subsequent protonation.
The reduced forms NADH and NADPH are conveniently regarded as hydride-donating reducing agents. We also noted that there were stereochemical features associated with these coenzymes.
During a reduction sequence, there is the stereospecific transfer of hydride from a prochiral center on the dihydropyridine ring, and it is delivered to the carbonyl compound also in a stereospecific manner.
In practice, NADPH is generally employed in reductive processes, whereas NAD+ is used in oxidations.
Should the oxidative process be the conversion then the coenzyme used as acceptor is usually a flavin nucleotide: flavin adenine dinucleotide (FAD) or flavin mononucleotide (FMN).
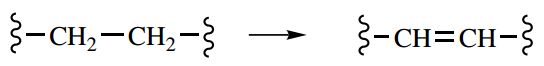
These entities are bound to the enzyme in the form of a flavoprotein, and take up two hydrogen atoms, represented in the figure as being derived by the addition of a hydride from the substrate and a proton from the medium.
Reductive sequences involving flavoproteins may be represented as the reverse reaction, where hydride is transferred from the coenzyme, and a proton is obtained from the medium.
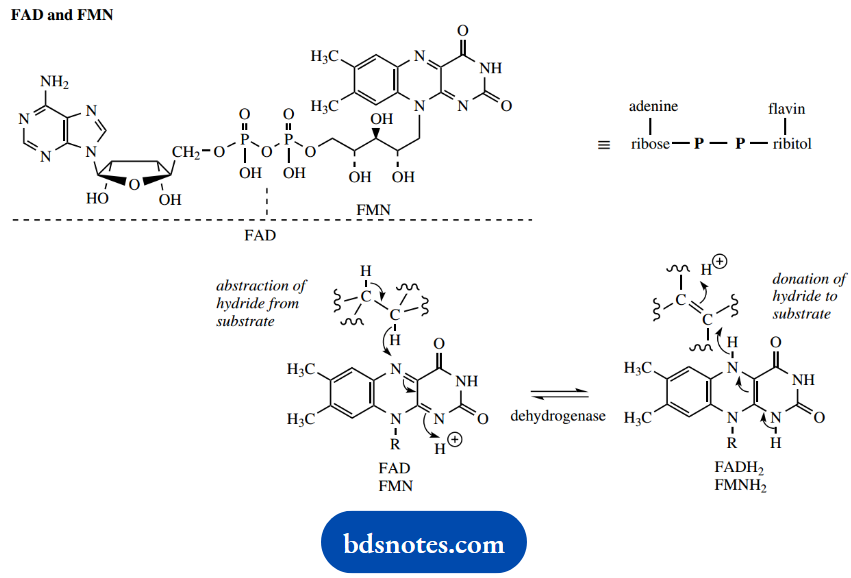
After the substrates containing either CH-OH or CH2-CH2 functional groups have been oxidized by the dehydrogenase enzyme-coenzyme system, energy abstraction from the oxidative transformation now depends upon reoxidation of the reduced coenzyme.
⇒ \(\left.\begin{array}{l}
\mathrm{NAD}(\mathrm{P}) \mathrm{H}+1 / 2 \mathrm{O}_2 \rightarrow \mathrm{NAD}(\mathrm{P})^{+}+\mathrm{H}_2 \mathrm{O} \\
\mathrm{FADH}_2+1 / 2 \mathrm{O}_2 \rightarrow \mathrm{FAD}+\mathrm{H}_2 \mathrm{O} \\
\mathrm{FMNH}_2+1 / 2 \mathrm{O}_2 \rightarrow \mathrm{FMN}+\mathrm{H}_2 \mathrm{O}
\end{array}\right\} \text { large negative } \Delta \mathrm{G}\)
This will be one of the processes shown, all of which have a large negative ΔG and are capable of harnessing this energy via the synthesis of ATP molecules. However, even these are not achievable directly, and the electron transport chain of oxidative phosphorylation is utilized.
Oxidative Phosphorylation And The Electron Transport Chain
The total oxidation of an organic compound using molecular oxygen as the electron acceptor has the potential to yield a very large amount of energy, sufficient for the synthesis of several molecules of ATP from ADP if there could be one efficiently coupled oxidation process.
It is quite unrealistic to achieve this in one step; instead, a multistage process termed oxidative phosphorylation is employed. This process removes packets of energy, more nearly corresponding to the amounts required for the synthesis of single ATP molecules from ADP.
Oxidation of a compound, per atom of oxygen used, can yield up to three molecules of ATP, representing an energy efficiency of about 50%. It takes place in the mitochondria via a sequence of redox (reduction-oxidation) reactions known as the electron transport chain or the respiratory chain and provides the principal source of ATP for an aerobic cell.
The electron transport chain involves a series of compounds acting together, achieving the removal of hydrogen equivalents from organic molecules and eventually reacting them with oxygen to form water.
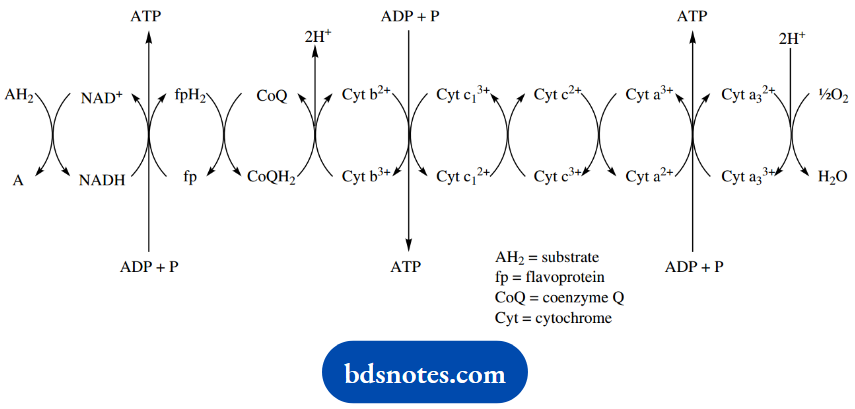
The first part of the chain involves the transfer of a pair of hydrogen atoms, whereas only electrons are transferred in the final stages of the pathway. The individual molecules involved are NAD+, flavoprotein, coenzyme Q (ubiquinone), and a number of cytochromes.
We have already seen the redox reactions of NAD+ and flavoproteins containing FAD or FMN. The term coenzyme Q or ubiquinone covers a range of structures (as shown), depending on the length of the hydrocarbon side-chain, which varies according to species. In humans, the redox carrier is coenzyme Q10 (n = 10).
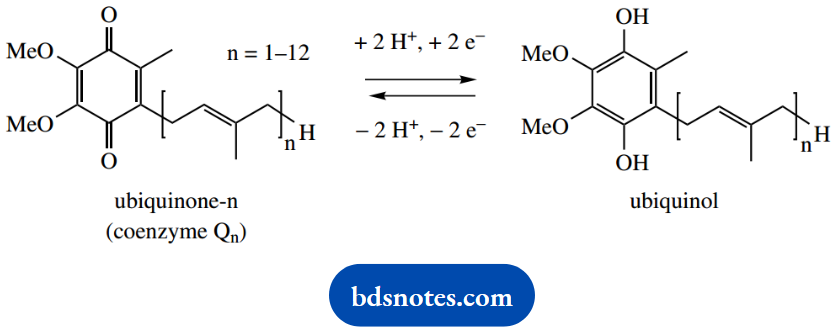
Ubiquinone is readily reduced to ubiquinol, a process requiring two protons and two electrons; similarly, ubiquinol is readily oxidized back to ubiquinone.
This redox process is important in oxidative phosphorylation, in that it links hydrogen transfer to electron transfer. The cytochromes are haem-containing proteins.
As we have seen, haem is an iron-porphyrin complex. Alternate oxidation-reduction of the iron between Fe2+ (reduced form) and Fe3+ (oxidized form) in the various cytochromes is responsible for the latter part of the electron transport chain.
The individual cytochromes vary structurally, and their classification (a, b, c, etc.) is related to their absorption maxima in the visible spectrum.
Most compounds oxidized by the electron transport chain donate hydrogen to NAD+, and then NADH is reoxidized in a reaction coupled with the reduction of a flavoprotein.
During this transformation, sufficient energy is released to enable the synthesis of ATP from ADP. The reduced flavoprotein is reoxidized via the reduction of coenzyme Q; subsequent redox reactions then involve cytochromes and electron transfer processes rather than hydrogen transfer.
In two of these cytochrome redox reactions, there is sufficient energy release to allow ATP synthesis. In due course, electrons are passed to oxygen, which is converted into water in the presence of protons.
The total process whereby hydrogen atoms are passed to NAD+ generates three molecules of ATP per pair of hydrogen atoms. However, substrates with the CH2-CH2 grouping that are oxidized by flavoproteins effectively bypass the first ATP generation step, so only produce two molecules of ATP per pair of hydrogen atoms.
The electron transport chain is vital to aerobic organisms. Interference with its action may be life-threatening.
Thus, cyanide and carbon monoxide bind to haem groups and inhibit the action of the enzyme cytochrome c oxidase, a protein complex that is effectively responsible for the terminal part of the electron transport sequence and the reduction of oxygen to water.
What has been achieved by the participation of coenzyme systems and the electron transport chain is twofold.
First, there is no need for the substrate AH2 to react with oxygen. Second, it provides common routes for the oxidation of many different organic compounds, rather than a specific route for every compound, a vast variety of which will be present in the normal diet.
Although it is somewhat simplistic, many of the non-oxidative reactions of intermediary metabolism can be viewed as additional chemical transformations designed to provide substrates containing either CH-OH or CH2-CH2 that may then be subjected to dehydrogenation.
The Glycolytic Pathway
The glycolytic pathway, or glycolysis, is a metabolic sequence in which glucose is broken down to pyruvic acid. The subsequent fate of pyruvate then depends upon whether or not the organism is aerobic or anaerobic: Under aerobic conditions, pyruvate is oxidized via oxidative phosphorylation; under anaerobic conditions, pyruvate is converted further into compounds such as lactate or ethanol, depending upon the organism.
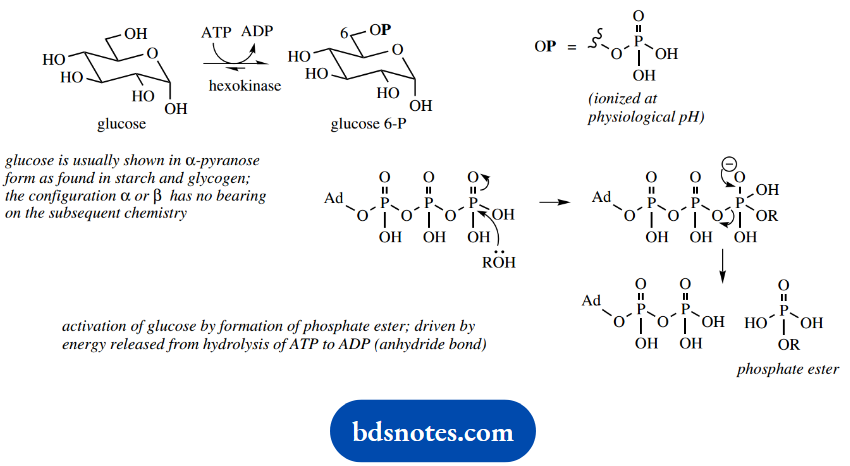
The first step in glycolysis is the phosphorylation of glucose to give the ester glucose 6-phosphate. The glucose starting material may well have come from hydrolysis of starch obtained in the diet, or by utilization of glycogen reserves.
This phosphorylation step is achieved by the reaction of the 6-hydroxyl with the anhydride ATP, during which process ATP is converted into ADP.
This process is driven by the energy contained in the anhydride function of ATP, and represents an expenditure of energy to get the metabolic process started, though the overall objective of glycolysis is to acquire energy via the synthesis of ATP molecules.
Glucose 6-phosphate is then isomerized to fructose 6-phosphate. This conversion of an aldose sugar to a ketose sugar is easy to rationalize in terms of keto-enol tautomerism.
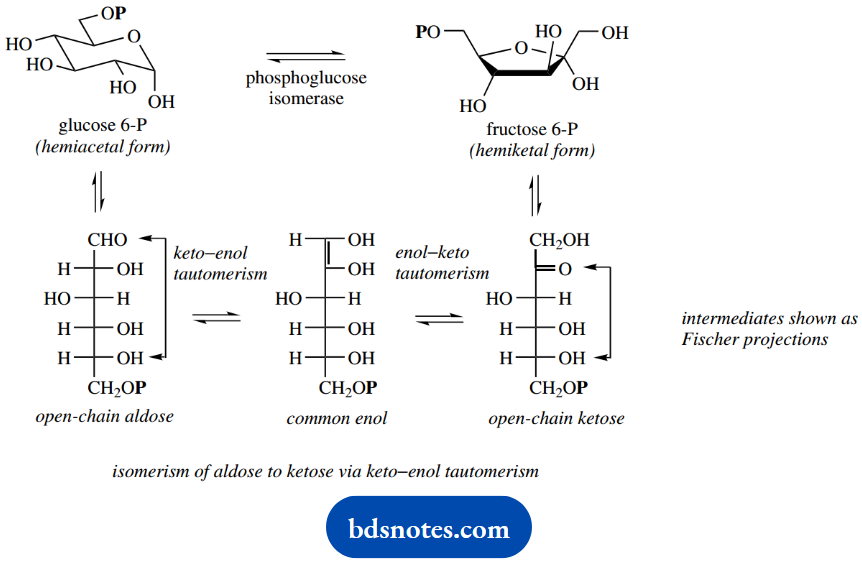
We should first consider the open-chain form of glucose 6-phosphate, rather than its pyranose hemiacetal form. The open-chain aldose has the requirements for enolization, namely a hydrogen α to the aldehyde carbonyl group.
Enolization produces in this case an enediol, which can revert to a keto form in two ways, i.e. reforming the open-chain aldose or, alternatively, producing the ketose fructose 6-phosphate. The enediol may be considered a common enol for the two enolization processes.
The open-chain form of fructose 6- phosphate may then form a hemiketal, as shown, generating a furanose ring.
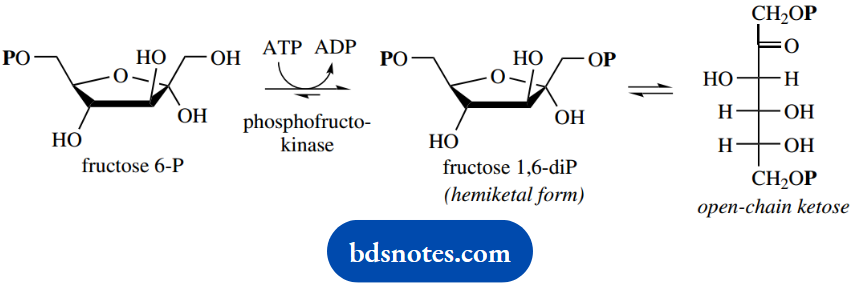
Further phosphorylation, again using ATP as in the first reaction, converts fructose 6-phosphate into fructose 1,6-diphosphate.
Again, there is the expenditure of energy by the use of ATP; we have now used two molecules of ATP, and there has been no net generation of energy. This represents a significant investment before any rewards are forthcoming.
For the subsequent reactions, we need to consider fructose 1,6-diphosphate in its open-chain form rather than the hemiketal originally drawn. Now follows the reverse aldol reaction catalyzed by aldolase, as we have already discussed in some detail elsewhere.
For a simple chemical interpretation, we can write this as involving enolate anions, either as leaving the group in the forward reaction or as a nucleophile in the reverse reaction, but the enzymic reaction is known to utilize enamine derivatives.
It is worth emphasizing again that organisms make use of this reaction both in its forward direction for carbohydrate metabolism and in its reverse direction for carbohydrate synthesis, according to requirements.
