Nucleophilic Reactions Involving Enolate Anions
Enols And Enolization
“What are nucleophilic reactions involving enolate anions? A detailed notes and Q&A guide”
Aldehydes and ketones, and other carbonyl compounds having hydrogen atoms on the α-carbon, exist in solution as equilibrium mixtures of two or more isomeric forms.
These isomers are termed the keto form, which is how we normally represent a carbonyl compound, and the enol form, which takes its name from the combination of double bonds and alcohol.
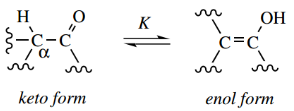
The interconversion of keto and enol forms is termed enolization, or keto-enol tautomerism.
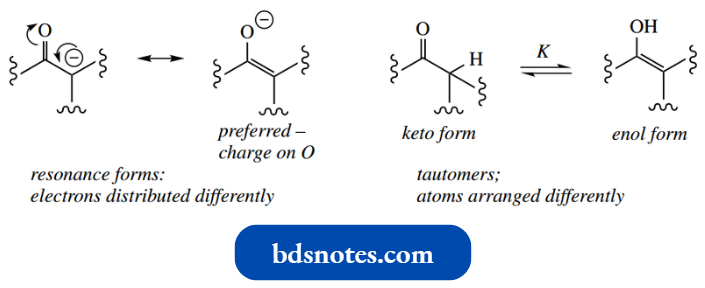
- The two isomeric structures are not resonance forms but are termed tautomers. Resonance forms have the same arrangement of atoms, but the electrons are distributed differently.
- Tau tomers have the atoms arranged differently, and tau tomerism is an equilibrium reaction between the isomeric forms.
- Thus, in the general case shown, the α-hydrogen in the keto tautomer disappears and the oxygen atom gains hydrogen to produce the hydroxyl of the enol system.
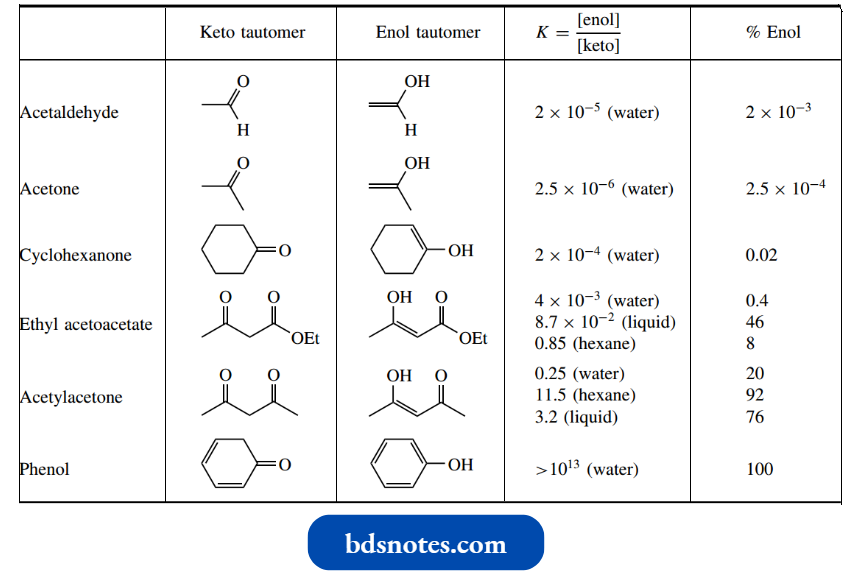
To indicate the importance of enolization, equilibrium constants for several substrates. These equilibrium constants are only approximate, and they depend very much on the solvents employed.
- Nevertheless, we can see that the equilibrium constant K = [enol]/[keto] is very small for substrates like acetaldehyde, acetone, and cyclohexanone, with only a few molecules in every million existing in the enol form.
- However, in ethyl acetoac estate, enol concentrations are measured in per cent ages, and in acetylacetone, the equilibrium constant indicates the enol form can be distinctly favoured over the normal keto form.
In hexane solution, only 8% of acetylacetone molecules remain in the keto form.
- Normally then, the keto form we have traditionally written for carbonyl compounds is very much favoured over the enol tautomer.
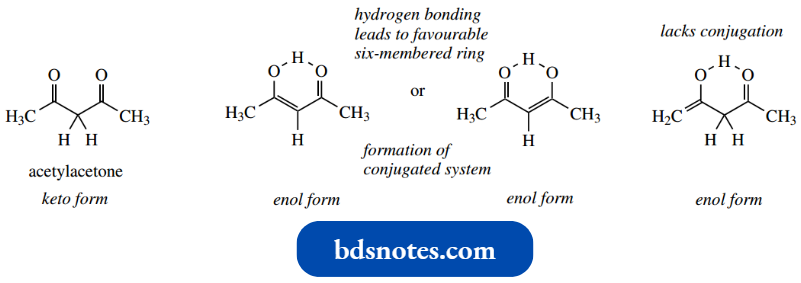
- The high contribution of enol forms in equilibrium mixtures of the 1,3- dicarbonyl compounds such as ethyl acetoacetate and acetylacetone are ascribed principally to additional stability conferred by the formation of a conjugated enone system, with further stabilization coming from the establishment of the hydrogen bonding in a favourable six-membered ring.
At the other extreme, as in the case of cyclohexadiene, the enol tautomer is the only contributing tautomer, since the enol form (phenol) benefits from the stabilization conferred by the aromatic ring system.
Keto Enol Equilibria:
“Understanding enolate anions through FAQs: Nucleophilic reactions explained”
“How do enolate anions act as nucleophiles in organic chemistry? FAQ answered”
It is important to note that, in 1,3-dicarbonyl compounds such as acetylacetone, enolization involves loss of the α-hydrogen between the two carbonyl groups and not the terminal α-hydrogens.
- Enolization involving the latter α-hydrogens would not generate conjugation stabilization; and despite the possibility of hydrogen bonding, this enol form is not favoured relative to the alternatives.
- Conjugation acid-catalysed tautomerism can only be achieved if the central α-hydrogens, those sandwiched between the two carbonyls, are involved.
- The interconversion of keto and enol forms may be catalysed by both acids and by a base.
In acid, this may be rationalized by a mechanism in which protonation of the carbonyl to give the conjugate acid is followed by loss of the α-proton.
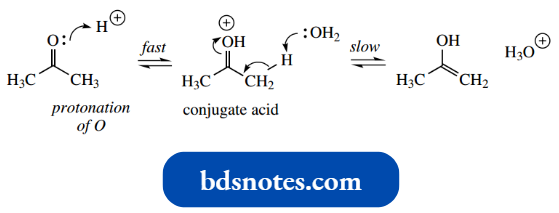
It is important to appreciate the role of the solvent in this transformation, removing and supplying protons, and to understand that tautomerism is not merely a transfer of a proton from the α-carbon to the carbonyl oxygen.
- The rate-determining step in tautomerism will be the removal of the α-hydrogen; protonation of the carbonyl (formation of the conjugate acid) can be considered rapid.
- In base, slow abstraction of the α-hydrogen by the base will be the first step, followed by rapid protonation of the conjugate base, again making use of the solvent for the removal and supply of protons.
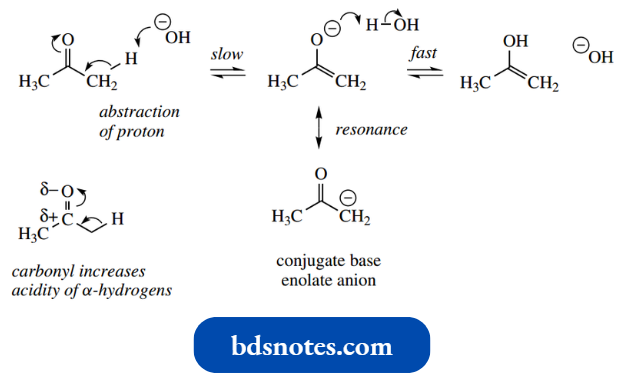
This process is thus exploiting the acidity associated with the α-hydrogens (pKa 19), which is considerably greater than that of the corresponding alkane (pKa 50).
- The effect of the adjacent carbonyl is to increase the acidity of the α-hydrogens.
- This is a direct consequence of the polarization of the carbonyl arising from the electronegativity of the oxygen atom.
- The conjugate base in this process is called an enolate anion and is stabilized by resonance.
- Of the two resonance forms of the enolate anion, that with the charge on the electronegative oxygen will be preferred over that with a charge on the carbon.
- Note the distinct difference between resonance as shown here, a redistribution of electrons, and tautomerism, as described above.
Tautomers are isomers in equilibrium and have the atoms arranged differently.
“Importance of studying nucleophilic reactions with enolate anions for chemistry students: Questions explained”
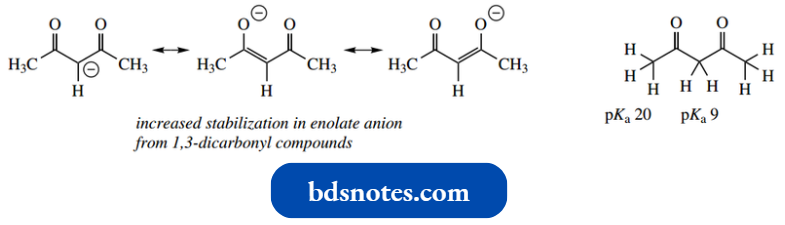
In 1,3-dicarbonyl compounds such as acetylace tone, the protons between the two carbonyls will be even more acidic (pKa 9), since there are now two carbonyl groups exerting their combined influence.
- It can also be seen that resonance in the enolate anion is even more favourable with two carbonyl atoms arranged differently groups.
- This increased stability is not achieved by removal of the terminal α-hydrogens, and in acety lacetone, these have pKa 20, comparable to that in acetone. Put another way, the treatment of acetylacetone with base preferentially removes a proton from the central methylene.
Enols And Enolization In The Glycolytic Pathway:
Enols and enolization feature prominently in some of the basic biochemical pathways.
- Biochemists will be familiar with the terminology enol as part of the name phosphoenolpyruvate, a metabolite of the glycolytic pathway. We shall here consider it in non-ionized form, i.e. phosphoenolpyruvic acid.
- As we have already noted, in the enolization between pyruvic acid and enol pyruvic acid, the equilibrium is likely to favour the keto form pyruvic acid very much.
- However, in phosphoenolpyruvic acid the enol hydroxyl is esterified with phosphoric acid, effectively freezing the enol form and preventing tautomerism back to the keto form.
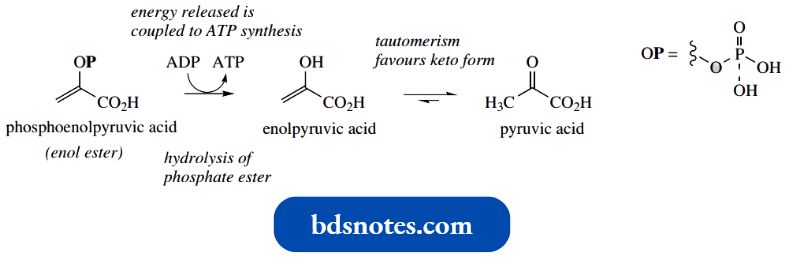
- Once the phosphate ester is hydrolysed, there is immediate rapid tautomerism to the keto form, which becomes the driving force for the metabolic transformation of phosphoenolpyruvic acid into pyruvic acid and explains the large negative free energy change in the transformation. This energy release is coupled to ATP formation.
- Tautomerism occurs elsewhere in the glycolytic pathway. The transformation of glyceraldehyde 3-phosphate into dihydroxyacetone phosphate involves two such keto–enol tautomerisms and proceeds through an enediol.
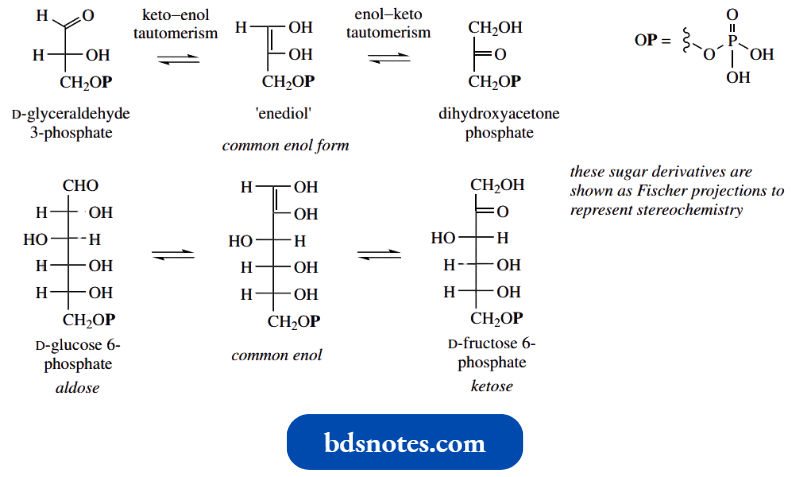
“Common challenges in understanding enolate anion mechanisms effectively: FAQs provided”
- This enediol can be regarded as a common enol tautomer for two different keto structures.
- In other words, there are two ways in which this enediol can tautomerize back to a keto form, and the reaction thus appears to shift the position of the carbonyl group.
- The reaction is enzyme-catalysed, which allows the normal equilibrium processes to be disturbed.
- It is nice to see this series of reactions being repeated in the glycolytic pathway, this time accounting for the transformation of glucose 6-phosphate into fructose 6-phosphate.
- Although the substrates are different, the reacting portion of the molecules is the same as that in the glyceraldehyde 3-phosphate to dihydroxyacetone phosphate transformation. Again, this is an enzyme-catalysed reaction.
Hydrogen Exchange:
The intermediacy of enols or enolate anions may be demonstrated by hydrogen exchange reactions.
- Both acid-catalysed and base-catalysed tautomerism mechanisms involve the removal of a proton from the α-carbon and the supply of a proton from the solvent to the carbonyl oxygen.
- Accordingly, this removal/supply of protons can be observed using isotopes of hydrogen, either radioactive tritium or the stable deuterium, which can be detected easily via NMR techniques.
- Thus, pentane-3-one can be deuterated using a large excess of D2O, with either acid (DCl) or base (NaOD) catalyst; the acid or base catalyst should also be deuterated to minimize dilution of the label.
After suitable equilibration, usually requiring prolonged heating, the α-positions will become completely labelled with deuterium.
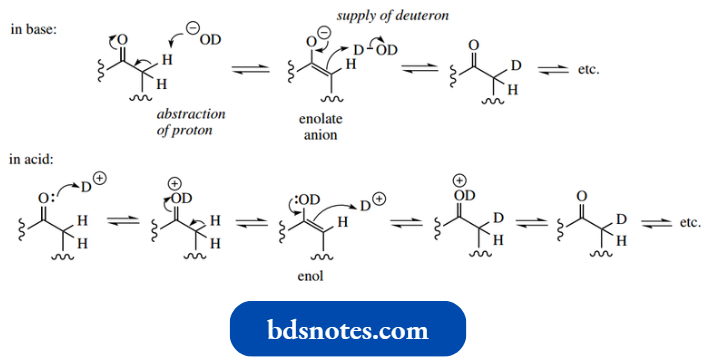
“Why is early learning of enolate anion chemistry critical for organic synthesis? Answered”
Two mechanisms are shown above. The base-catalysed mechanism proceeds through the enolate anion. The acid-catalysed process would be formulated as involving an enol intermediate.
- Note that the terminal hydrogens in pentane-3-one are not exchanged, since they do not participate in the enolization process.
- Of course, it is also possible to re-exchange the labelled hydrogens by a similar process using an excess of ordinary water, a process that might be exploited to determine or confirm the position of labelling in a deuterium-labelled substrate.
- Although this section has been termed hydrogen exchange, it is important to realize that we could also visualize this simply as an enolate anion acting as a base.
This is also true of the next section, and in some of the following sections we shall encounter enolate anions acting as nucleophiles.
Nucleophilic Reactions Involving Enolate Anions Racemization:
The process of hydrogen exchange shown above has implications if the α-carbon is chiral and has hydrogen attached.
- Removal of the proton will generate a planar enol or enolate anion, and regeneration of the keto form may then involve a supply of protons from either face of the double bond, so changing a particular enantiomer into its racemic form.
- Reacquiring a proton in the same stereochemical manner that it was lost will generate the original substrate, but if it is acquired from the other face of the double bond it will give the enantiomer, i.e. together making a racemate.
Note that removal and replacement of protons at the other α-carbon, i.e. the methyl, will occur, but has no stereochemical consequences.