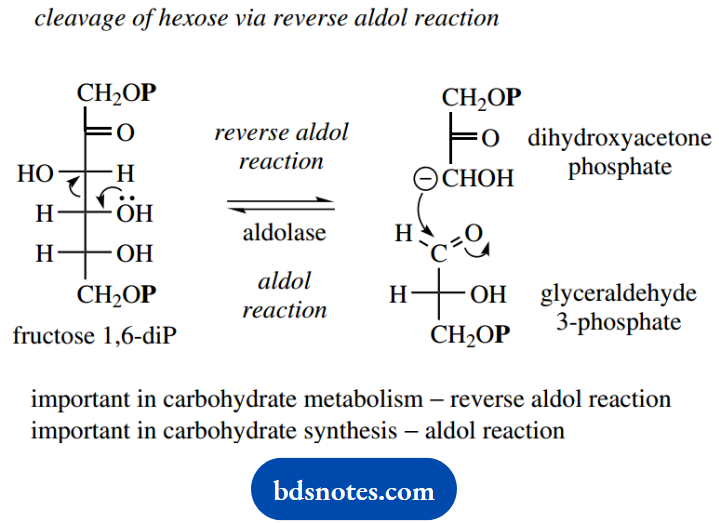
The reverse aldol reaction results in the form- is not on the direct pathway, and is converted into a tion of dihydroxyacetone phosphate and glycersecond molecule of glyceraldehyde 3-phosphate by aldehyde 3-phosphate. Dihydroxyacetone phosphate is the enzyme triose phosphate isomerase.
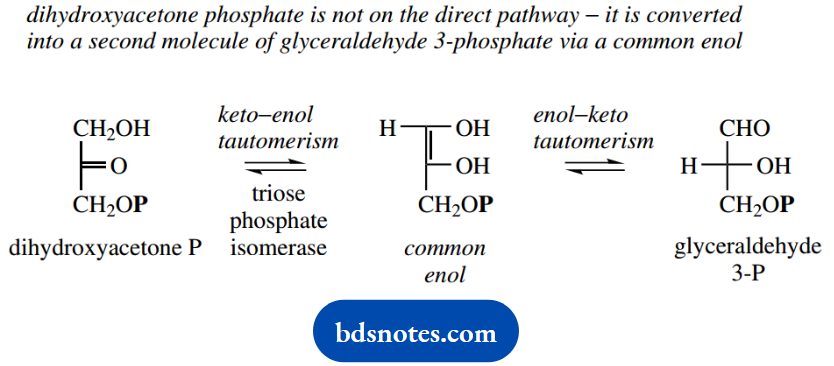
This is achieved by two keto-enol tautomerism reactions and a common enol. Mechanistically, it is identical to the isomerization of glucose 6-phosphate to fructose 6-phosphate seen earlier in the sequence, so we can move on to the next step of the pathway.
In this step, the aldehyde group of glyceraldehyde 3-phosphate appears to be oxidized to an acid, which becomes phosphorylated, and hydrogen is passed to NAD+, which becomes reduced to NADH. We shall see shortly that the fate of this NADH is quite significant.
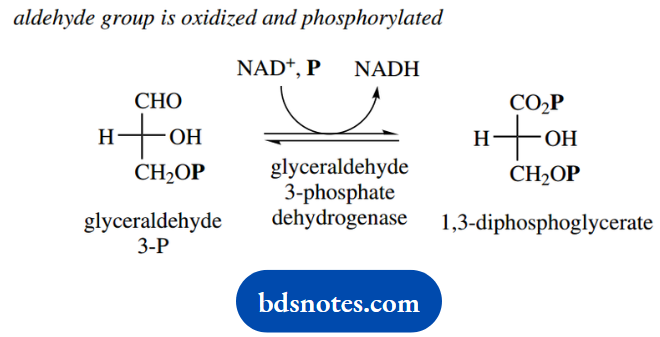
At first glance, this oxidation-phosphorylation reaction seems rather obscure. It becomes much more logical when we see that the enzyme achieves this via a multi-stage process.
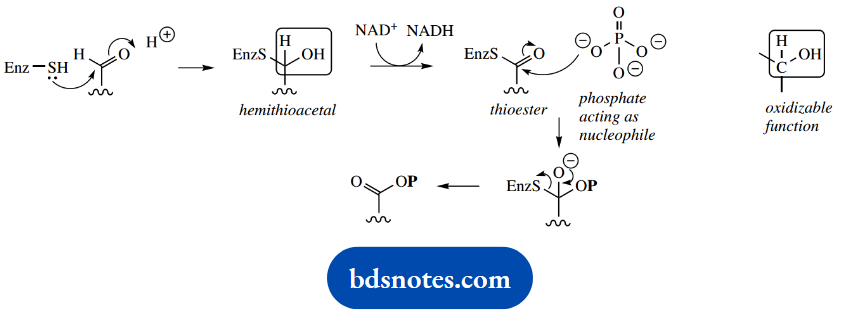
Critical to the reaction is the involvement of a thiol group on the enzyme. This reacts with the aldehyde group of the substrate glyceraldehyde 3-phosphate to form a hemithioacetal. It is this intermediate that reacts with NAD+ since it contains an oxidizable CH-OH function.
The product is then a thioester. The thioester is attacked by a phosphate nucleophile, and a stepwise sequence involving thiol-containing enzyme since it contains a good leaving group, EnzS-, the reaction product is released from the enzyme as 1,3- diphosphoglycerate.
If we look at the structure of 1,3-diphosphoglycerate, we can see that it is actually an anhydride, albeit a mixed anhydride of carboxylic acid and phosphoric acid. Accordingly, we expect it to be fairly reactive toward nucleophiles, and indeed it is.
It is sufficiently reactive that hydrolysis liberates enough energy to synthesize ATP from ADP.
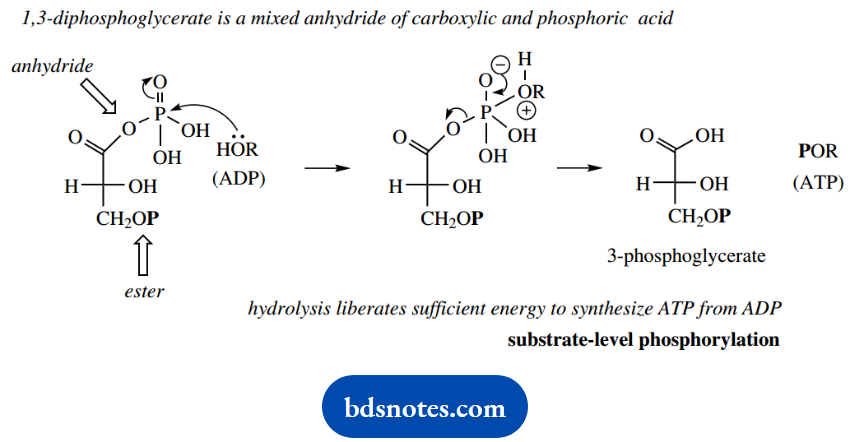
ATP synthesis is achieved by ADP acting as the nucleophile towards this mixed anhydride, attacking the P=O bond, with the carboxylate being the leaving group.
Note that this reaction is favored, whereas the alternative possibility involving hydrolysis of the phosphate ester does not occur.
This is precisely what we would predict knowing the different reactivities of anhydrides and esters.
This direct synthesis of ATP by a process in which ADP acquires an additional phosphate from a suitable donor molecule is often termed substrate-level phosphorylation, differentiating it from ATP synthe¬sis that is achieved through oxidative phosphorylation.
After donating its phosphate group to ADP, 1,3- diphosphoglycerate is converted into 3-phospho- glycerate. This reaction is followed by enzymic modification to 2-phosphoglycerate.
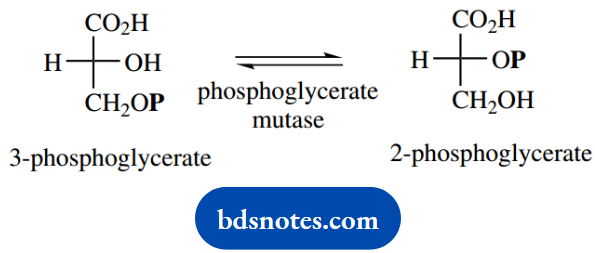
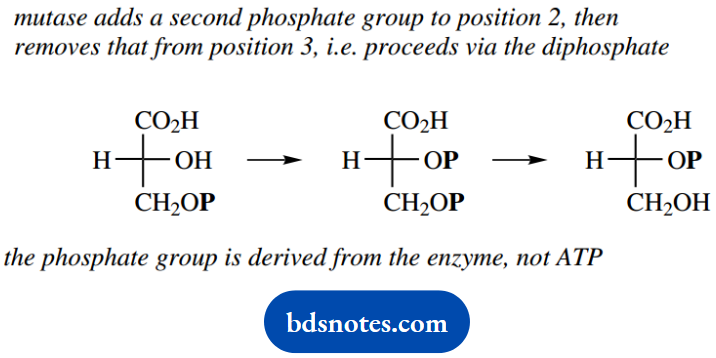
Although this reaction is catalyzed by a mutase, which perhaps suggests this is a rearrangement reaction, there is no transfer of the phosphate group to the adjacent hydroxyl.
Instead, this reaction proceeds via an intermediate diphosphate, so we are actually seeing a phosphorylation-dephosphorylation or esterification-hydrolysis sequence. There is an unusual aspect of this extra phosphorylation, and that is that no ATP is involved.
Instead, the new phosphate group is derived from the enzyme itself. Although this is unexpected, it does avoid another energy-requiring reaction and the use of precious ATP.
Then follows an elimination reaction, in which water is removed from 2-phosphoglycerate to yield phosphoenolpyruvate.
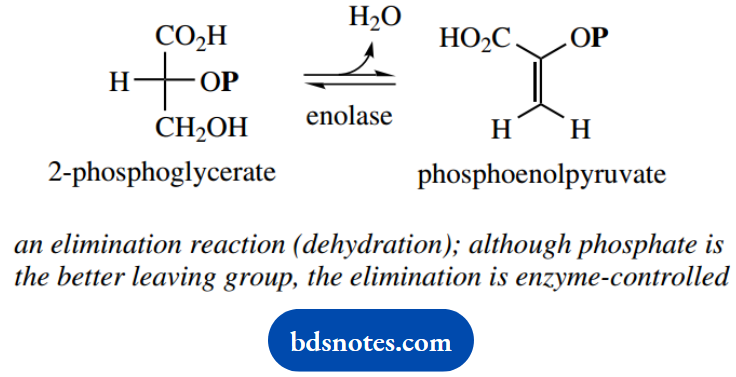
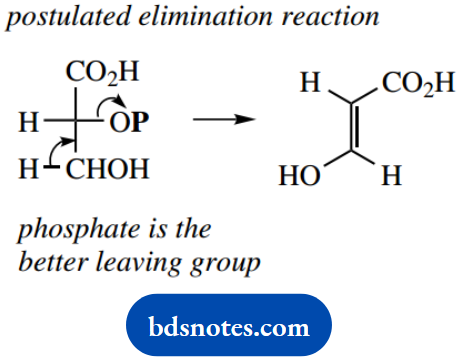
This reaction is catalyzed by an enzyme called enolase; though this may appear quite straightforward, it is chemically unusual. Eliminations depend upon the presence of a suitable leaving group, and by far the better-leaving group in 2-phosphoglycerate is the phosphate.
We might predict that the product from an elimination reaction on 2-phosphoglycerate would logically be the alternative enol system. That this does not occur indicates and emphasizes the enzyme’s special contribution to the reaction.
The product phosphoenolpyruvate is able to donate its phosphate group directly to ADP, resulting in ATP synthesis.
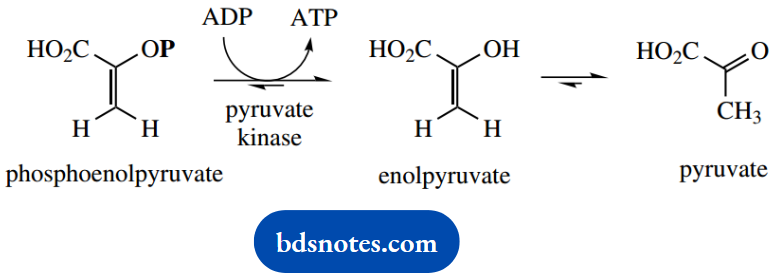
Substrate-Level Phosphorylation
Although phosphoenolpyruvate is only an enol ester, hydrolysis gives an unfavoured enol; tautomerism to the keto form is the driving force for the reaction and results in a large negative ΔG
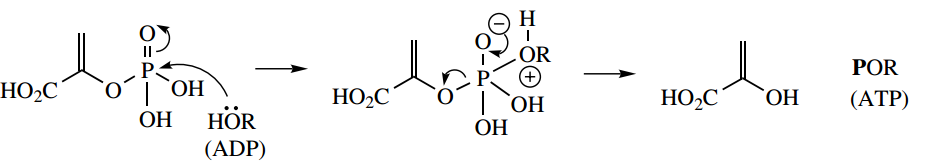
This is another example of substrate-level phosphorylation but differs from the earlier example that involved hydrolysis of a mixed anhydride. Here, we have merely the hydrolysis of an ester, and thus a much lower release of energy.
In fact, with 1,3- diphosphoglycerate, we specifically noted the difference in reactivity between the anhydride and ester groups. So how can this reaction lead to ATP synthesis? The answer lies in the stability of the hydrolysis product, enolpyruvic acid.
Once formed, this enol is rapidly isomerized to its keto tautomer, pyruvic acid, with the equilibrium heavily favoring the keto tautomer.
The driving force for the substrate-level phosphorylation reaction is actually the position of equilibrium in the subsequent tautomerization.
This completes the glycolytic pathway; well, almost. To maintain the operation of the pathway, the NAD+ used in the conversion of glyceraldehyde 3-phosphate into 1,3-diphosphoglycerate must be regenerated from its reduced form NADH, since only small amounts of the coenzyme will be available to the organism.
If the organism is aerobic, then it is possible to use the oxidative phosphorylation processes to regenerate NAD+ from NADH, and in so doing also achieve the synthesis of ATP.
However, for anaerobes, or for aerobes under temporary anaerobic conditions, pyruvate synthesized in the last reaction is modified further to achieve this end.
For example, certain organisms use NADH to reduce pyruvate to lactate and regenerate NAD+. This process might occur in actively exercised muscles, when there is a temporary shortage of oxygen, leading to a build-up of lactic acid and ensuing cramp pains.
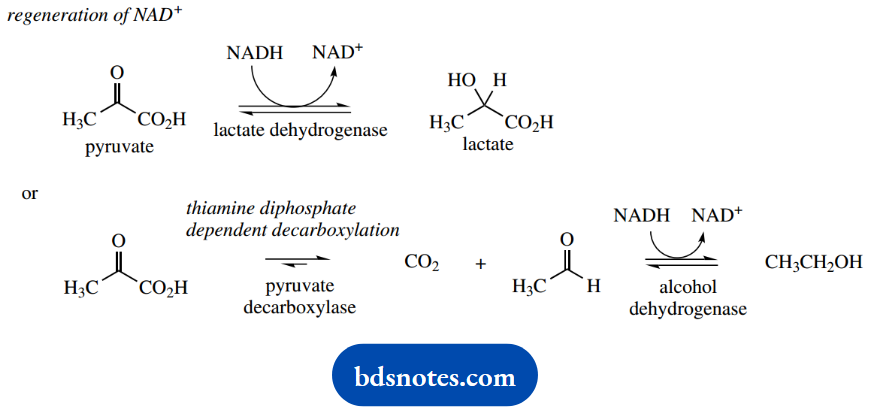
Other organisms are equipped to produce ethanol, by employing a thiamine diphosphate-dependent decarboxylation of pyruvate to acetaldehyde and NAD+ is regenerated by reducing the acetaldehyde to ethanol.
This is a characteristic of baker’s yeast and forms the essential process for both bread making (production of CO2) and the brewing industry (formation of ethanol).
The glycolytic pathway is crucial to anaerobes for ATP production; this is reflected in the fact that ATP synthesis is achieved via substrate-level phosphorylation, and does not depend on the availability of oxidative phosphorylation.
The energy yield from glycolysis for the anaerobic decomposition of glucose to 2 mol of lactic acid may be calculated as follows:
- 2 mol of ATP are used up in phosphorylations;
2 mol of ATP are gained per half molecule of glucose, i.e. a total of 4 mol ATP; - Net yield from glucose → 2 mol lactic acid = 2 mol ATP.
The Krebs Cycle
The Krebs cycle is sometimes still referred to as the citric acid cycle, citric acid being one of the intermediates involved, and even the tricarboxylic acid cycle, in that several of the intermediates are triads.
As the name suggests, the process is a cycle, so that there is a reasonably constant pool of intermediates functioning in an organism, and material for degradation is processed via this pool of intermediates.
Overall, though, the material processed does not increase the size of the pool. The compound that enters the cycle is the thioester acetyl-coenzyme A (acetyl-CoA).
We have just seen that anaerobic organisms metabolize pyruvate from the glycolytic pathway by various means, but that the prime objective is to reoxidize NADH to NAD+.
In aerobic organisms, reoxidation of NADH is achieved via oxidative phosphorylation, generating ATP in the process, and there is no longer any need to sacrifice pyruvate for this purpose.
Accordingly, pyruvate from glycolysis is converted into acetyl-CoA by a process known as oxidative decarboxylation. but, for the moment, it can be represented by the equation.

The whole process is multi-step, and catalyzed by the pyruvate dehydrogenase enzyme complex, which has three separate enzyme activities.
During the transformation, an acetyl group is effectively removed from pyruvate, and passed via carriers of thiamine diphosphate (TPP) and lipoic acid eventually to coenzyme A, a complex material whose principal functional group involved in metabolic reactions is a thiol.
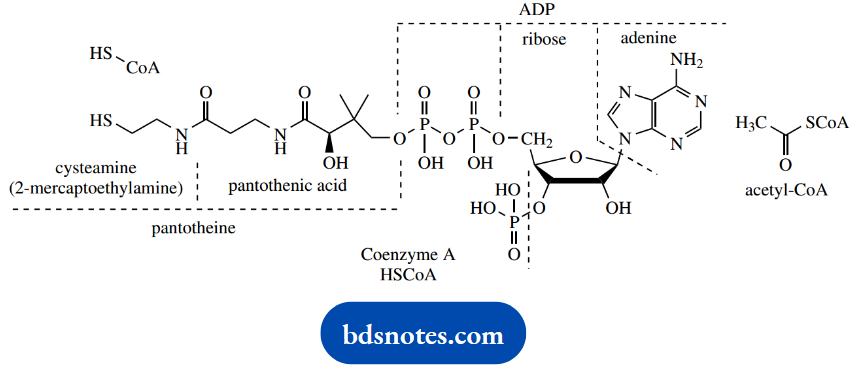
The requirement for NAD+ is to reoxidize the lipoic acid carrier. It is worth mentioning that the pyruvate → acetaldehyde conversion we considered at the end of the glycolytic pathway involves the same initial sequence, and pyruvate decarboxylase is another thiamine diphosphate-dependent enzyme.
Acetyl-CoA is a thioester of acetic acid with coenzyme A. It is a remarkably common intermediate in many metabolic degradative and synthetic pathways, for which the reactivity of the thioester function plays a critical role.
There are two major sources of the acetyl-CoA entering the Krebs cycle: glycolysis via the oxidative decarboxylation of pyruvate and fatty acid degradation. There are other minor sources of acetyl-CoA, including the metabolism of amino acids from protein.
The Krebs cycle intermediate that reacts with acetyl-CoA is oxaloacetate, and this reacts via an aldol reaction, giving citryl-CoA. However, the enzyme citrate synthase also carries out hydrolysis of the thioester linkage, so that the product is citrate; hence the terminology ‘citric acid cycle’.
The hydrolysis of the thioester is actually responsible for disturbing the equilibrium and driving the reaction to completion.
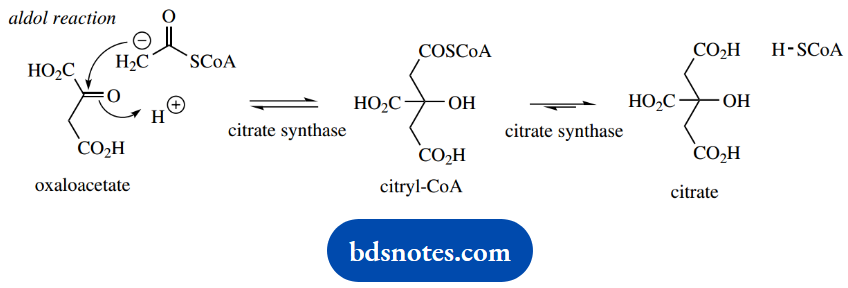
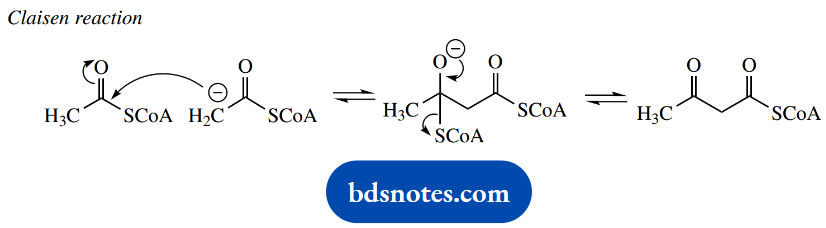
The aldol reaction is easily rationalized, with acetyl-CoA providing an enolate anion nucleophile that adds to the carbonyl of oxaloacetate – easily rationalized, but surprising.
Oxaloacetate is more acidic than acetyl-CoA, in that there are two carbonyl groups flanking the methylene. If one were to consider a potential base-catalyzed reaction between these two substrates, then logic suggests that oxaloacetate would be preferentially converted into an enolate anion nucleophile.
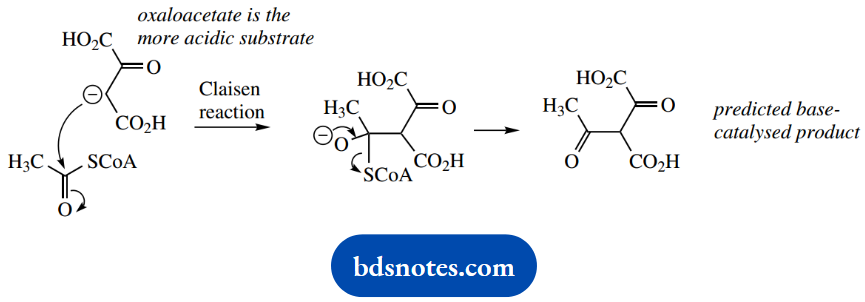
This could then attack the carbonyl of acetyl-CoA, but via a Claisen reaction, since there is a thiolate leaving group.
That citrate synthase achieves an aldol reaction (as shown) reflects that the enzyme active site must have a basic residue appropriately positioned to abstract a proton from acetyl-CoA allowing it to act as the nucleophile.
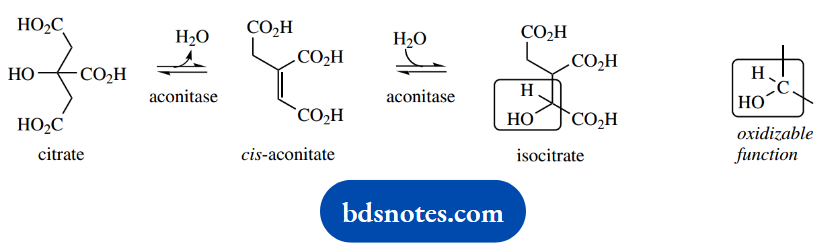
Citrate is subsequently isomerized to isocitrate; this involves dehydration and rehydration via the intermediate cis-aconitate. Both reactions are catalyzed by the single enzyme aconitase. They may be considered simply as acid-catalyzed elimination followed by acid-catalyzed addition reactions.
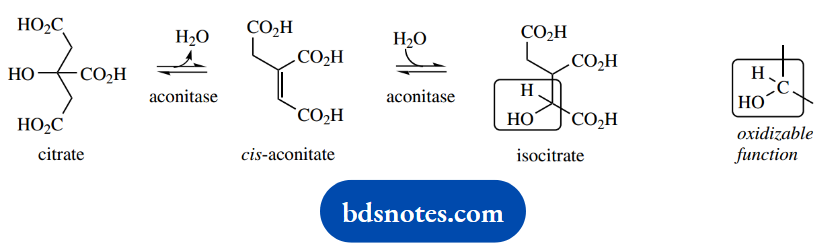
Worthy of note in this reaction is that citrate displays prochirality. The methylene carbons may be considered prochiral, in that enzymic elimination of a proton is likely to be entirely stereospecific.
In addition, the apparently equivalent side chains on the central carbon are also prochiral and going to be positioned quite differently on the enzyme.
This means that only one of these side chains is involved in the dehydration-rehydration sequence, and it can be shown from labeling studies that the side chain modified is not the one that was recently derived from acetyl-CoA as a nucleophile.
In isocitrate, there is a CHOH group that is available for oxidation via the coenzyme NAD+ and the enzyme isocitrate dehydrogenase. NADH will then be reoxidized via oxidative phosphorylation, and lead to ATP synthesis.
The oxidation product from isocitrate is oxalosuccinate, β-ketoacid that easily decarboxylates through an intramolecular hydrogen-bonded system.
Although thermal (non-enzymic) decarboxylation would probably occur readily, it turns out that the enzyme isocitrate dehydrogenase also catalyzes this reaction.
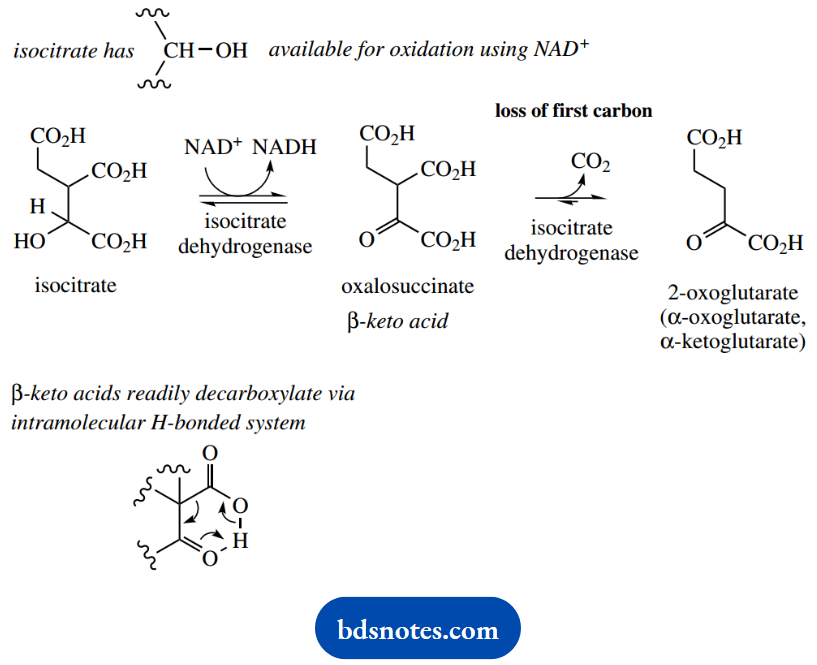
The product is 2-oxoglutarate, sometimes referred to as α-oxoglutarate or α-ketoglutarate. Note specifically that we have just lost one of the carbon atoms. Oxaloacetate (a C4 compound) reacted with acetyl-CoA (C2) to give citrate (C6), and this reaction now gives us a C5 compound; to complete the cycle and get back to C4, we shall need to lose another carbon atom.
This is achieved in the next reaction catalyzed by 2-oxoglutarate dehydrogenase.
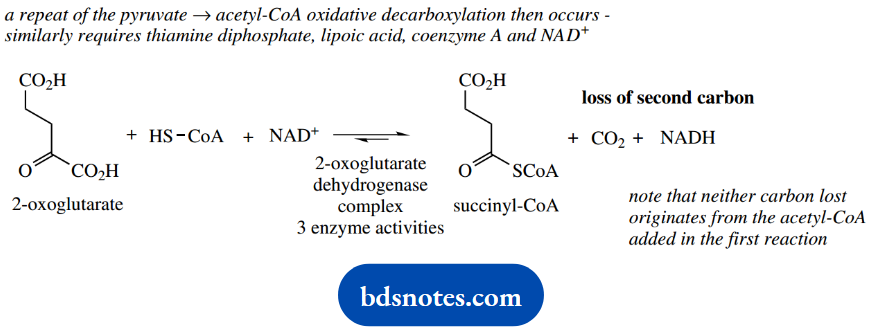
Now this reaction is effectively a repeat of the pyruvate → acetyl-CoA oxidative decarboxylation we saw at the beginning of the Krebs cycle. It similarly requires thiamine diphosphate, lipoic acid, coenzyme A, and NAD+.
A further feature in common with that reaction is that 2-oxoglutarate dehydrogenase is also an enzyme complex comprised of three separate enzyme activities.
2-Oxoglutarate is thus transformed into succinyl-CoA, with the loss of a further carbon as CO2, and producing NADH that can be exploited in ATP synthesis via oxidative phosphorylation.
Note that, because of the prochirality in citric acid and subsequent enzymic selectivity, neither of the carbon atoms lost in the two decarboxylations originates from the acetyl-CoA molecule added in the first reaction, the aldol addition.
These carbons are not lost until further cycles of the pathway have been completed.
The product succinyl-CoA is able to participate in ATP synthesis as an example of substrate-level phosphorylation – we met some other examples in the glycolytic pathway.
Essentially, hydrolysis of succinyl-CoA liberates sufficient energy that it can be coupled to the synthesis of ATP from ADP.
However, guanosine triphosphate (GTP) is the nucleoside phosphate produced, rather than ATP. ATP is then produced indirectly from GTP.
There appears to be no obvious reason why this reaction should be coupled to the synthesis of GTP, rather than to the direct synthesis of ATP; the other product of the reaction is succinate.
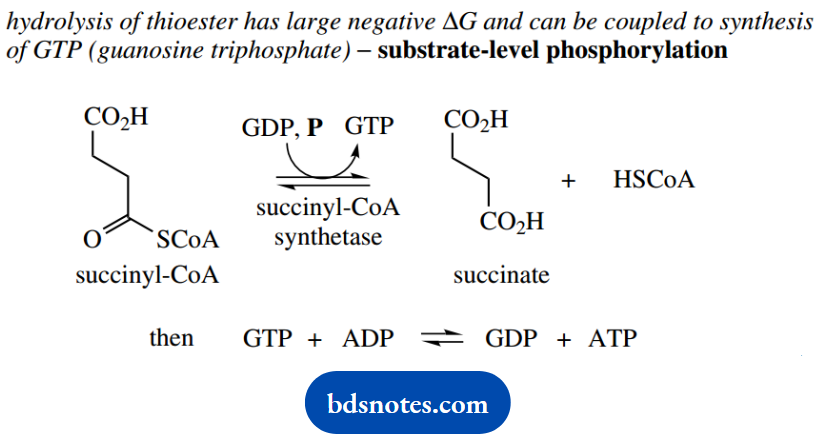
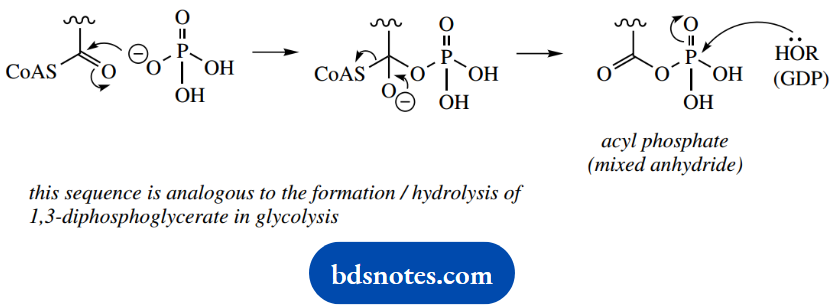
When we investigate this substrate-level phosphorylation reaction in detail, we find it also involves a molecule of phosphate. Phosphate reacts initially with succinyl-CoA, converting the thioester into an acyl phosphate, which is, of course, a mixed anhydride.
It is actually hydrolysis of this mixed anhydride that can be coupled to nucleoside triphosphate synthesis, and it is fitting to compare this with the formation and hydrolysis of 1,3-diphosphoglycerate in the glycolytic pathway.
As we move on in the Krebs cycle, the next reaction is the oxidation of the CH2-CH2 grouping in succinate to give the unsaturated diacid fumarate. We have already looked at this type of oxidation and seen that it involves a dehydrogenase enzyme coupled to a flavin nucleotide coenzyme.