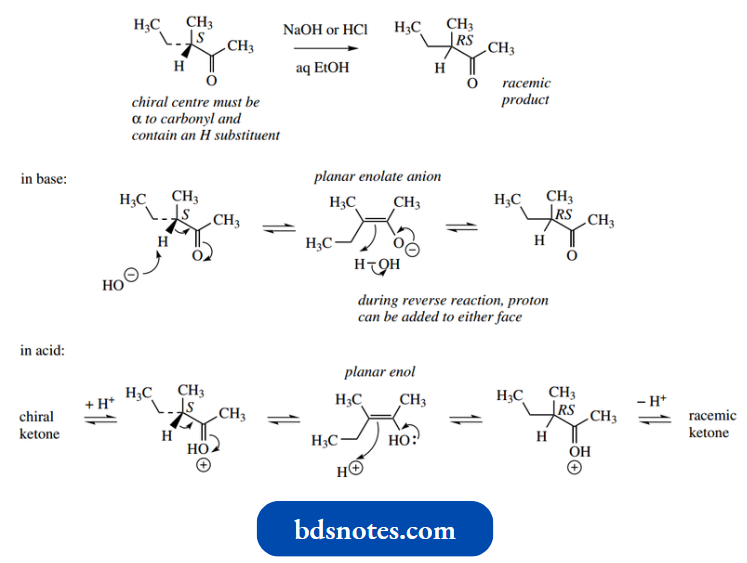
“Factors influencing success with nucleophilic reactions involving enolate anions: Q&A”
The chiral centre must be α to the carbonyl and must contain a hydrogen substituent.
If there is more than one chiral centre in the molecule with only one centre α to the carbonyl, then the other centres will not be affected by enolization, so the product will be a mixture of diastereoisomers of the original compound rather than the racemate.
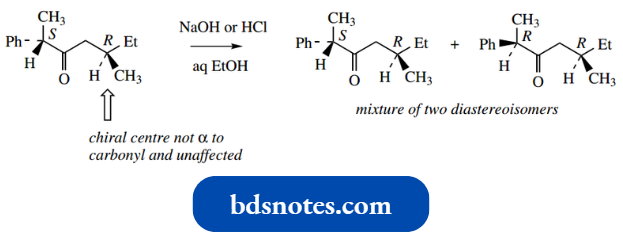
Sometimes, other features in the molecule may facilitate the formation of the enol or enolate.
Thus, in the ketone shown below, conjugation of the enol double bond with the aromatic ring system helps to stabilize the enol tautomer; therefore, enolization and racemization occur more readily.
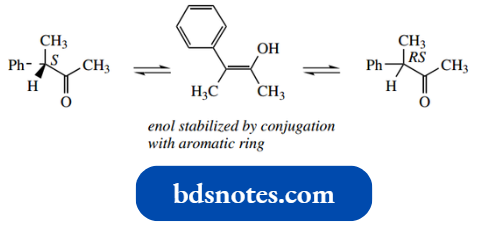
It should be noted that the rate of racemization (or the rate of hydrogen exchange) is the same as the rate of enolization since the protonation reaction is fast.
Hence, the rate is typical of a bimolecular process and depends upon two variables, the concentration of carbonyl compound and the concentration of acid (or base).
Rate = k[C=O][acid]
or
Rate = k[C=O][base]
where C=O is the carbonyl substrate and k is the rate constant.
Interconversion Of Monoterpene Stereoisomers Through Enolization:
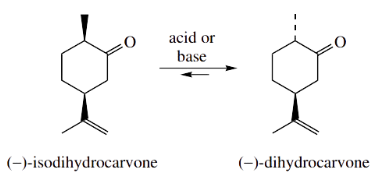
On heating with either acid or base, the monoterpene ketone isodihydrocarvone is largely converted into one product only, its stereoisomer dihydrocarvone.
“Steps to explain types of nucleophilic reactions with enolate anions: Aldol vs Michael addition: Notes guide”
There are two chiral centres in isodihydrocarvone, but only one of these is adjacent to the carbonyl group and can participate in enolization.
- Under normal circumstances, we might expect to generate an equimolar mixture of two diastereoisomers.
- This is because two possible configurations could result from the chiral centre α to the carbonyl, whereas the other centre is going to stay unchanged.
- We might thus anticipate the formation of a 50:50 mixture of isodihydrocarvone and dihydrocarvone.
- That the product mixture is not composed of equal amounts of isodihydrocarvone and dihydrocarvone can be rationalized by considering stereochemical factors, particularly the conformations adopted by the two compounds, which turn out to favour the product over the starting material.
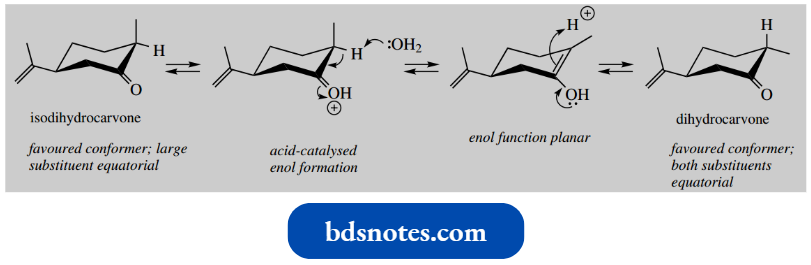
The favoured conformation of isodihydrocarvone has the large isopropenyl substituent equatorial.
- On forming the enol (or enolate anion), it will adopt the conformation in which both substituents are equatorial (or equatorial-like).
- To revert to a keto tautomer might then involve acquiring a proton from either side of the planar enol/enolate.
- However, there is going to be a distinct preference for forming the more favoured product that has two equatorial substituents.
- This is dihydrocarvone. The equilibrium mixture set up thus contains predominantly dihydrocarvone, rather than an equal mixture of two diastereoisomers.
The second chiral centre contains a large group, and its stereochemical preference effectively dictates the chirality at the second centre, and thus the nature of the product.
Nucleophilic Reactions Involving Enolate Anions Conjugation:
When an enol tautomer reverts to a keto tau timer, it must acquire a proton, and we have already seen that it may be acquired from different faces of the double bond, giving two types of stereochemistry.
- In the example described, the stereochemistry of the product was effectively dictated by the existing chirality at a second centre.
- Now we can see a further variant, in that the stability of the product dictates that an alternative carbon in the enol tautomer receives the proton. This relates to conjugation in the product.
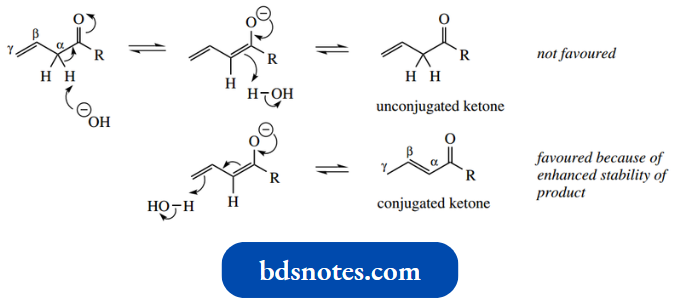
A β,γ-unsaturated carbonyl compound exposed to acid or base is usually converted rapidly into an α,β-unsaturated carbonyl derivative. This isomerization is easily interpreted by considering enolization.
“Role of enolate formation in base-catalyzed reactions: Questions answered”
Removal of an α-proton from a β,γ-unsaturated ketone generates an enolate anion, and this might be transformed back to the β,γ-unsaturated compound by deprotonation at the α-position.
- However, this does not occur because the enolate anion now has conjugated double bonds, and we can propose an alternative mechanism for deprotonation, invoking the conjugation and protonating at the γ-position.
- This protonation is preferred, in that the product is now a conjugated ketone and, therefore, energetically favoured over the non-conjugated ketone. Since all the reactions are equilibria, eventually a more stable product will result.
Conversion Of Pregnenolone Into Progesterone:
An important transformation in steroid biochemistry is the conversion of pregnenolone into progesterone.
- Progesterone is a female sex hormone, a progestogen, but this reaction is also involved in the production of corticosteroids such as hydrocortisone and aldosterone.
- The reaction also occurs in plants, and features in the formation of cardioactive glycosides, such as digitoxin in foxglove.