For this reaction, the coenzyme is FAD. The reduced form of FAD can then be reoxidized to FAD via oxidative phosphorylation, generating energy in the form of ATP in the process.
The sequence continues with hydration, and addition and results in the formation of oxaloacetate, which of water, to produce malate, which completes the cycle and regenerates the substrate to an oxidizable CHOH group. Oxidation involves NAD+, which reacts with further acetyl-CoA.
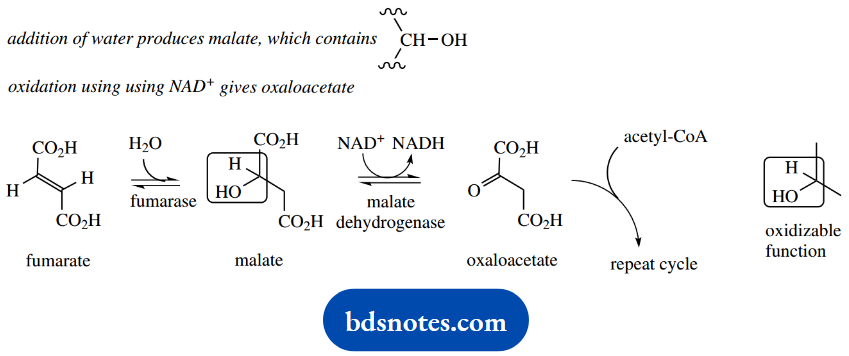
This sequence of reactions, namely oxidation of CH2-CH2 to CH=CH, then hydration to CH2-CHOH, followed by oxidation to CH2-CO, is a sequence we shall meet again in the P-oxidation of fatty acids.
The first oxidation utilizes FAD as a coenzyme and the second NAD+. In both cases, participation in the oxidative phosphorylation system allows the regeneration of the oxidized coenzyme and the subsequent generation of energy in the form of ATP.
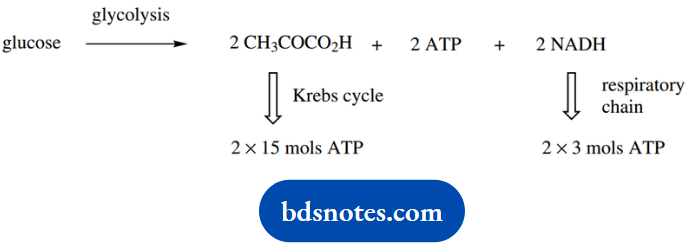
The energy yield from the Krebs cycle by the aerobic breakdown of pyruvate may be calculated as follows. overall:
CH3COCO2H + 3 H2O → 3 CO2 + 5 x 2H
- five pairs of hydrogen atoms are available for oxidation;
- four pairs are passed to NAD+ and via the respiratory chain yield 4 x 3 = 12 mol ATP;
- one pair is passed to FAD and via the respiratory chain yields 2 mol ATP;
- there is also the gain of one ATP via GTP;
- therefore, there will be a total yield of 15 mol ATP.
By combining the glycolytic pathway, the Krebs cycle, and oxidative phosphorylation, the energy yield from the aerobic degradation of glucose will be
Total = 38 mols ATP
The total yield of 38 mol ATP by aerobic degradation of glucose may not be achieved under all circumstances, but it is, nevertheless, considerably more efficient than that from the anaerobic breakdown, namely 2 mol ATP.
Oxidation Of Fatty Acids
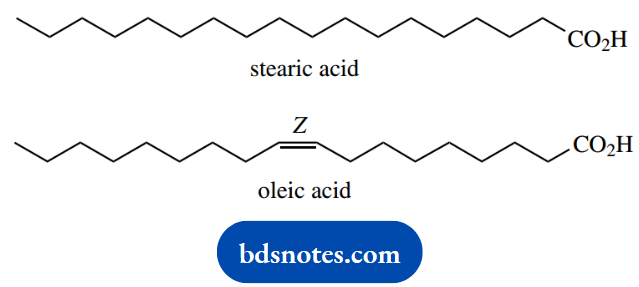
Fat degradation provides a major source of energy for most organisms. Fats are esters of glycerol with long-chain fatty acids and are hydrolyzed by the action of enzymes called lipases. This gives the alcohol portion, glycerol, together with a range of fatty acids, such as stearic acid.
Most fats taken in the diet provide a range of fatty acids of varying chain length and different levels of unsaturation, according to the source. Because of their biosynthetic origin, the vast majority of fatty acids have an even number of carbon atoms.
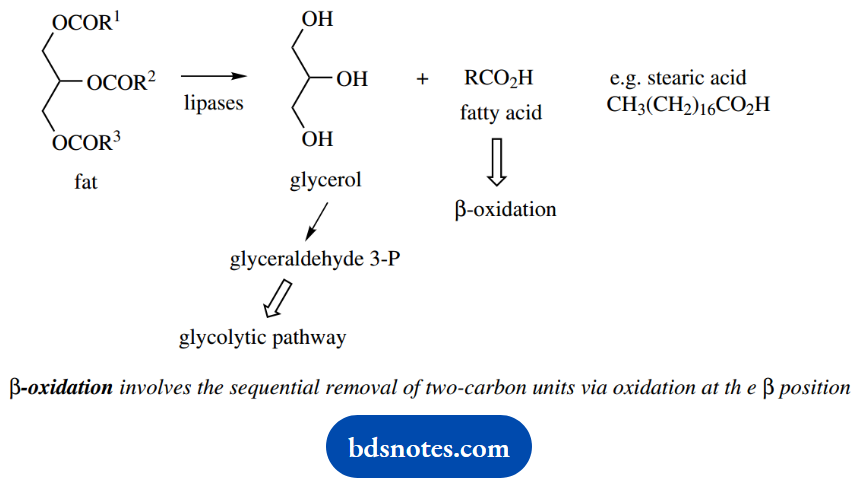
Glycerol provides a minor source of energy, in that it can be modified readily to glyceraldehyde 3- phosphate, one of the intermediates in the glycolytic pathway.
The fatty acids are metabolized by a process termed β-oxidation, which involves the sequential removal of two-carbon units via oxidation at the β- position. The process for saturated fatty acids will now be described.
Metabolism Of Saturated Fatty Acids
The free fatty acid needs activating before it can be metabolized. This is achieved by conversion into its thioester by esterification with coenzyme A.
We have already seen that thioesters are reactive entities, and it is reasonable, therefore, to suppose that such activation will cost energy. It is achieved in a two-stage reaction catalyzed by a single enzyme, an acyl-CoA synthetase. Energy is supplied in the form of ATP.
The fatty acid is initially converted into an acyl- AMP derivative by attack of the carboxylate as a nucleophile onto the P=O system of ATP, with loss of diphosphate as a leaving group.
This reaction is far from favorable, and the equilibrium is disturbed by subsequent pyrophosphatase-catalyzed hydrolysis of diphosphate into two molecules of phosphate.
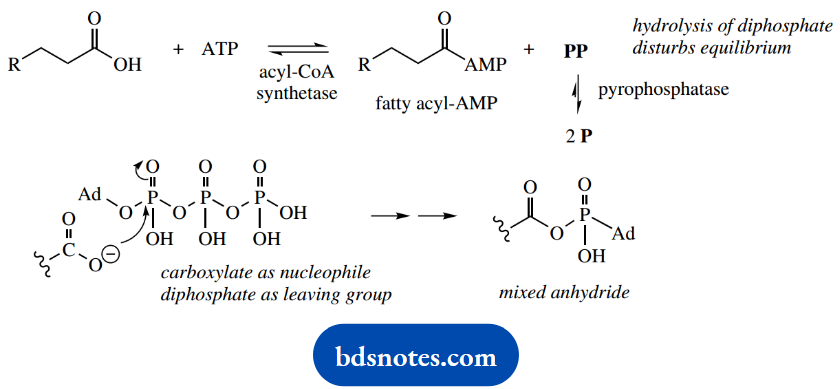
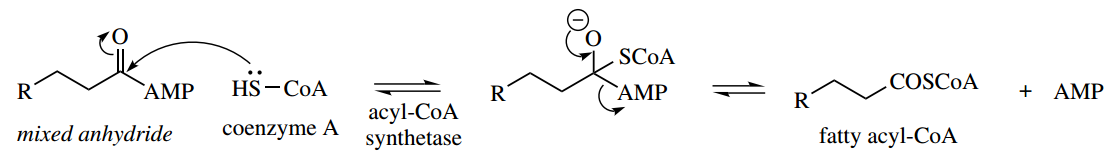
This means that the energy demands (ATP → AMP) are equivalent to two ATP → ADP transformations. However, the product fatty acyl-AMP is actually a reactive mixed anhydride and may be attacked by the thiol group of coenzyme A, giving the required thioester. We have met an analogous series of reactions in non-ribosomal peptide biosynthesis.
Oxidation at the β-position is then achieved by the same sequence of dehydrogenation, hydration, and dehydrogenation reactions that we have seen earlier in the succinate → fumarate → malate → oxaloacetate transformations in the Krebs cycle.
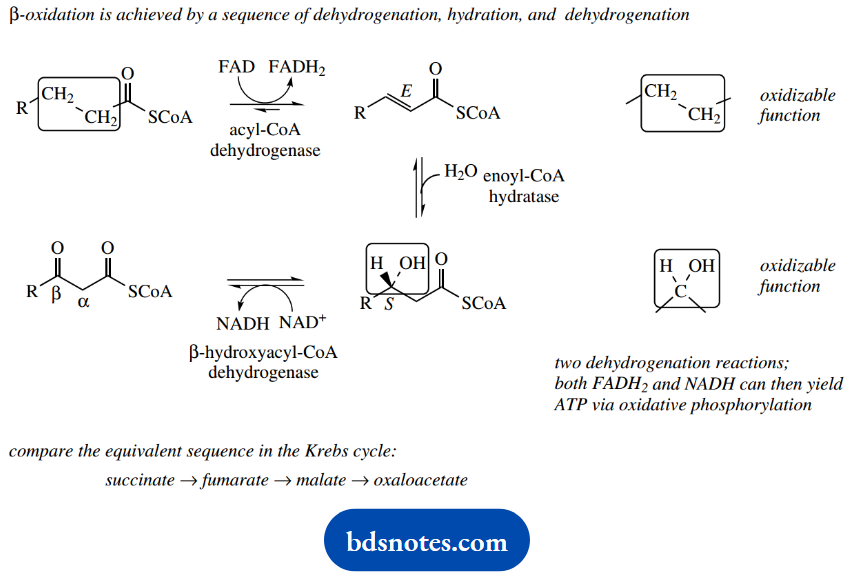
Because of the enzyme specificity in the hydration step, the new carbonyl group is introduced P to the original thioester carbonyl.
The sequence includes two dehydrogenation reactions and involves both FAD and NAD+ as coenzymes. The reduced forms of these coenzymes can be reoxidized by means of oxidative phosphorylation, and can, therefore, yield ATP.
Although the reactions just described form the basis of β-oxidation, the terminology β-oxidation when applied to fatty acid metabolism is usually understood to include the next step, the sequential chain shortening.
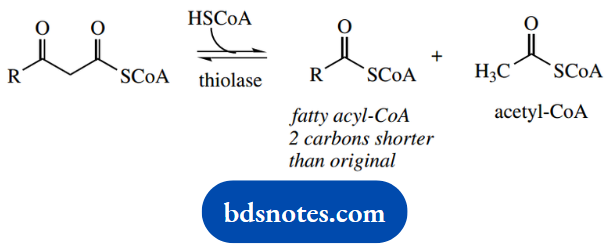
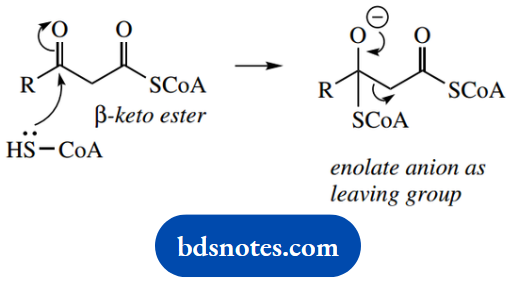
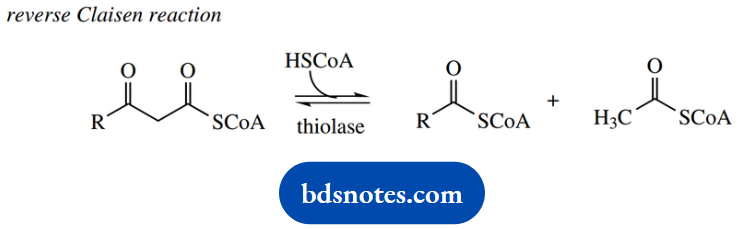
There follows cleavage of acetyl-CoA from the end of the chain via a reverse Claisen reaction. This requires the use of a molecule of coenzyme A as a nucleophile, with the loss of the enolate anion of acetyl-CoA as the leaving group.
The net result is the production of a new fatty acyl-CoA that is two carbons shorter than the original and a molecule of acetyl-CoA that can be metabolized via the Krebs cycle.
Thus, a fatty acid such as stearic acid (C18), after activation, can undergo the P-oxidation and chain-shortening process eight times, producing nine molecules of acetyl-CoA for further metabolism.
stearic acid can undergo P-oxidation / chain shortening 8x
CH3(CH2)16CO2H —► 9 CH3CO-SCoA ⇒ Krebs cycle
The overall energy yield from β-oxidation may thus be calculated as follows:
- Each sequence of β-oxidation involves the passage of one pair of hydrogen atoms to FAD (which yields 2 mol ATP via the respiratory chain) and one pair to NAD+ (which yields 3 mol ATP via the respiratory chain).
- Stearoyl-CoA thus produces 8 x 5 = 40 mol ATP from eight β-oxidations.
- The nine acetyl-CoA moles generated will yield 9 x 12 = 108 mol ATP via the Krebs cycle.
- However, the activation of stearic acid to stearoyl- CoA is achieved by the reaction ATP → AMP, which is the effective loss of 2 mol ATP; nevertheless, only one activation step is necessary per fatty acid.
- The total yield is thus 40 + 108 — 2 = 146 mol ATP.
Comparison Of Fat And Carbohydrate As Energy Stores
It is instructive to compare the energy yield from three molecules of glucose (C18) with that from one molecule of stearic acid (also C18).
- We saw that aerobic degradation of each molecule of glucose via glycolysis and the Krebs cycle gave 38 mol ATP; three molecules would thus give 3 x 38 = 114 mol ATP.
- Stearic acid gives 146 mol ATP. The higher energy yield per carbon atom from fatty acid compared with carbohydrates reflects its higher level of reduction, which consequently allows more oxidation.
- Thus, fat is logically the preferred storage molecule for carbohydrates. This is borne out in practice.
- A 70 kg man would typically have fat reserves of about 7 kg, equivalent to his energy needs for 1 month, and carbohydrate reserves of about 0.35 kg, equivalent to his energy needs for only about 1 day.
- This is undoubtedly why low-carbohydrate diets have proved so effective for rapid weight loss. As soon as the reserves of carbohydrates are used up, the body resorts to metabolizing fat for its energy needs. This continues whenever carbohydrate intake is limited.
It should also be appreciated that although carbohydrates can readily be converted into fat, fat is not readily converted into carbohydrates in animals. Fat metabolism produces acetyl-CoA, which is then usually metabolized completely via the Krebs cycle.
Metabolism Of Unsaturated Fatty Acids
Much of the fat taken in via the diet will contain unsaturated fatty acids, particularly that portion that originates from plant material. For example, the fats in olive oil contain up to 85% oleic acid (C18 unsaturated), and only relatively small amounts of saturated fatty acids.
Animal fats have a much higher proportion of saturated fatty acid derivatives, but they still contain a substantial level of unsaturated fatty acids.
The fatty acid analysis of butterfat, for example, shows it contains about 28% oleic acid, and most of the remainder is composed of saturated fatty acids: 13% stearic acid (C18), 29% palmitic acid (C16), 12% myristic acid (C14), and other shorter- chain saturated fatty acids.
The vast majority of natural unsaturated fatty acids have one or more double bonds with the Z or cis configuration.
The metabolism of unsaturated fatty acids is similar to that of the saturated compounds just described, but additional enzymic reactions are necessary.
Thus, oleoyl-CoA, the CoA ester of oleic acid, will undergo β-oxidation three times, until the C12 derivative is reached. The 3,4-Z-double bond in this compound now prevents the normal dehydrogenation step that should introduce a 2,3-double bond.
As a result, the normal degradative process stops until this compound is isomerized to the normal intermediate with a 2,3-E-double bond by the action of an isomerase enzyme.
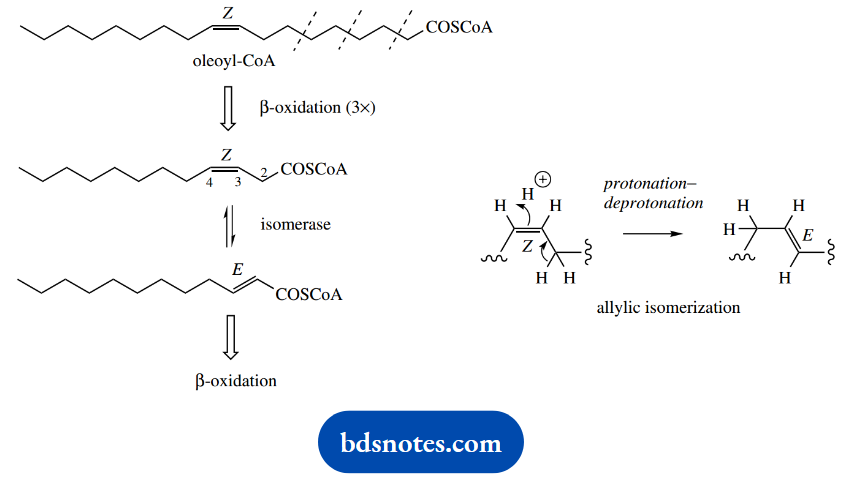
By means of this additional step, the P-oxidation process can then continue as normal. The energy yield will be only slightly less than that for stearoyl-CoA since there is the omission of transfer of one pair of hydrogen atoms to FAD, and consequently loss of 2 mol ATP.
Of course, the double bond in the starting ester may end up in the correct position for the P-oxidation processes, but it turns out that the usual Z or cis configuration of this double bond is wrong for normal enzymes.
Although hydration of the Z double bond occurs, the configuration of the hydroxy derivative is wrong for the subsequent dehydrogenase, so an inversion to the required configuration is achieved by the action of an epimerase enzyme. β-Oxidation processes can then continue normally.
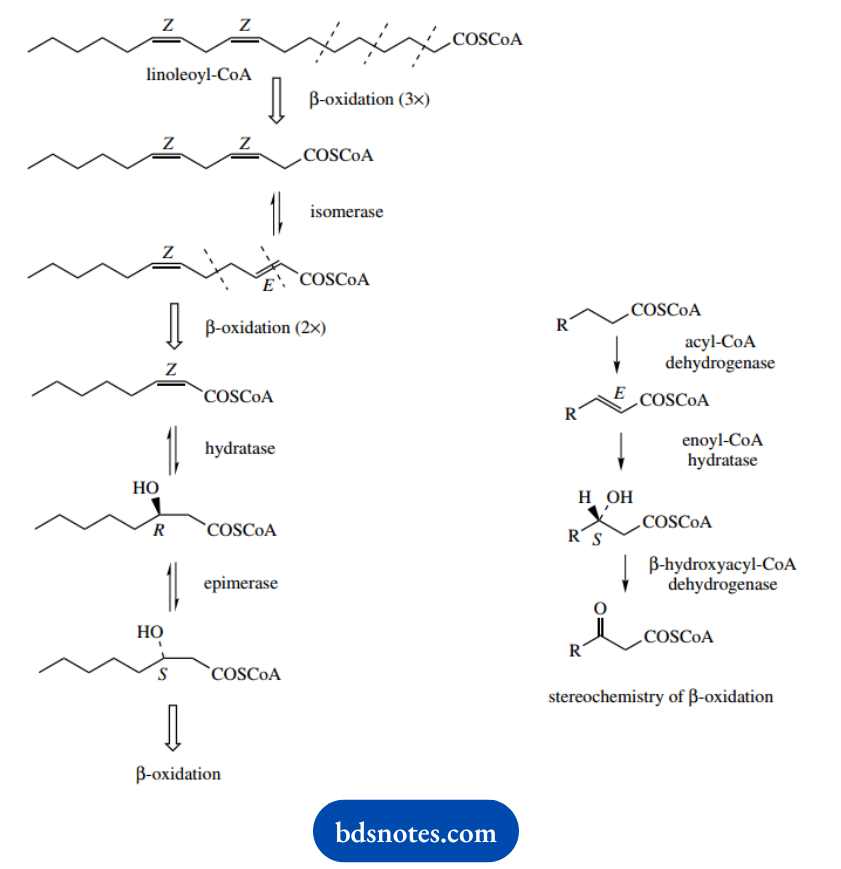
These processes are shown for the CoA ester of linoleic acid, the most common of the polyunsaturated acids.
Synthesis Of Fatty Acids
Fatty acid synthesis provides an organism with a means of storing energy in the form of an organic reverse Claisen reaction molecule that can be degraded by oxidative reactions whenever necessary.
In principle, fatty acid synthesis is the reverse of fatty acid metabolism, though there are some fundamental differences, which are quite logical when we consider the chemical reactivity of the intermediate reagents.
Fatty acid degradation involves a reverse Claisen reaction Therefore, we could consider using the Claisen reaction in fatty acid synthesis.
However, a more favorable pathway is used, employing a more reactive nucleophile. Rather than using the enolate anion derived from acetyl-CoA, nature uses the enolate anion derived from malonyl-CoA. Malonyl-CoA is obtained from acetyl-CoA by means of an enzymic carboxylation reaction, incorporating CO2 (usually from the soluble form bicarbonate).
Now CO2 is a particularly unreactive material, so this reaction requires the input of energy (from ATP) and the presence of a suitable coenzyme, biotin, as the carrier of CO2.
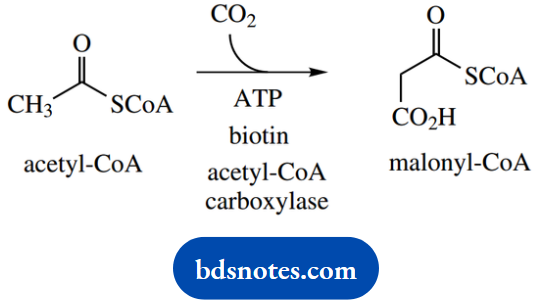
The conversion of acetyl-CoA into malonyl-CoA increases the acidity of the α-hydrogens since the acidic protons are flanked by two carbonyl groups, and thus it is easier to generate a nucleophile for the Claisen condensation. We should relate this to the use of diethyl malonate rather than ethyl acetate as a nucleophile.
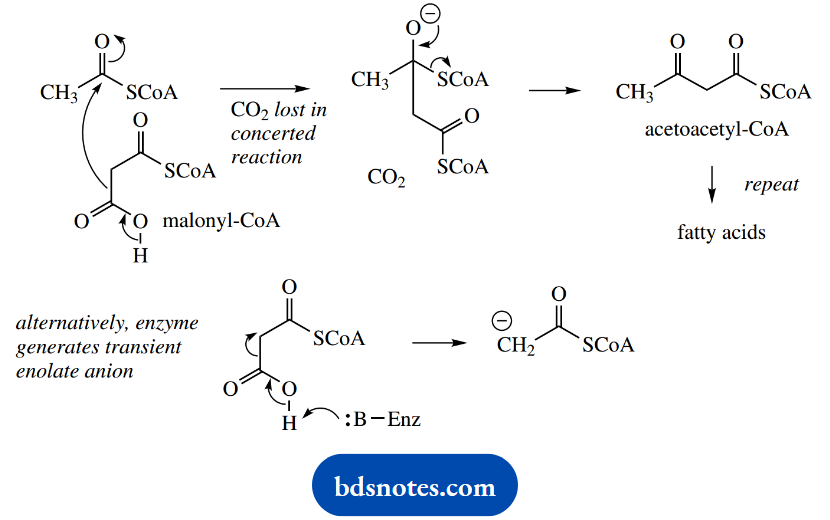
The Claisen reaction can now proceed smoothly, but nature introduces another little twist. The carboxyl group introduced into malonyl-CoA is simultaneously lost by a decarboxylation reaction during the Claisen condensation.
Accordingly, we now see that the carboxylation step helps to activate the α- carbon and facilitate Claisen condensation, and the carboxyl is immediately removed on completion of this task.
An alternative rationalization is that decarboxylation of the malonyl ester is used to generate the acetyl enolate anion without any requirement for a strong base.
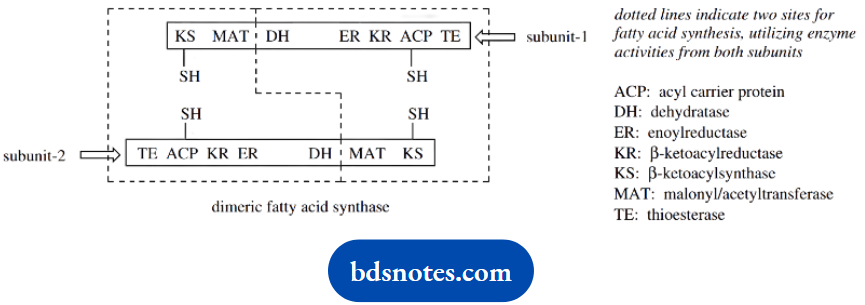
The processes of fatty acid biosynthesis are catalyzed by the enzyme fatty acid synthase. In animals, this is a multifunctional protein containing all of the catalytic activities required, whereas in bacteria and plants, it is an assembly of enzymes that can be separated.
Acetyl-CoA and malonyl-CoA themselves are not involved in the condensation step: they are converted into enzyme-bound thioesters.
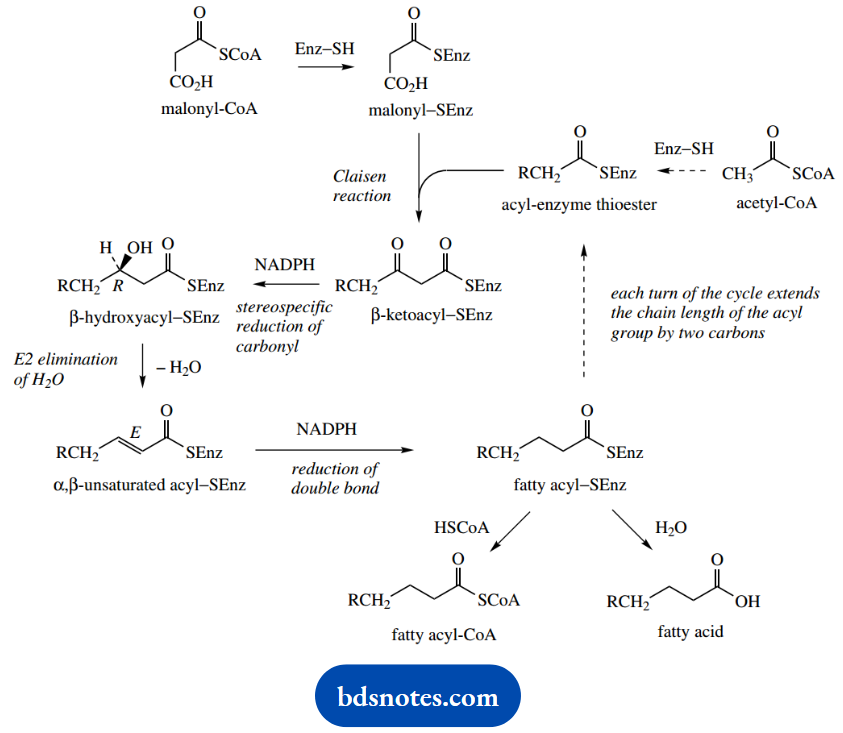
The Claisen reaction follows, giving the acetoacetyl thioester (β-ketoacyl-SEnz; R=H), which is reduced stereospecifically to the corresponding P- hydroxy ester, consuming NADPH in the reaction. Then follows the elimination of water, giving the E (trans) α,β-unsaturated ester.
Reduction of the double bond again utilizes NADPH and generates a saturated acyl-SEnz (fatty acyl-SEnz; R=H) that is two carbons longer than the starting material.
This can feed back into the system, condensing again with malonyl thioester, and going through successive reduction, dehydration, and reduction steps, gradually increasing the chain length by two carbons for each cycle, until the required chain length is obtained.
At that point, the fatty acyl chain can be released as a fatty acyl-CoA or as a free acid. The chain length actually elaborated is probably controlled by the specificity of the thioesterase enzymes that subsequently catalyze release from the enzyme.
Note that the reduction, dehydration, and reduction steps are essentially the reverse of the oxidation, hydration, and oxidation steps in fatty acid metabolism, though the enzymes and coenzymes involved are different.
Fatty Acid Synthase
Fatty acid synthesis is catalyzed in animals by the enzyme fatty acid synthase, which is a multifunctional protein containing all of the catalytic activities required.
Bearing in mind the necessity to provide a specific binding site for the various substrates involved, and then the fairly complex sequence of reactions carried out, it raises the question of just how it is possible for this process to be achieved at the enzymic level.
Nature has devised an elaborate but satisfyingly simple answer to this problem.
The fatty acid synthase protein is known to contain an acyl carrier protein (ACP) binding site, and also an active-site cysteine residue in the β-ketoacyl synthase domain.
Acetyl and malonyl groups are successively transferred from coenzyme A esters and attached to the thiol groups of Cys and ACP.
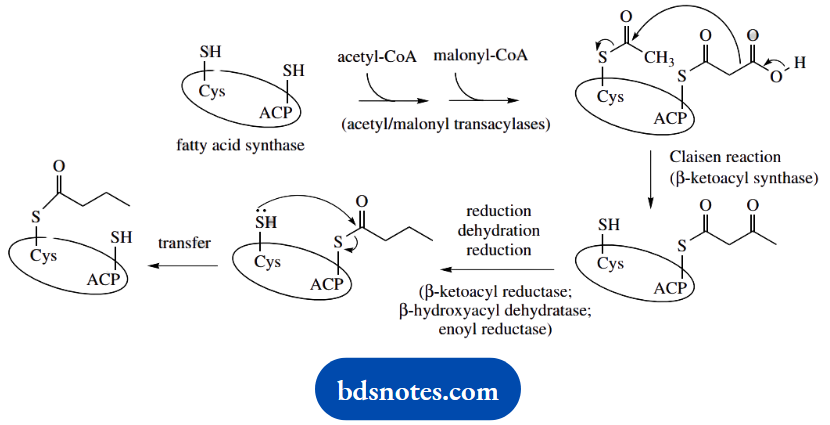
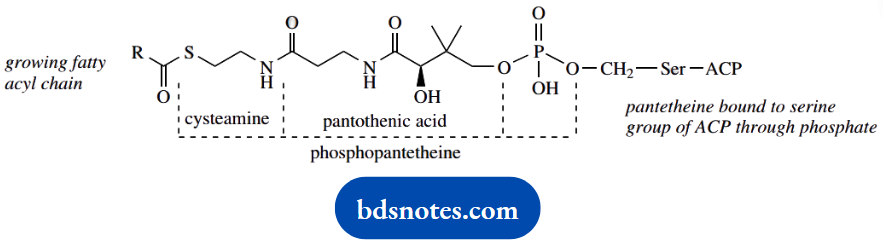
The Claisen condensation occurs, and the processes of reduction, dehydration, and reduction then occur whilst the growing chain is attached to ACP. The ACP carries a phosphopantetheine group exactly analogous to that in coenzyme A (pantothenic acid: vitamin B5).
This phosphopantetheine group provides a long flexible arm, enabling the growing fatty acid chain to reach the active site of each enzyme in the complex, and allowing the different chemical reactions to be performed without releasing intermediates from the enzyme.
The chain is then transferred to the thiol of Cys, and the process can continue.
Making the process even more efficient, animal fatty acid synthase is a dimeric protein containing two catalytic centers, and it is able to generate two growing chains at the same time.
The monomeric subunits are also arranged head to tail so that the acyl group of one unit actually picks up a malonyl extender from the other unit.