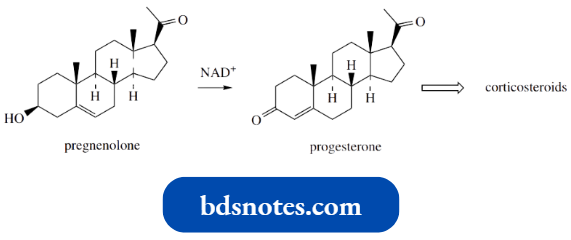
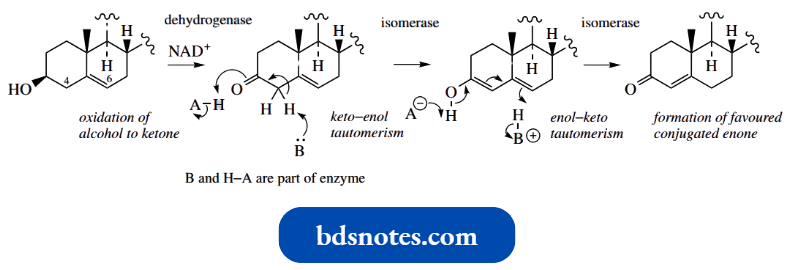
This enzymic conversion involves two enzymes, a dehydrogenase and an isomerase. The dehydrogenase component oxidizes the hydroxyl group on pregnenolone to a ketone and requires the oxidizing agent cofactor NAD+.
The isomerase then carries out two tautomerism reactions, enolization to a dienol followed by production of the more stable conjugated ketone.
“How does aldol condensation proceed via enolate anions? FAQ explained”
The enzyme provides a base (B:) and an acid (A–H) via appropriate amino acid side chains on the enzyme to facilitate proton removal and supply.
- A fascinating aspect is that the proton removed from the methylene (steroid position 4) by the base is then donated back to position 6. The base is suitably positioned to serve both sites in the steroid.
An exactly analogous enzymic transformation is encountered during the formation of oestrogen and androgen sex hormones, for example., estradiol and testosterone respectively, where dehydroepiandrosterone is oxidized to androstenedione.

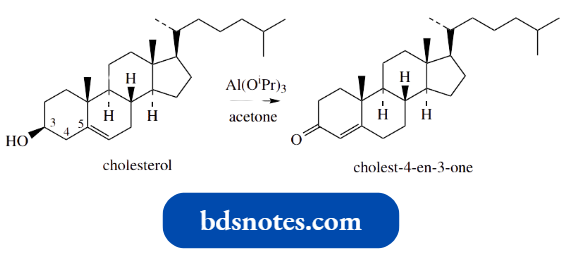
The isomerization reaction is also encountered in chemical manipulations of steroids.
- Thus, many natural steroids contain a 5-en-3-ol combination of functionalities, for example., cholesterol.
Treatment of cholesterol with an oxidizing agent (aluminium isopropoxide is particularly suitable) leads to cholest-4-en-3-one, the tautomerism occurring spontaneously under the reaction conditions.
Halogenation:
Aldehydes and ketones undergo acid- and base-catalysed halogenation in the α position. This is also dependent on enolization or the formation of enolate anions.
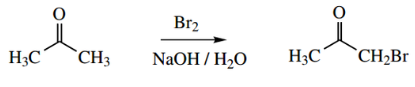
Thus, bromination of acetone may be achieved by using bromine in sodium hydroxide solution, and this is rationalized mechanistically through the formation of the enolate anion, which then attacks the polarized bromine electrophile.
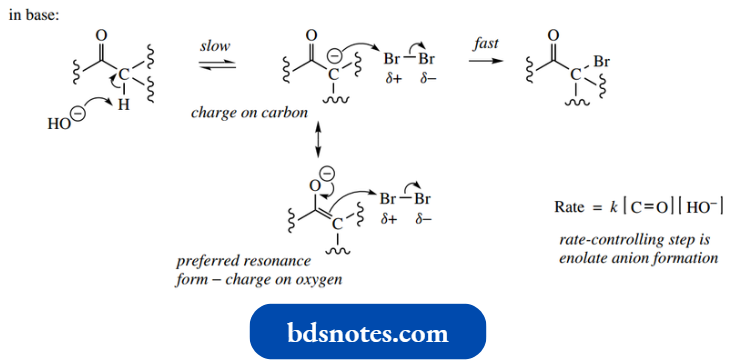
“Early warning signs of gaps in understanding enolate anion basics: Common questions”
There are two ways of representing this, according to which resonance form of the enolate anion is used.
- Although the preferred resonance form (charge located on the oxygen atom) should be used as the nucleophile because carbon is acting as the nucleophile and a new C–Br bond is formed, the less-favoured resonance form is frequently employed in mechanistic pathways.
- This makes mechanism drawing rather easier but is technically incorrect.
- Kinetic data show us that the rate of reaction is dependent upon two variables, i.e. the carbonyl in acid:
- substrate concentration and the concentration of base. These are the two components necessary for the formation of the enolate anion, which is the slow step in the sequence.
- After the formation of the enolate anion, nucleophilic attack on bromine is rapid; therefore, the bromine concentration does not figure in the rate equation. A related mechanism can be drawn for acid-catalysed halogenation.
Again, the halogen concentration does not figure in the rate equation, and the rate of enolization controls the rate of reaction.
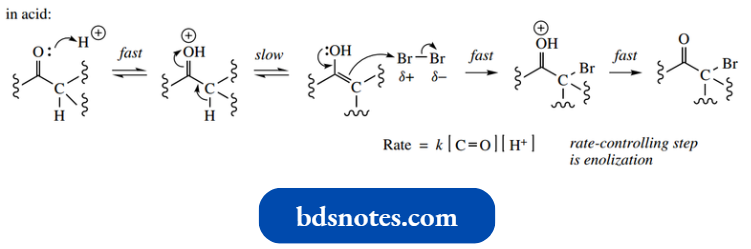
If we wish to synthesize a monohalogenated product, then we have to use an acid-catalysed reaction; base catalysis leads to multiple halogenations. This relates to the acidity of intermediates.
- Thus, each successive halogenation introduces an electron-withdrawing substituent, which increases acidity and facilitates enolate anion formation.
- On the other hand, an electron-withdrawing halogen substituent destabilizes the protonated carbonyl compound and consequently disfavours enolization.
base-catalysed reaction: consider acidity and enolate anion formation
acid-catalysed reaction: consider basicity and conjugate acid formation
Alkylation Of Enolate Anions
Though this topic is treated here under a separate heading, alkylation of enolate anions is nothing other than enolate anions acting as carbanion nucleophiles in SN2 reactions.
- We deferred this topic from, at that stage we had not encountered the concept of enols and enolate anions.
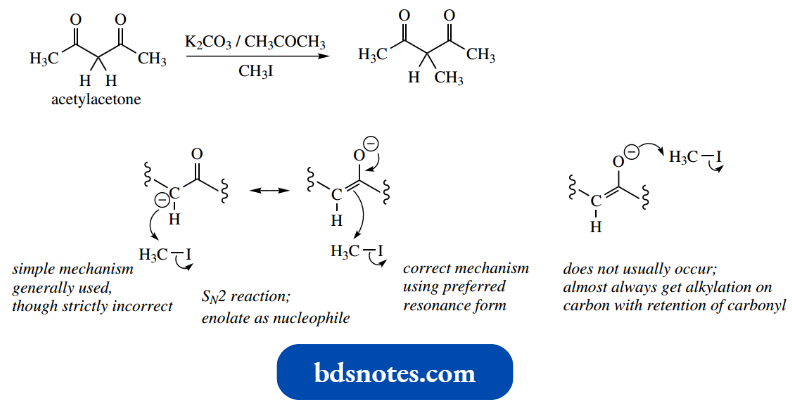
By treating the 1,3-dicarbonyl compound acuity acetone with methyl iodide in the presence of potassium carbonate, one observes alkylation at the central carbon.
“Asymptomatic vs symptomatic effects of ignoring enolate anion principles: Q&A”
This is easily rationalized via the initial formation of an enolate anion under the basic conditions, followed by an SN2 reaction on the methyl iodide.
- The enolate anion is the nucleophile and iodide is displaced as the leaving group.
- The enolate anion could be drawn with a charge on carbon or oxygen; the latter is preferred, as discussed above, in that the charge is preferentially located on the electronegative oxygen atom.
- It is feasible, therefore, that either carbon or oxygen could be the nucleophilic atom, and we might expect more chance of oxygen participating.
- Despite this, it is observed that, in almost all cases, alkylation occurs on carbon, not on oxygen, so it does not present a problem.
Two mechanisms could be drawn for the reaction, depending on whether the enolate anion has a charge on the carbon or oxygen.
- Since carbon is eventually the nucleophilic centre, it is permissible to use the carbanion version of the enolate (as, in general, we shall do), though this is strictly not correct, and purists would use the alternative version starting with charge on the oxygen.
- Now for some interesting features of the reaction, though they become fairly obvious with a little thought.
- First, the central methylene contains the more acidic protons (pKa 9) since it is flanked by two carbonyls, so the enolate anion formed involves this carbon.
In other words, alkylation occurs on the central carbon of acetylacetone, not on the terminal carbons.
- Second, it is possible to use carbonyl compounds such as acetone as a solvent without these reacting under the reaction conditions.
- Acetone will have similar acidity (pKa 19) to the acetyl groups of acetylacetone, so likewise will not form an enolate anion under conditions that only ionize the central methylene of a 1,3-dicarbonyl compound.
- Furthermore, the product formed still contains an acidic proton on a carbon flanked by two carbonyls, so it can form a new enolate anion and participate in a second SN2 reaction.
- The nature of the product will thus depend on electrophile availability.
With 1 mol of methyl iodide, a monomethylated compound will be the predominant product, whereas with 2 mol of methyl iodide, the result will be mainly the dimethylated compound.
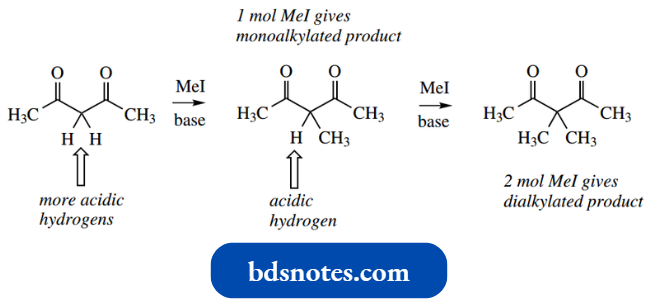
A further twist is that it is possible to use this reaction to insert two different alkyl groups.
This requires treating first with 1 mol of an alkylating agent, allowing the reaction to proceed, then supplying 1 mol of a second, but different, alkylating agent.

Of course, minor products might be produced, including monoalkylated products and dialkylated products (in which the two alkyl groups are the same), depending on the conditions and how near to completion the reaction proceeds.
- Note that we cannot use aryl halides in these reactions; rear side attack is impossible and we do not get SN2 reactions at sp2-hybridized carbon.
- 1,3-Dicarbonyl compounds, like acetylacetone, are reasonably acidic (pKa 9) and the formation of enolate anions is achieved readily. Potassium carbonate is basic enough to ionize acetylacetone in the above example.
However, if we are presented with a substrate having only a single carbonyl group, for example.,. acetone (pKa 19), then it follows that we must use a stronger base to remove the correspondingly less acidic protons. Strong bases that might be used include sodium hydride and sodium amide.
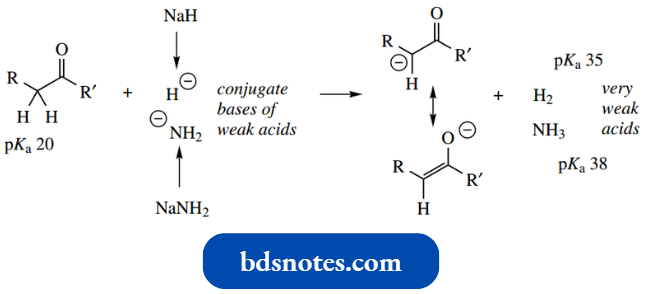
“Can targeted interventions improve outcomes using enolate anion knowledge? FAQs provided”
These compounds ionize and act as sources of hydride and amide ions respectively, which can remove α-protons from carbonyl compounds.
- These ions are the conjugate bases of hydrogen and ammonia respectively, compounds that are very weak acids indeed.
- What becomes important here is that enolate anion formation becomes essentially irreversible; the enolate anion formed is insufficiently basic to be able to remove a proton from either hydrogen or ammonia.
- This is in marked contrast to the earlier examples of enolate anion formation that were reversible. We now have a means of preparing the enolate anion, rather than relying upon an equilibrium reaction.
Accordingly, reactions are usually done in two stages, preparation of the enolate anion followed by the addition of the alkylating agent electrophile.
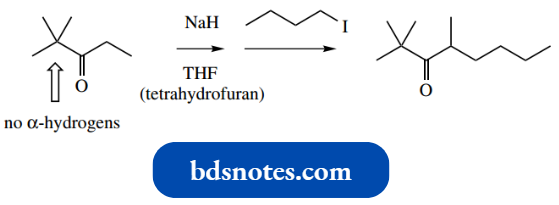
In the example shown, alkylation of the ketone is readily accomplished using such a two-stage process with 1 mol of alkyl halide.
- Note that the specificity of this reaction relies on one of the α-carbons having no acidic hydrogens so that only one enolate anion can be formed.
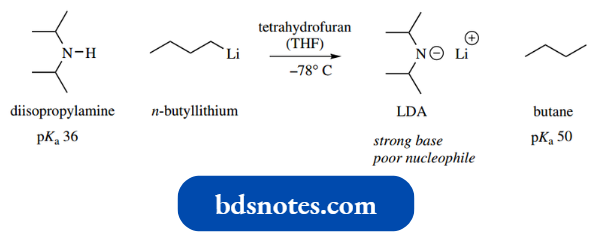
- Another strong base routinely employed in synthetic procedures to prepare enolate anions is lithium diisopropylamide (LDA).
- The diisopropyl amide anion is formed by removing a proton from diisopropylamine using the organometallic derivative n-butyllithium.
- Because of the highly reactive nature of n-butyllithium (it reacts explosively with air) this reaction has to be conducted in an oxygen-free atmosphere and at a very low temperature.
The ionization works because although the acidity of diisopropyl lamine is not great (pKa 36), the other product formed, i.e. butane, is significantly less acidic (pKa 50). The reaction is essentially irreversible.
When the carbonyl compound is added to this base, the abstraction of a proton and formation of the enolate anion follow, as seen with sodium hydride or sodium amide above.
- Again, this reaction is essentially irreversible because the other product is the weak base diisopropylamine (pKa 36).
- So far, there does not seem any particular advantage in using LDA rather than sodium hydride or sodium amide, and the manipulations required are very much more difficult and dangerous.
- The real benefit is that LDA is a very strong base, and because of its quite large size it is also a relatively poor nucleophile.
This reduces the number of competing reactions that might occur where nucleophilicity competes with basicity.
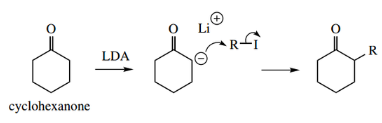
In symmetrical structures such as cyclohexanone, ionization at α-positions occurs readily and allows the preparation of alkylated products.
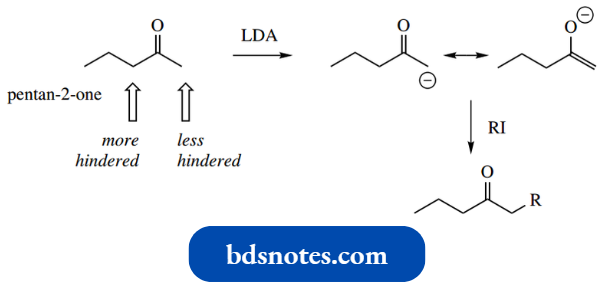
- In unsymmetrical structures, the sheer size of LDA as a base may allow selectivity by preferential removal of certain α-protons.
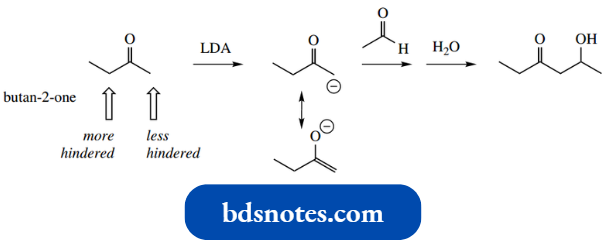
- Thus, the ketone pentane-2-one will undergo preferential removal of a proton from the terminal methyl in the generation of an enolate anion.
This allows selective alkylation to be achieved.
“Differential applications of thermodynamic vs kinetic enolates: Notes explained”
Addition–Dehydration: The Aldol Reaction
We now have examples of the generation of enolate anions from carbonyl compounds, and their potential as nucleophiles in simple SN2 reactions.
- However, we must not lose sight of the potential of a carbonyl compound to act as an electrophile.