It is interesting that the sequence of enzyme activities along the protein chain of the enzyme complex does not correspond with the order in which they are employed.
A similar approach is employed in the formation of peptides such as peptide antibiotics.
In marked contrast to the ribosomal biosynthesis of proteins, where a biological production line interprets the genetic code, many natural peptides are known to be synthesized by a more individualistic sequence of enzyme-controlled processes, in which each amino acid is added as a result of the specificity of the enzyme involved.
The many stages of the whole process appear to be carried out by a multi-functional enzyme termed a non-ribosomal peptide synthase.
Pantothenic acid bound to the enzyme as pantothenic is used to carry the growing peptide chain through its thiol group.
The long ‘pantothenic arm’ allows different active sites on the enzyme to be reached in the chain assembly process, a process remarkably analogous to the fatty acid synthase mechanism.
Amino Acids And Transamination
The synthesis of amino acids depends upon the ami- nation of the Krebs cycle intermediate 2-oxoglutarate to glutamate, a process of reductive amination. This can occur when a high concentration of ammonium ions is available and involves NADH or NADPH as a reducing agent.
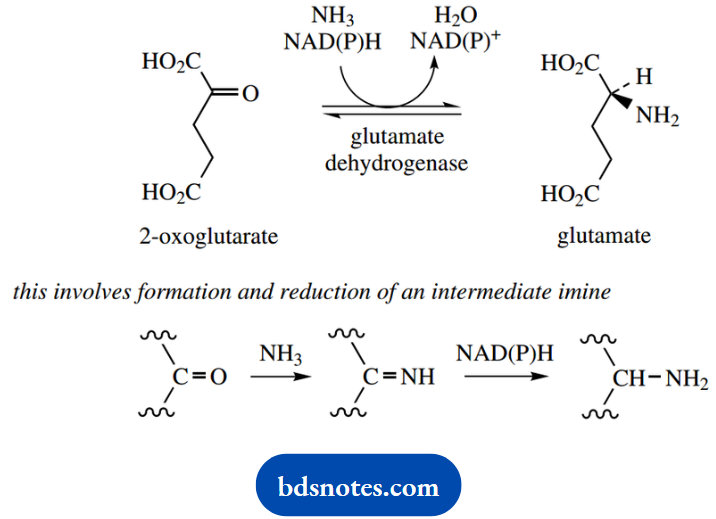
The reaction involves the formation of an imine through the reaction of ammonia with the ketone, followed by the reduction of this imine. As we noted earlier, nicotinamide coenzymes may also participate in imine reductions as well as aldehyde/ketone reductions, further emphasizing the imine-carbonyl analogy.
The reverse reaction, removal of ammonia from glutamate, is also of importance in amino acid catabolism.
Glutamate can then participate in the formation of other amino acids via the process called transamination. Transamination is the exchange of the amino group from an amino acid to a keto acid and provides the most common process for the introduction of nitrogen into amino acids and for the removal of nitrogen from them.
The reaction is catalyzed by a transaminase enzyme, and the coenzyme pyridoxal phosphate (PLP) is required.
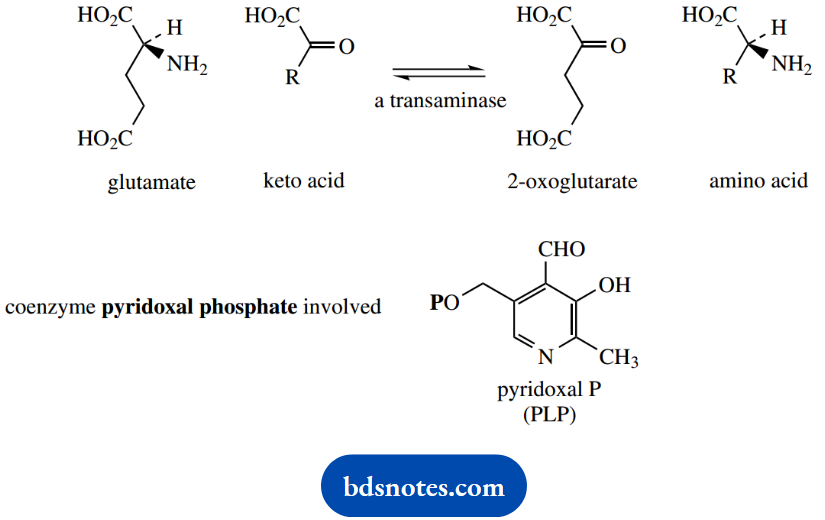
The process initially features the formation of an imine intermediate (aldimine) using the amine group of the amino acid with the aldehyde group of PLP. The imine function formed is conjugated with the aromatic pyridine ring.
Accordingly, protonation of the pyridine nitrogen (as would occur at physiological pH) makes the α-hydrogen of the original amino acid considerably more acidic.
This can be removed, in a process rather like that seen with conjugated carbonyl compounds. This generates a dihydropyridine ring system; the process is effectively imine-enamine tautomerization, though in an extended conjugated system.
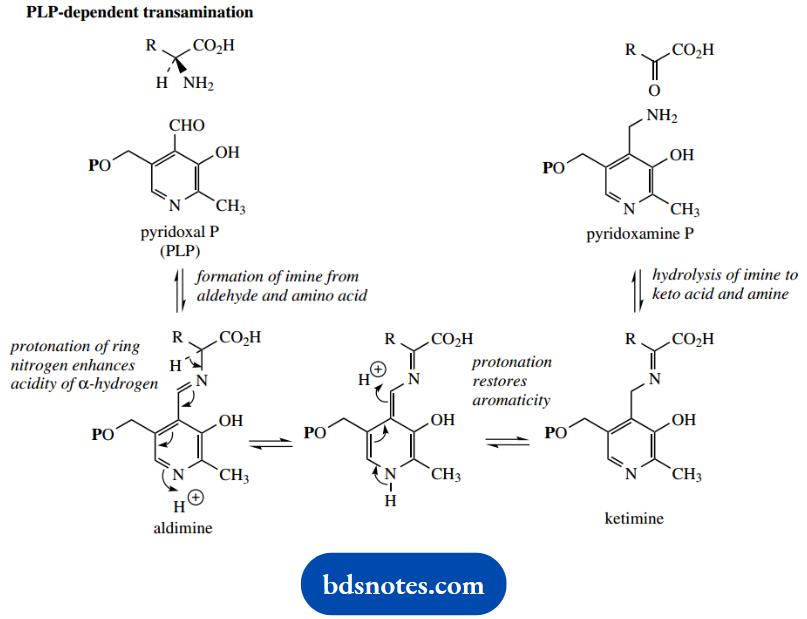
Deprotonation then produces a new imine (ketamine) and also restores aromaticity in the pyridine ring. However, because of the conjugation, it allows protonation at a position that is different from where the proton was originally lost.
The net result is that the imine double bond has effectively moved to a position adjacent to its original position. Hydrolysis of this new imine group generates a keto acid and pyridoxamine phosphate.
The remainder of the sequence is now a reversal of this process. This now transfers the amine function from pyridoxamine phosphate to another keto acid.
The glutamic acid-2-oxoglutaric acid couple features as the usual donor-acceptor molecules for the amino group, and glutamate transaminase is thus the most important of the transaminases.
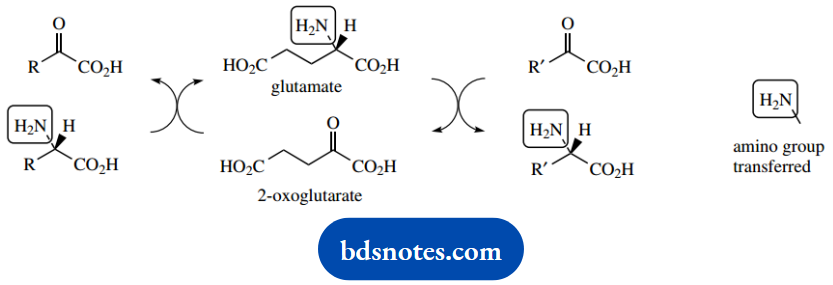
Transamination allows the amino group to be transferred from glutamic acid to a suitable keto acid. In the reverse mode, the amino group can be transferred from an amino acid to 2-oxoglutaric acid.
Equilibrium constants for the reactions catalyzed by transaminases are close to unity, so the reactions proceed readily in either direction.
It now becomes possible, as shown, to transfer the amino group of one amino acid, which may be readily available, to provide another amino acid, which could be in short supply.
This has obvious advantages over the process seen for glutamate synthesis via the reductive amination of 2-oxoglutarate, in that it no longer requires the intervention of free ammonia.
We thus have the situation that some organisms are able to carry out the fixation of ammonia via reductive amination, whereas others manipulate via transamination the amino acid structures obtained from protein in the diet.
PLP-dependent Reactions
We have just noted the role that pyridoxal phosphate plays as a coenzyme (cofactor) in transamination reactions. Pyridoxal 5′-phosphate (PLP) is crucial to a number of biochemical reactions.
PLP, together with a number of closely related materials that are readily converted into PLP, for example, pyridoxal, pyridoxine, and pyridoxamine, are collectively known as vitamin B6, which is essential for good health.
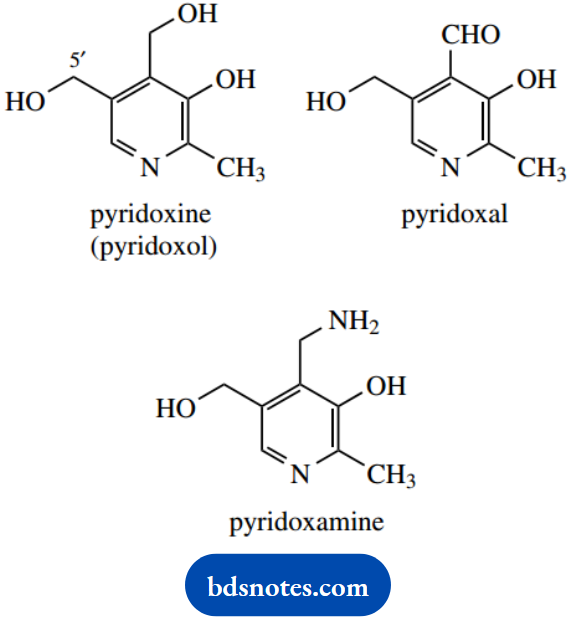
At neutral pH, PLP is considerably ionized, so that the phenol group loses a proton and the pyridine nitrogen is protonated. Of course, the phosphate will also be ionized.
These ionic centers facilitate binding to the enzyme, but, for clarity, they are omitted from the mechanisms shown. However, the positively charged nitrogen is essential for the cofactor’s chemical reactivity, and we need to invoke it in the mechanisms.
In transamination, we have seen the formation of an imine, in which the protonated nitrogen acts as an electron sink, making the α-hydrogen of the original amino acid acidic and facilitating its removal.
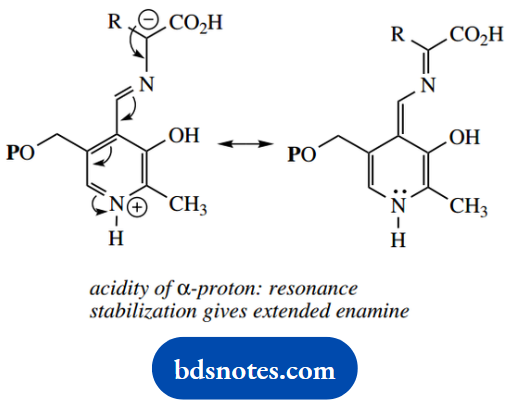
The reversal of this process could potentially occur with deprotonation from either face of the C=N double bond, and a mixture of aldimines would result, leading to the generation of a racemic amino acid. This accounts for the mode of action of PLP-dependent amino acid racemase enzymes.
Of course, the enzyme controls the removal and supply of protons; this is not a random event. One important example of this reaction is the alanine racemase, employed by bacteria to convert L-alanine into D-alanine for cell wall synthesis.
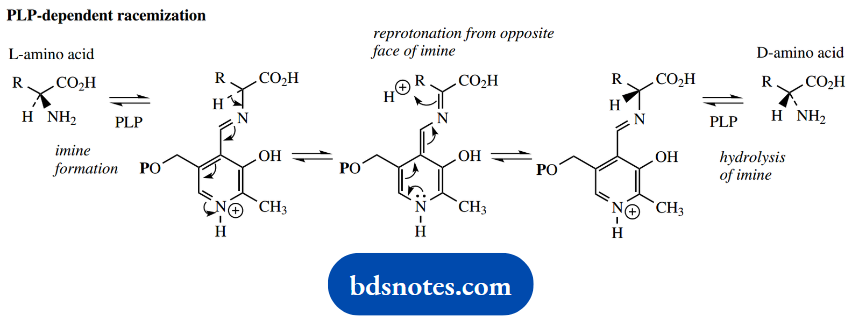
In transamination and racemization reactions, we have seen the loss of a proton from the aldimine, i.e. breaking of bond a. Let us now consider the two alternatives, namely the breaking of bonds b or c, to explore further the scope for PLP-dependent reactions.
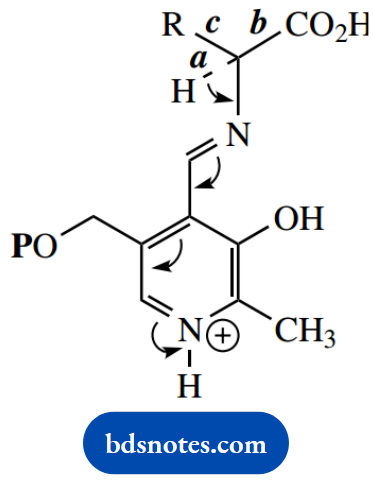
Breaking of bond b accounts for PLP-dependent decarboxylations. Decarboxylation of the intermediate aldimine is facilitated in the same way as the loss of a proton in the transamination sequence.
The protonated nitrogen acts as an electron sink, and the conjugated system allows the loss of the carboxyl proton, with subsequent bond breaking and loss of CO2.
The resultant imine may subsequently be hydrolyzed, releasing an amine (the decarboxylated amino acid) and regenerating PLP. There are many examples of decarboxylation of amino acids.
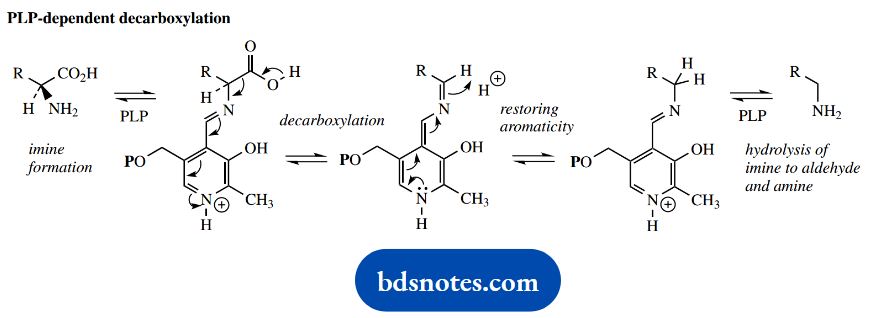
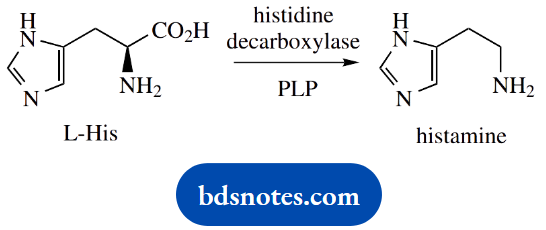
PLP- Dependent Amino Acid Decarboxylations
An important example of PLP-dependent amino acid decarboxylation is the conversion of histidine into histamine. Histamine is often involved in human allergic responses, for example, to insect bites or pollens.
Stress stimulates the action of the enzyme histidine decarboxylase and histamine is released from mast cells. Topical antihistamine creams are valuable for pain relief, and oral antihistamines are widely prescribed for nasal allergies such as hay fever.
Major effects of histamine include dilation of blood vessels, inflammation and swelling of tissues, and narrowing of airways. In serious cases, life-threatening anaphylactic shock may occur, caused by a dramatic fall in blood pressure.