- This section, the aldol reaction, is concerned with the enolate anion aldol reaction formation of nucleophiles attacking carbonyl electrophiles to give addition compounds, though it is usual for such addition compounds to then lose water, i.e. addition–dehydration.
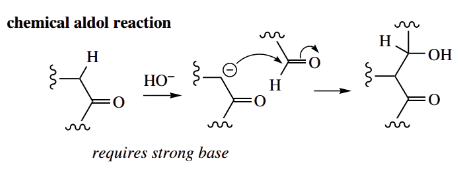
- The namesake aldol reaction is the formation of an additional compound, aldol, from two molecules of acetaldehyde, when this aldehyde is treated with aqueous sodium hydroxide.
The terminology aldol comes from the functional groups in the product, aldehyde and alcohol.
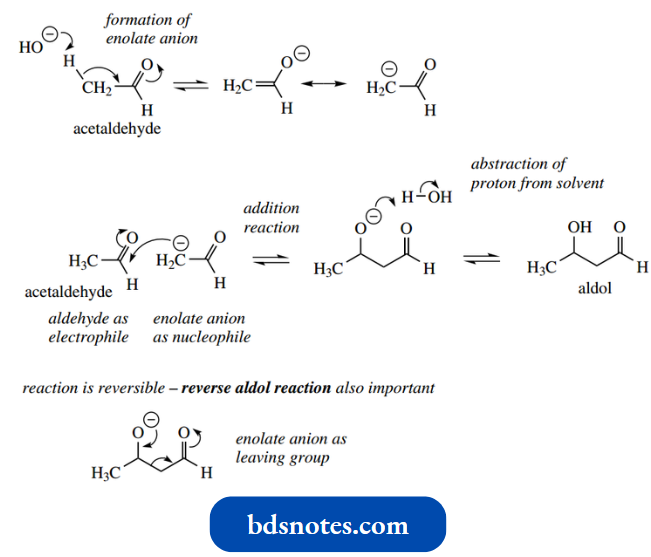
This is easily formulated as the production of an enolate anion followed by a nucleophilic attack of this anion onto the carbonyl group of a second molecule of acetaldehyde.
- Aldol is then produced when the addition anion abstracts a proton from solvent. The reaction is reversible, and it is usually necessary to disturb the equilibrium by some means.
- Removal rather messy mixture of products containing at least four different components. This is because both starting materials might feature as nucleophiles or as electrophiles.
In the reverse reaction, the addition anion reforms the carbonyl group by expelling the enolate anion as a leaving group. This reverse aldol reaction is sufficiently important in its own right, and we shall meet examples.
- Note that, as we saw with simple aldehyde and ketone addition reactions, aldehydes are better electrophiles than ketones.
- This arises from the extra alkyl group in ketones, which provides a further inductive effect and extra steric hindrance.
Accordingly, the aldol reaction is more favourable with aldehydes than with ketones. With ketones, it is essential to disturb the equilibrium in some way.
- The aldol reaction as formulated above involves two molecules of the starting substrate. However, by a consideration of the mechanism, one can see that different carbonyl compounds might be used as nucleophiles or electrophiles.
- This would be termed a mixed aldol reaction or crossed aldol reaction. However, if one merely reacted, say, two aldehydes together under basic conditions, one would get a rather messy mixture of products containing at least four different components.
- This is because both starting materials might feature as nucleophiles or as electrophiles.
Mixed Aldol Reaction
⇒ \(\mathrm{RCH}_2 \mathrm{CHO}+\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CHO} \longrightarrow 4 \text { products }\)
Nucleophile And Electrophilic
⇒ \(\mathrm{RCH}_2 \mathrm{CHO}+\mathrm{RCH}_2 \mathrm{CHO}\)
⇒ \(\mathrm{RCH}_2 \mathrm{CHO}+\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CHO}\)
⇒ \(\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CHO}+\mathrm{RCH}_2 \mathrm{CHO}\)
⇒ \(\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CHO}+\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CHO}\)
For the mixed aldol reaction to be of value in synthetic work, it is necessary to restrict the number of combinations. This can be accomplished as follows.
- First, if one of the materials has no α-hydrogens, then it cannot produce an enolate anion, and so cannot function as the nucleophile.
- Second, in aldehyde plus ketone combinations, the aldehyde is going to be a better electrophile, so reacts preferentially in this role.
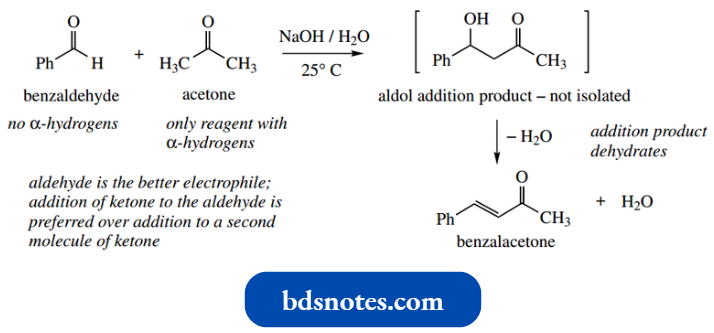
- A simple example of this approach is the reaction of benzaldehyde with acetone under basic conditions.
Such reactions are synthetically important as a means of increasing chemical complexity by forming new carbon-carbon bonds.
“Steps to apply enolate anion chemistry in organic synthesis: Design vs optimization: Notes guide”
Benzaldehyde has no α-hydrogens, so it cannot be converted into an enolate anion to become a nucleophile. Acetone has α-hydrogens, so it can form an enolate anion and become a nucleophile.
- We now have two possible electrophiles, i.e. one an aldehyde and the other a less reactive ketone.
- The preferred reaction is thus acetone as an enolate anion nucleophile, with benzaldehyde as the preferred electrophile, giving the additional product shown.
- This is not isolated, since it readily dehydrates to give the unsaturated ketone benzalacetone.
- The additional product from aldol reactions frequently dehydrates by heating in acid or in base to give the corresponding α,β-unsaturated carbonyl compound.
Under basic conditions, this occurs readily, even though hydroxide is a poor leaving group, because of the acidity of the α-proton and the conjugation stabilization in the product.
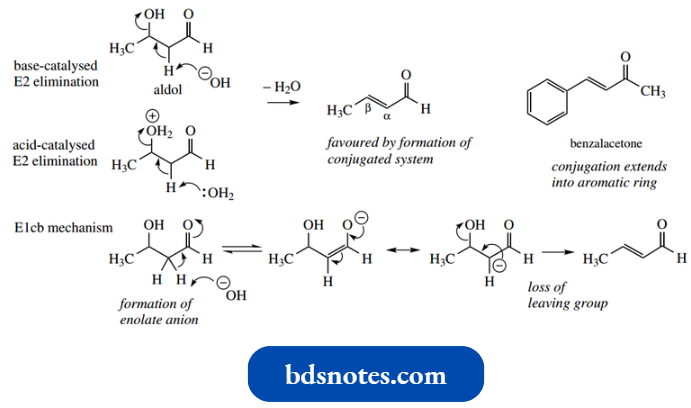
There is evidence that this is not an E2 mechanism under basic conditions, but a so-called E1cb mechanism.
- This stands for elimination–unimolecular–conjugate base, and proceeds via initial removal of the acidic proton to give the conjugate base (enolate anion).
- The reaction is unimolecular because it is the loss of a leaving group from the conjugate base which is the rate-determining step. Removal of the acidic proton is faster than loss of the hydroxide ion.
- Since E1cb reactions are rare (this is the only one we shall consider), we deliberately chose not to include it under general elimination reactions.
- The conditions of the reaction are often sufficient to cause dehydration of the addition product as it is formed, and it is normally extremely difficult to isolate the addition product.
It turns out that the addition reaction (equilibrium) is slow, whereas the elimination reaction (non-reversible) is faster.
- This usually disturbs the equilibrium in an aldol reaction, especially if the product is stabilized by even further conjugation, as in the case of benzalacetone above, where the benzene ring also forms part of the conjugated system.
- An alternative approach to mixed aldol reactions, and the one usually preferred, is to carry out a two-stage process, forming the enolate anion first using a strong base like LDA.
- The first step is essentially irreversible, and the electrophile is then added in the second step.
- An aldol reaction between butane-2-one and acetaldehyde exemplifies this approach.
Note also that the large base LDA selectively removes a proton from the least-hindered position, again restricting possible combinations.