The catecholamines noradrenaline (norepinephrine) and adrenaline (epinephrine) are amines derived via decarboxylation of amino acids. Noradrenaline is a mammalian neurotransmitter and adrenaline, the ‘fight or flight’ hormone, is released in animals from the adrenal gland as a result of stress.
These compounds are synthesized by successive hydroxylation and N-methylation reactions on dopamine.
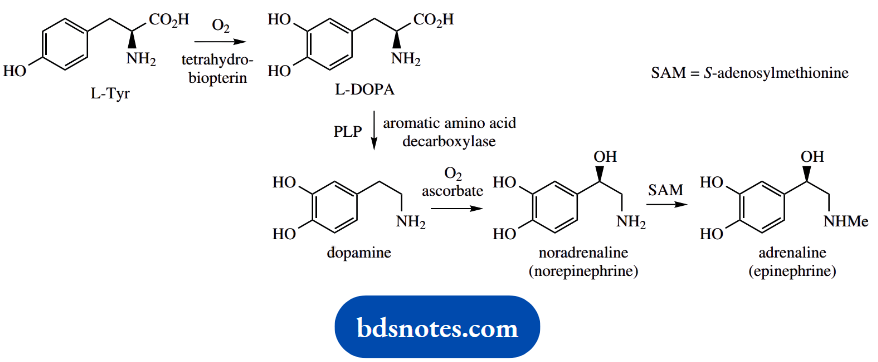
Dopamine is the decarboxylation product of DOPA, dihydroxyphenylalanine, and is formed in a reaction catalyzed by DOPA decarboxylase. This enzyme is sometimes referred to as aromatic amino acid decarboxylase, since it is relatively non-specific in its action and can catalyze the decarboxylation of other aromatic amino acids, for example, tryptophan and histidine.
DOPA is itself derived by aromatic hydroxylation of tyrosine, using tetrahydrobiopterin as a cofactor.
The neurotransmitter 5-hydroxytryptamine (5-HT, serotonin) is formed from tryptophan by hydroxylation and then decarboxylation, paralleling the tyrosine → dopamine pathway.
The non-specific enzyme aromatic amino acid decarboxylase again catalyzes the decarboxylation.
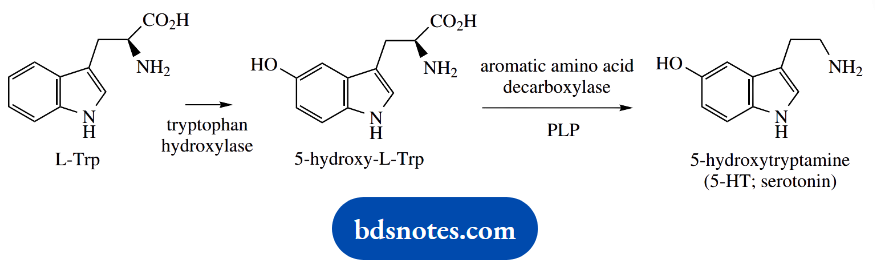
5-HT is a neurotransmitter found in cardiovascular tissue, the peripheral nervous system, blood cells, and the CNS. It mediates many central and peripheral physiological functions, including contraction of smooth muscle, vasoconstriction, food intake, sleep, pain perception, and memory, a consequence of it acting on several distinct receptor types.
Although 5-HT may be metabolized by monoamine oxidase, platelets, and neurons possess a high-affinity 5-HT re-uptake mechanism. This mechanism may be inhibited by the widely prescribed antidepressant drugs termed selective serotonin reuptake inhibitors (SSRI), for example, fluoxetine (Prozac®), thereby increasing levels of 5-HT in the CNS.
Yet another neurotransmitter, γ-aminobutyric acid or GABA, is formed by PLP-dependent decarboxylation of an amino acid, in this case, glutamic acid.
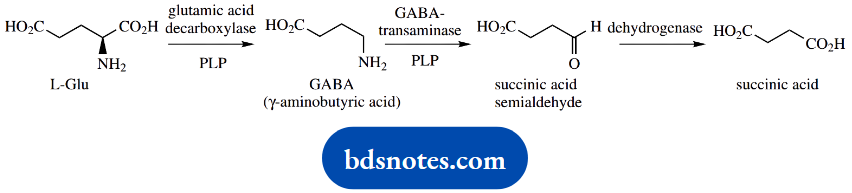
GABA acts as an inhibitory transmitter in many different CNS pathways. It is subsequently destroyed by a transamination reaction in which the amino group is transferred to 2-oxoglutaric acid, giving glutaric acid and succinic semialdehyde.
This also requires PLP as a cofactor. Oxidation of the aldehyde group produces succinic acid, a Krebs cycle intermediate.
The breaking of bond c is going to be less common than deprotonation or decarboxylation. In most amino acids, R is an alkyl group, so there is little chance of losing R as a cation. Indeed, the only occasions on which we can break bond c are when R is hydroxymethyl (as in serine) or a similar grouping (as in threonine).
In both cases, bond breaking is facilitated by the hydroxyl, in that the lone pair can feed into the conjugated system. The result in the case of serine is the loss of formaldehyde, whereas, in the case of threonine, it is the loss of acetaldehyde.
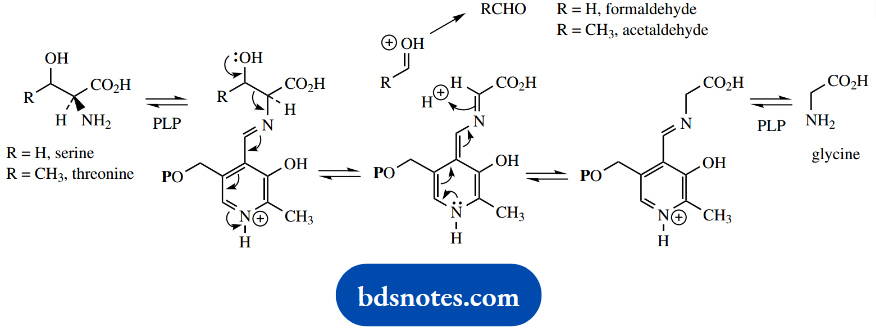
The fragment attached to pyridoxal will be the same in both cases and after hydrolysis, it is released as the amino acid glycine.
In case this seems a bit complicated, consider the reverse reaction, which would be an attack of an electron-rich system onto the carbonyl group of an aldehyde, i.e. an aldol reaction. Therefore, what we are seeing here is merely a reverse aldol-type reaction.
It is interesting to see how different products are formed according to which of the three different bonds is cleaved in the aldimine derived from an amino acid and PLP.
There is one other point to ponder though. What determines the type of cleavage that occurs?
The answer must lie in the enzyme and how it binds the substrate, and it merely becomes a consideration of stereochemistry. By considering the shape of the aldimine, we see that the pyridine ring and the adjacent C=N double bond must be planar to achieve maximum orbital overlap and conjugation.
Electrons from the bond that is broken should feed smoothly into this planar conjugated system. This requires the bond to be positioned at right angles to the plane and, therefore, parallel to the p orbitals.
As shown in the accompanying diagrams, rotation about the N-C bond positions the vulnerable group towards the exterior face of the enzyme so that it can be attacked.
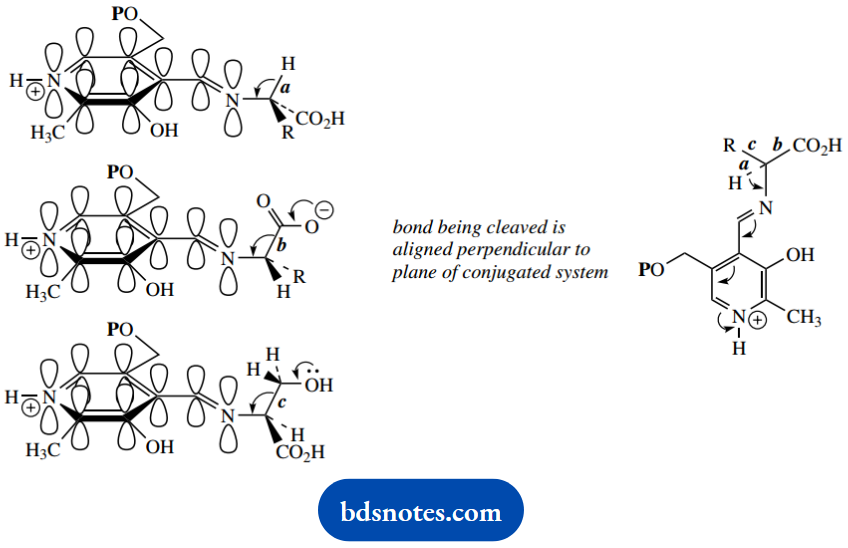
Do appreciate that one enzyme will not catalyze all three types of reaction. We need different enzymes to accomplish a particular reaction on a particular substrate.
Rotation about the N-C bond positions the vulnerable group for reaction; it also positions the R and/or CO2– groups so that they can interact and bind to the enzyme, thus providing specificity.
One further point for the sake of accuracy; we have omitted it to simplify the mechanistic features.
PLP does not exist as the free aldehyde when it is bound to the enzyme, but actually uses the aldehyde group in its binding.
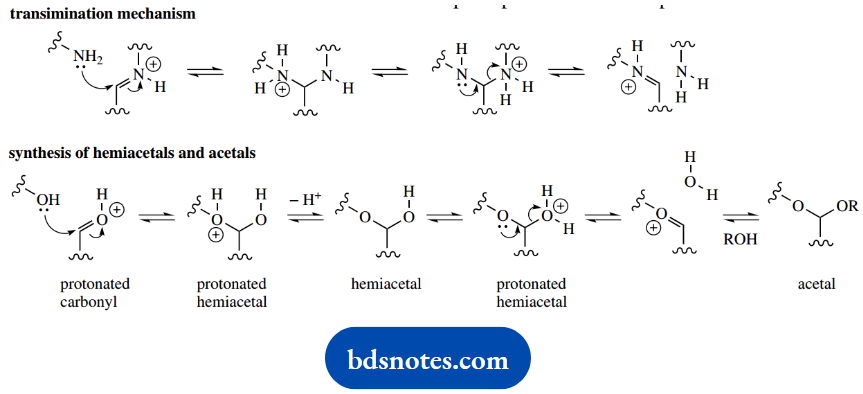
An imine linkage is formed between the aldehyde and the primary amine group of a lysine residue in the enzyme active site. When the substrate RNH2 binds to the enzyme to produce the intermediates shown above, it achieves this by a transamination reaction.
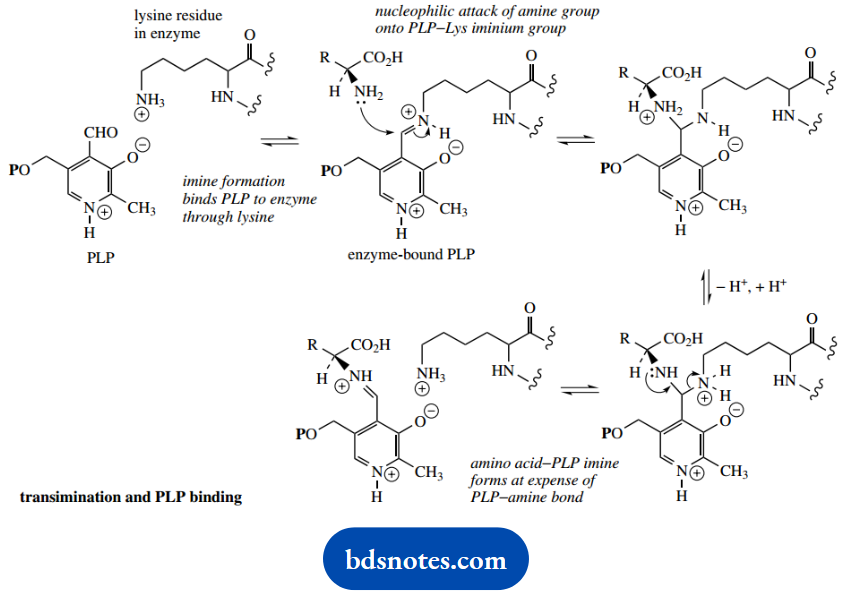
A similar transamination, in the reverse sense, takes place at the end of the reaction sequence to displace the product but still retains PLP bound to the enzyme via the lysine group.
Should the chemistry seem a little complicated, remind yourself of the mechanism for hemiacetal and acetal formation; that featured an oxygen analogy for the transimination sequence.
If we had included this additional series of reactions in the above descriptions, it would have obscured the simple principles of the PLP-dependent reactions.
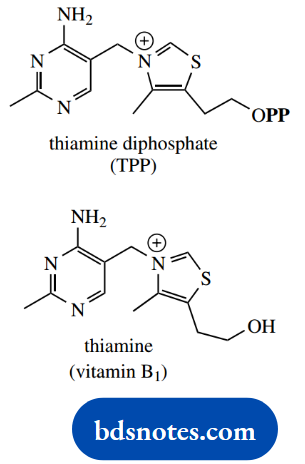
TPP-Dependent Reactions
Thiamine diphosphate (TPP) is the coenzyme for the pyruvate dehydrogenase complex that catalyzes the oxidative decarboxylation of pyruvate to acetyl- CoA and thus links the glycolytic pathway to the Krebs cycle. Later in the Krebs cycle, TPP is the cofactor for the 2-oxoglutarate dehydrogenase complex, which catalyzes a similar reaction on 2-oxoglutarate.
In the glycolytic pathway, the pyruvic acid → acetaldehyde conversion also features TPP. A further enzyme, transketolase, has the unexpected property of transferring a two-carbon fragment between carbohydrates in the pentose phosphate pathway, and TPP is again involved.
The biosynthetic pathways to two amino acids, valine, and isoleucine, also involve TPP-dependent enzymes. All of these reactions employ TPP as a carrier of an acyl anion equivalent.
TPP thus plays a very important role in carbohydrate metabolism. The parent alcohol thiamine is one of the B group vitamins, namely vitamin B1; dietary deficiency leads to the condition beriberi, characterized by neurological disorders, loss of appetite, fatigue, and muscular weakness.
The conversion of pyruvic acid into acetyl-CoA is conveniently written according to the equation

This simple equation conceals a quite complex reaction sequence involving not just TPP, but other coenzymes, including lipoic acid. Let us first inspect the nature of TPP.
With its pyrimidine ring and diphosphate grouping, TPP looks rather like a nucleotide, but the central ring system is a thiazole rather than a sugar.
This heterocyclic ring is alkylated on nitrogen and is thus a thiazolium salt. This plays a key role in the reactivity of TPP.
The proton in the thiazolium ring is relatively acidic (pKa about 18) and can be removed by even weak bases to generate the carbanion or ylid. A ylid (also ylide) is a species with positive and negative charges on adjacent atoms; this ylid is an ammonium ylid with extra stabilization from the sulfur atom.