Aldol And Reverse Aldol Reactions In Biochemistry: Aldolase, Citrate Synthase:
Both the aldol and reverse aldol reactions are encountered in carbohydrate metabolic pathways in biochemistry.
- One reversible transformation can be utilized in either carbohydrate biosynthesis or carbohydrate degradation, according to a cell’s particular requirement.
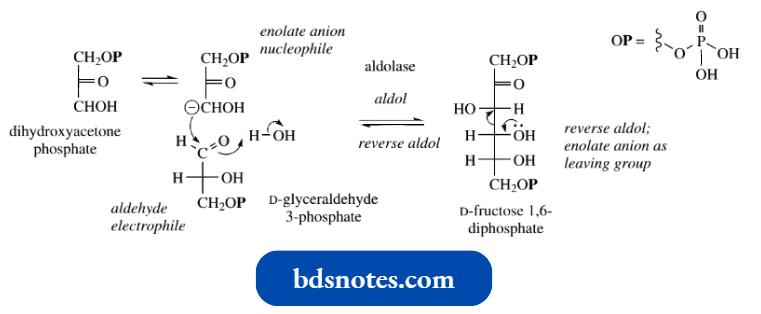
- D-Fructose 1,6-diphosphate is produced during carbohydrate biosynthesis by an aldol reaction between dihydroxyacetone phosphate, which acts as the enolate anion nucleophile, and D-glyceraldehyde 3-phosphate, which acts as the carbonyl electrophile; these two starting materials are also interconvertible through keto-enol tautomerism, as seen earlier.
The biosynthetic reaction may be simplified mechanistically as a standard mixed aldol reaction, where the nature of the substrates and their mode of coupling is dictated by the enzyme. The enzyme is called aldolase.
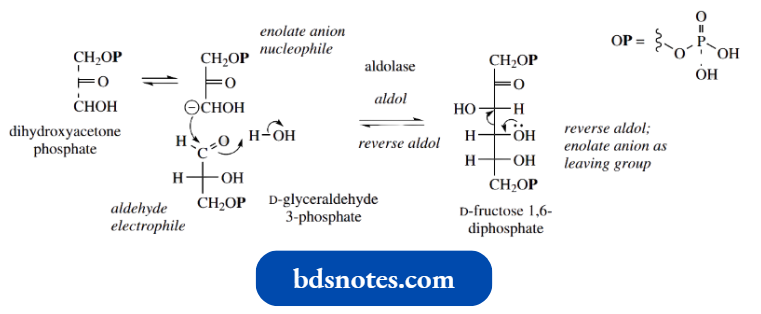
During carbohydrate metabolism in the glycolytic pathway, fructose 1,6-diphosphate is cleaved to give dihydroxyacetone phosphate and glyceraldehyde 3-phosphate.
- This is a reverse aldol reaction, in which a carbonyl group is formed at the expense of carbon-carbon bond cleavage with expulsion of an enolate anion leaving group.
- The additional functional groups present in the substrates would seriously limit any base-catalysed chemical aldol reaction between these substrates, but this reaction is enzyme-mediated, allowing reaction at room temperature and near-neutral conditions.
The aldol and reverse aldol reactions just described accommodate the chemical changes observed, though we now know that nature uses a slightly different approach via enamines.
- This does not significantly alter our understanding of the reactions, but it does remove the requirement for a strong base and also accounts for the bonding of the substrate to the enzyme.
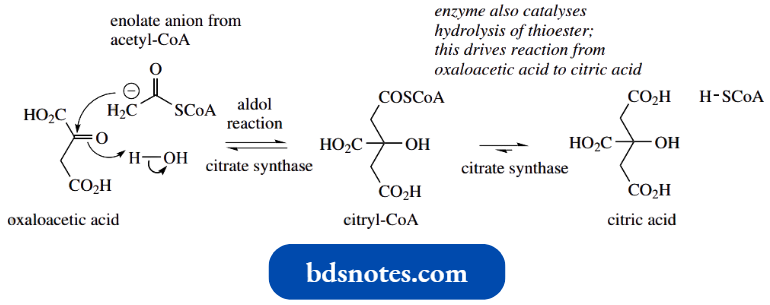
- A similar aldol reaction is encountered in the Krebs cycle in the reaction of acetyl-CoA and oxaloacetic acid. This yields citric acid and is catalysed by the enzyme citrate synthase.
This intermediate provides the alternative terminology for the Krebs cycle, namely the citric acid cycle.
The aldol reaction is easily rationalized, with acetyl-CoA providing an enolate anion nucleophile that adds to the carbonyl of oxaloacetic acid. We shall see later that esters and thioesters can also be converted into enolate anions.
“Role of enolate anions in forming carbon-carbon bonds: Questions answered”
One interesting feature here is that both acetyl-CoA and oxaloacetic acid have the potential to form enolate anions and that oxaloacetic acid is more acidic than acetyl-CoA, in that the two carbonyl groups are flanking the methylene.
- That citrate synthase achieves the aldol reaction as shown reflects that the enzyme active site must have a basic residue appropriately positioned to abstract a proton from acetyl-CoA rather than oxaloacetic acid, thus allowing acetyl-CoA to act as the nucleophile.
- The obvious product of the aldol reaction would be the thioester citryl-CoA. However, the enzyme citrate synthase also carries out hydrolysis of the thioester linkage, so that the product is citric acid; hence the terminology.
The hydrolysis of the thioester is responsible for disturbing the equilibrium and driving the reaction to completion.
- We should also consider occasions when there are two carbonyl groups in the same molecule.
- We then have the possibility of an intramolecular aldol reaction, and this offers a convenient way of synthesizing ring systems.
Rings with five or six carbons are particularly favoured. Thus, treatment of octan-2,7-dione with base gives good yields of the cyclopentene derivative shown.
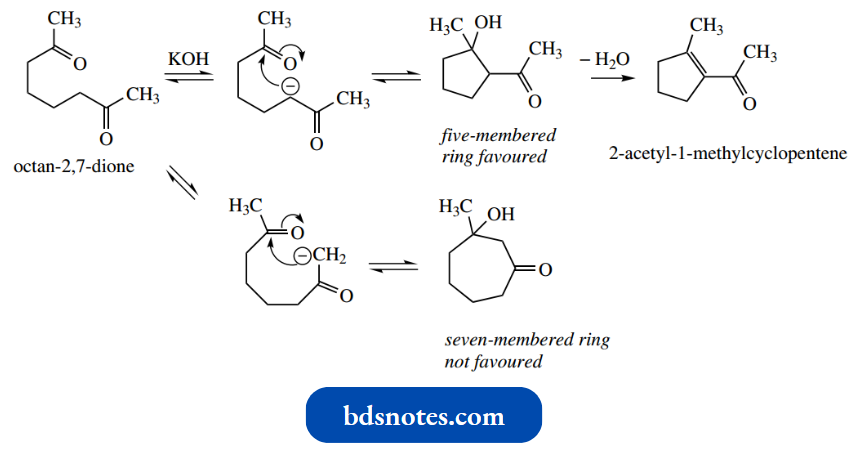
The reaction is readily formulated. Note that there are two potential products from the aldol addition, one of which is five-membered and the other seven-membered.
- The five-membered product is more favourable than the seven-membered one simply based on ring strain.
- However, if both products form, they will be in equilibrium as shown. It is the next step, dehydration, that drives the six-membered ring and seven-membered ring not favoured the reaction giving the more stable product, the cyclopentene.
- Any seven-membered addition product can then equilibrate to give more of the five-membered compound.
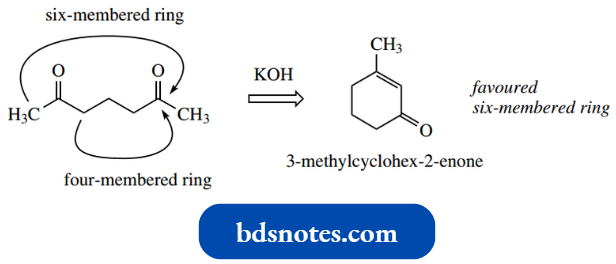
A similar reaction with heptane 2,6-dione would lead to the methylcyclohexane product, and not the sterically unfavourable four-membered ring alternative.
Note also that if the substrate has both aldehyde and ketone functions the aldehyde will act as the electrophile.
The ketoaldehyde shown forms the one product in good yield, there now being restrictions on preferred ring size and the regiochemistry of the mixed aldol reaction.