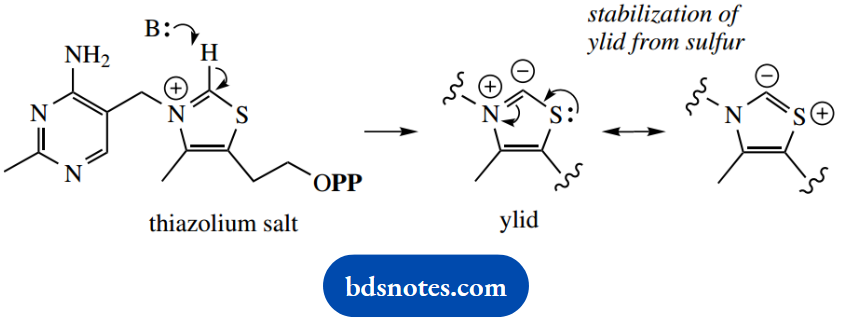
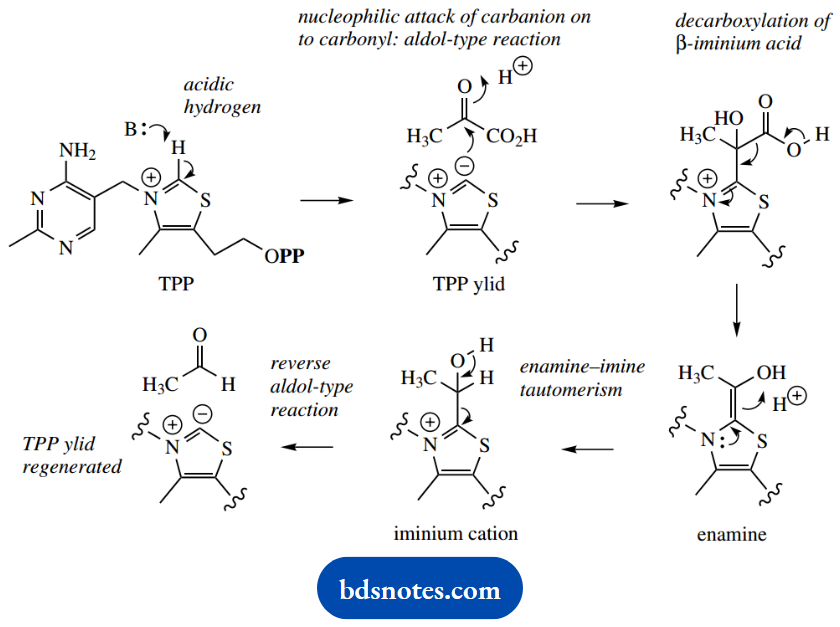
This ylid can act as a nucleophile and is also a reasonable leaving group. In addition, the carbonyl group of pyruvic acid is followed by decarboxylation, the positive nitrogen in the ring acting as an electron sink.
The resulting molecule is an enamine, but because of the neighboring heteroatoms, it is extremely electron-rich. It accepts a proton, and this achieves tautomerism of the enamine to the iminium ion.
This is followed by a reverse aldol reaction, which also regenerates the ylid as the leaving group. This sequence would thus accommodate the pyruvic acid → acetaldehyde conversion catalyzed by pyruvate decarboxylase in the glycolytic pathway.
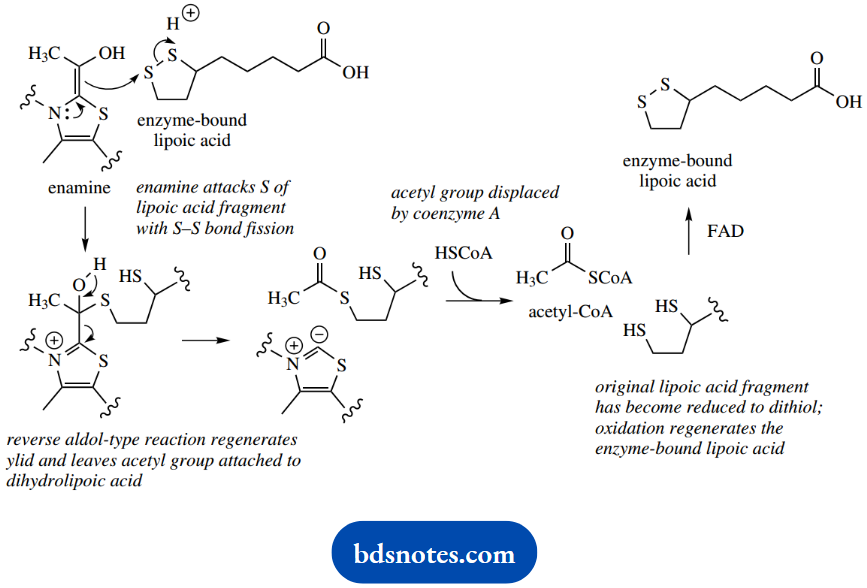
In oxidative decarboxylation of pyruvate to acetyl- CoA, the enzyme-bound disulfide-containing coen¬zyme lipoic acid is also involved. The electron-rich enamine intermediate, instead of accepting a proton, is used to attack sulfur in the lipoic acid moiety.
This leads to fission of the S-S bond, and thereby effectively reduces the lipoic acid fragment. Regen¬eration of the TPP ylid via the reverse aldol-type reaction leaves the acetyl group bound to the dihydrolipoic acid as a thioester.
This acetyl group is then released as acetyl-coA by displacement with the thiol coenzyme A. The bound dihydrolipoic acid frag¬ment must then be reoxidized to restore its function.
An exactly equivalent reaction is encountered in the Krebs cycle in the conversion of 2-oxoglutaric acid into succinyl-coA.
Each of the complexes pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase actually contains three enzyme activities. We can now readily appreciate what the separate activities might be.
In the case of pyruvate dehydrogenase, the individual activities are pyruvate dehydrogenase (requires cofactor TPP), dihydrolipoamide acyltransferase (requires cofactors lipoic acid and coenzyme A), and dihydrolipoamide dehydrogenase (requires cofactors FAD and NAD+).
The requirement for NAD+ is to reoxidize FADH2 after regeneration of lipoic acid. The ‘lipoamide’ terminology indicates that the lipoic acid is enzyme bound through its carboxylic acid group to an amino group of lysine via an amide linkage.
At the end of the first paragraph in this section, we stated ‘All these reactions employ TPP as a carrier of an acyl anion equivalent’. An acyl anion is an unlikely species since we would consider locating a negative charge on the carbon of a carbonyl group as definitely unfavorable.
However, the following scheme should emphasize how we can consider that TPP is effectively removing and transferring an acetyl anion equivalent in the above reactions.
TPP And Acyl Anion Equivalents
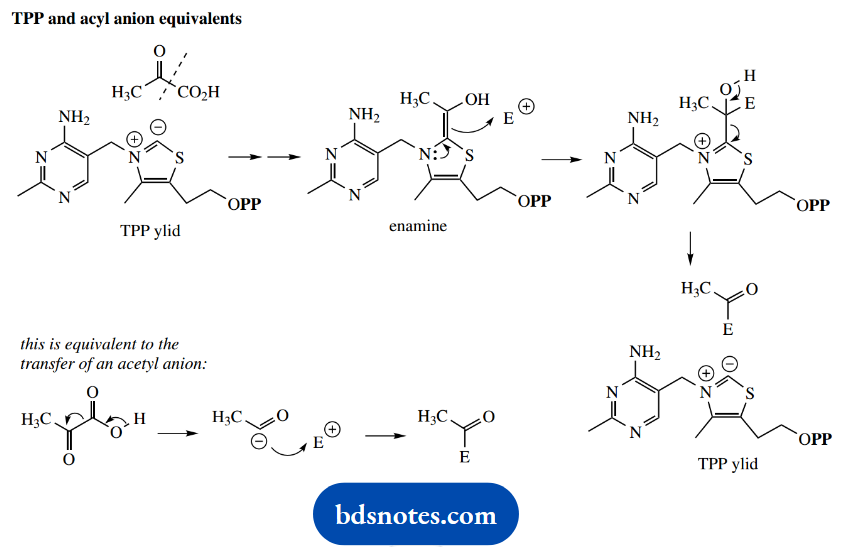
A slightly different acyl anion equivalent is transferred in transketolase reactions, and this anion is then used in a subsequent aldol reaction.
Transketolase removes a two-carbon fragment from keto sugars such as xylulose 5-phosphate (alternatively fructose 6-phosphate or sedoheptulose 7-phosphate) through the participation of the thiamine diphosphate ylid.
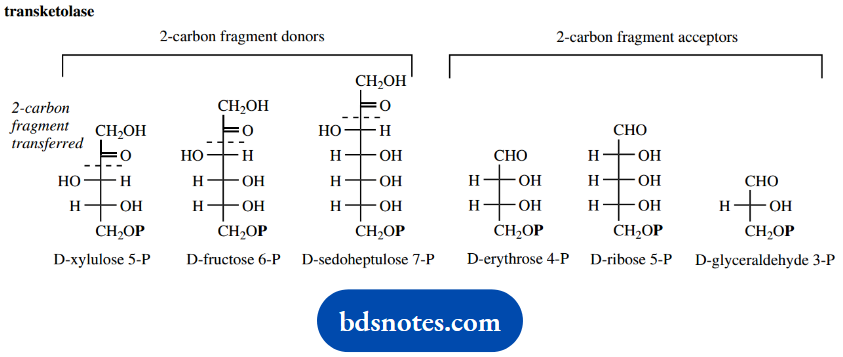
The nucleophilic attack of this ylid onto the ketone carbonyl results in an addition product that then fragments by a reverse aldol reaction. This generates a chain-shortened aldose, for example, glyceraldehyde 3-phosphate from xylulose 5-phosphate, and the two-carbon acyl anion equivalent attached to TPP.
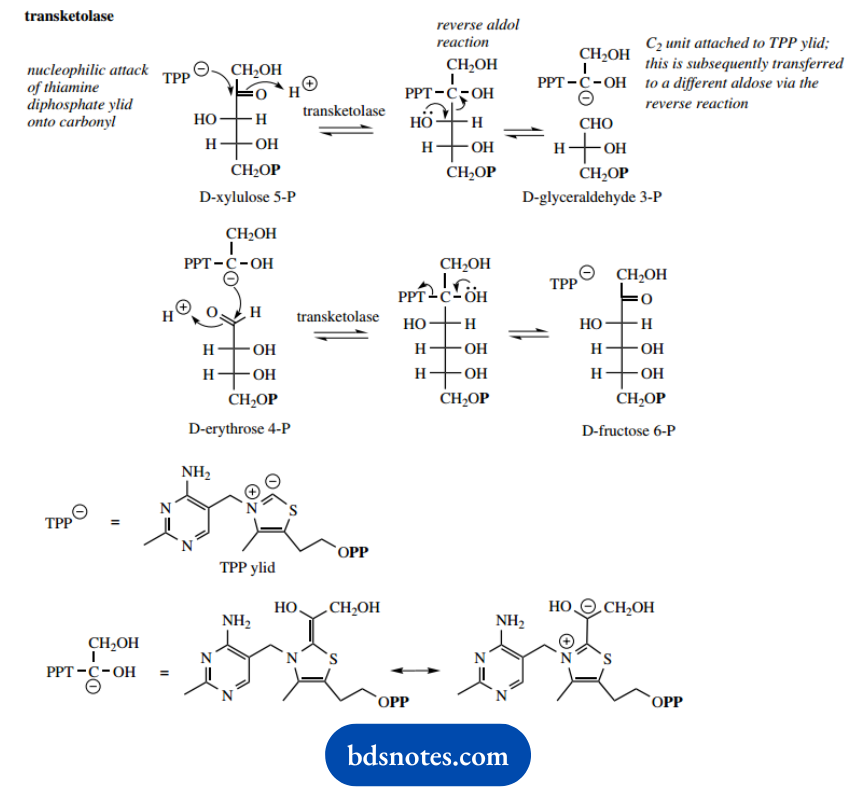
Then, in what is formally a reverse of this reaction, this carbanion equivalent can attack another aldose, such as erythrose 4-phosphate, extending its chain length by two carbons.
Transketolase is crucial to metabolism in creating a link between the pentose phosphate pathway and glycolysis.
Reactions Catalysed By Transketolase Enzymes:

An additional enzyme that transfers C3 rather than C2 units is called transaldolase, but, in common with aldolase, this enzyme utilizes an imine-enamine mechanism through an imine link with lysine, and does not involve TPP.
Biotin-Dependent Carboxylations
We have briefly noted the role of biotin when we considered the biosynthesis of fatty acids. Biotin is a carrier of carbon dioxide and is involved in carboxylation reactions.
In fatty acid biosynthesis, we noted how acetyl-CoA was transformed by carboxylation into the more effective nucleophilic agent malonyl-CoA, thus facilitating the Claisen reaction.
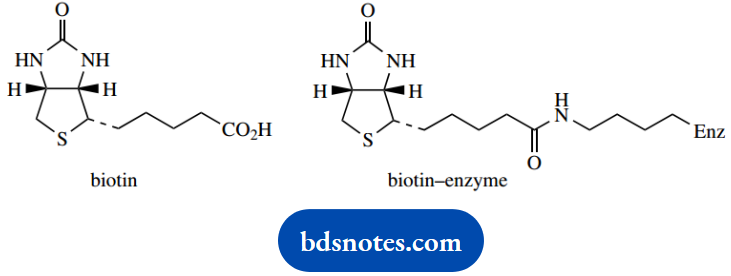
Structurally, biotin (vitamin H) is composed of two fused five-membered heterocycles, a cyclic urea, and a cyclic sulfide (tetrahydrothiophene).
Carbon dioxide is a normally unreactive material, and in combination with biotin requires the input of energy (from ATP). Carbon dioxide is usually present as the soluble form of bicarbonate, and this reacts with ATP to form a mixed anhydride, as part of the reaction catalyzed by the carboxylase.
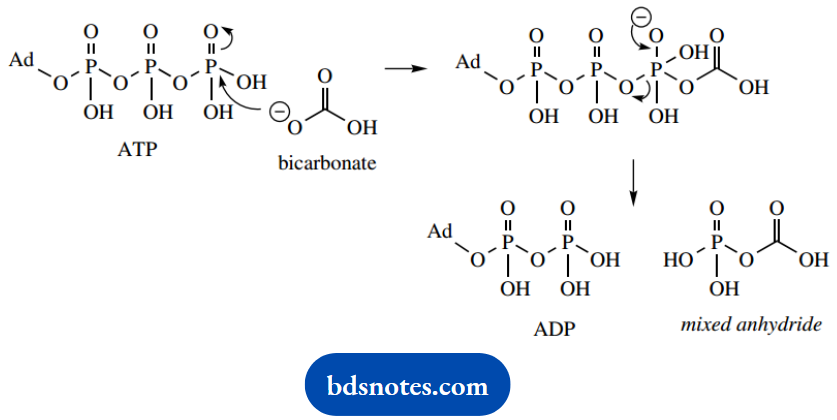
This mixed anhydride carboxylates the coenzyme in a biotin-enzyme complex. Biotin is bound to a lysine residue in the enzyme as an amide. The carboxylation reaction is effectively a nucleophilic attack of the cyclic urea on the mixed anhydride.
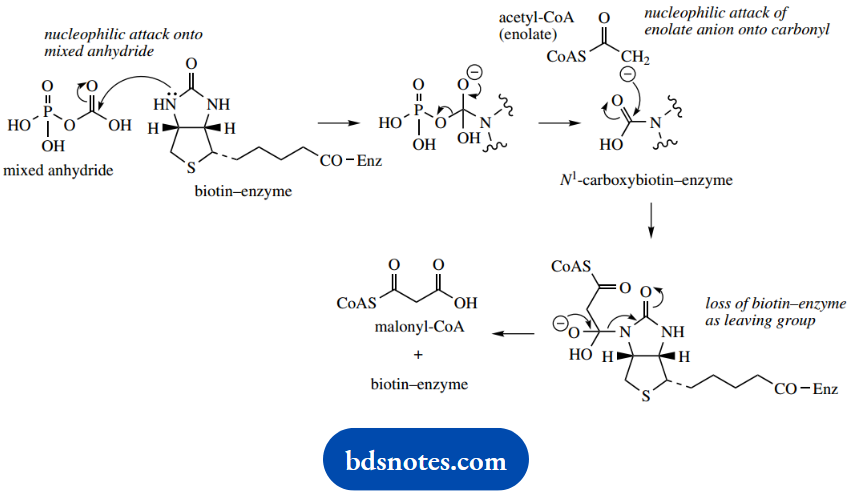
In what can be considered a reversal of this sequence, the acetyl-CoA acts as the nucleophile and is carboxylated to malonyl-CoA with displacement of the biotin–enzyme system.
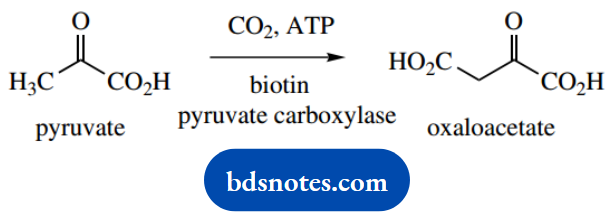
Fixation of carbon dioxide by biotin–enzyme complexes is not unique to acetyl-CoA, and another important example occurs in the generation of
oxaloacetate from pyruvate in the synthesis of glucose from non-carbohydrate sources (gluconeogenesis).
This reaction also allows replenishment of Krebs cycle intermediates when compounds are drawn off for biosynthetic purposes, for example, amino acid synthesis.
Leave a Reply