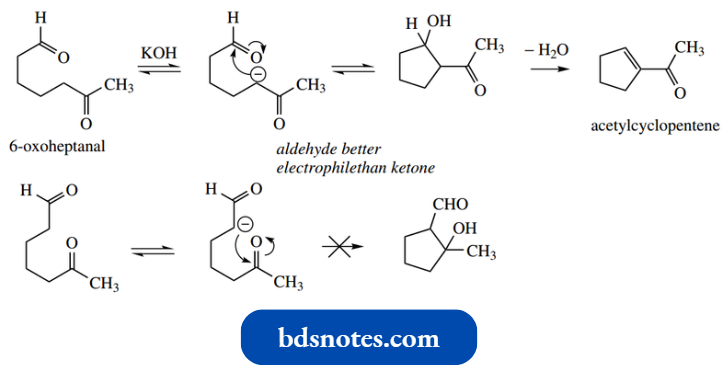
If a five- or six-membered ring can form, then intramolecular aldol reactions usually occur more rapidly than the corresponding intermolecular reactions between two molecules of substrate. This provides a very useful route to cyclic compounds.
Other Stabilized Anions As Nucleophiles: Nitriles And Nitromethane
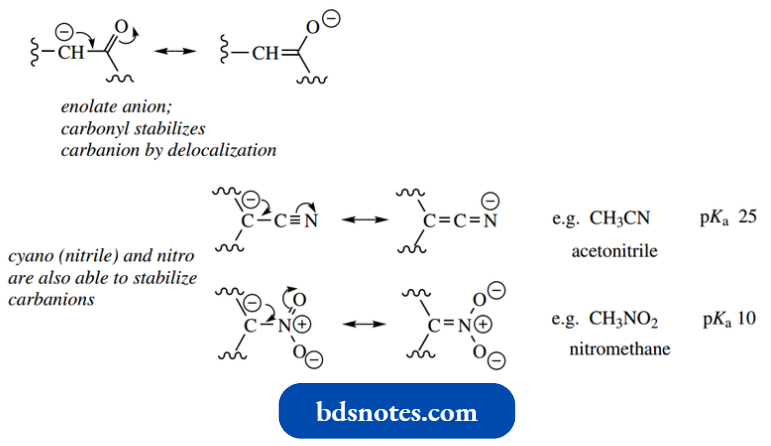
An enolate anion behaves as a carbanion nucleophile, the carbonyl group stabilizing the anion by delocalization of charge.
- Both cyano (nitrile) and nitro groups can fulfil the same role as a carbonyl by stabilizing a carbanion, so we see similar enhanced acidity of α-protons in simple nitrile and nitro compounds.
- pKa values for nitriles are about 25, whereas aliphatic nitro compounds have pKa of about 10. Nitro compounds are thus considerably more acidic than aldehydes and ketones (pKa about 20).
Accordingly, it is possible to generate analogues of enolate anions containing cyano and nitro groups and to use these as nucleophiles towards carbonyl elec trophiles in aldol-like processes. Simple examples are shown.
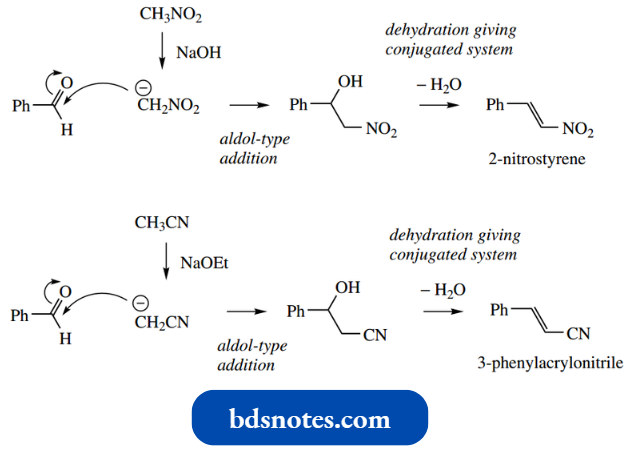
“How does Michael addition utilize enolate anions? FAQ explained”
As with many aldol reactions, addition is usually followed by the elimination of water, generating a conjugated system with the cyano or nitro group. The presence of extended conjugation through aromatic substituents enhances this process.
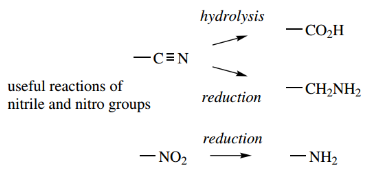
These reactants introduce either nitrile or nitro groups into the product. These groups may be converted into carboxylic acids or amines, as shown.
Enamines As Nucleophiles:
We met enamines as products from addition–elimination reactions of secondary amines with aldehydes or ketones.
Enamines are formed instead of imines because no protons are available on nitrogen for the final deprotonation step, and the nearest proton that can be lost from the iminium ion is that at the β-position.
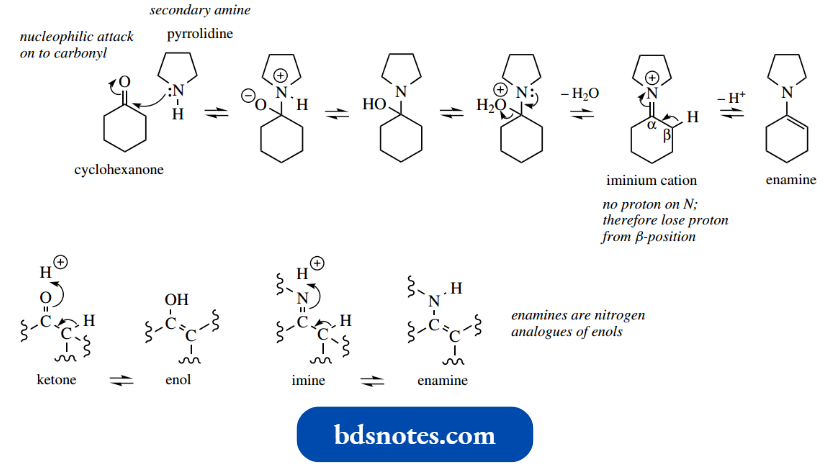
“Early warning signs of complications from ignoring enolate anion protocols: Common questions”
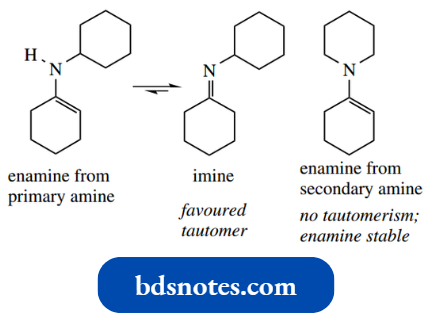
There is a distinct relationship between keto-enol tautomerism and the iminium–enamine interconvert H sion; it can be seen from the above scheme that enamines are nitrogen analogues of enols.
- Their chemical properties reflect this relationship. It also leads us to another reason why enamine formation is a property of secondary amines, whereas primary amines give imines with aldehydes and ketones.
- Enamines from primary amines enamine from primary amine imine favoured enamine from secondary amine would undergo rapid conversion into the more stable imine tautomers (compare enol and keto tautomers); this isomerization cannot occur with enamines from secondary amines, and such enamines are, therefore, stable.
The most prominent property of enamines is that the β-carbon can behave as a carbon nucleophile.
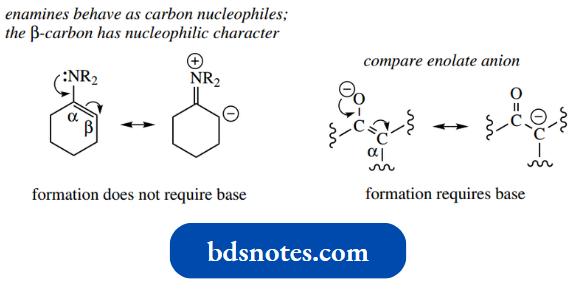
This resonance form can then act as a nucleophile, in much the same way as an enolate anion can. However, there is a marked difference, and this is what makes enamines such useful synthetic intermediates.
- Generation of an enolate anion requires the treatment of a carbonyl compound with a base, sometimes a very strong base.
- This is a consequence of resonance; the overlap of lone pair electrons from the nitrogen provides an iminium system, with the negative counter-charge on the β-carbon.
- The formation of the enamine resonance form is a property of the enamine and requires no base. A simple SN2 alkylation reaction serves as an example.
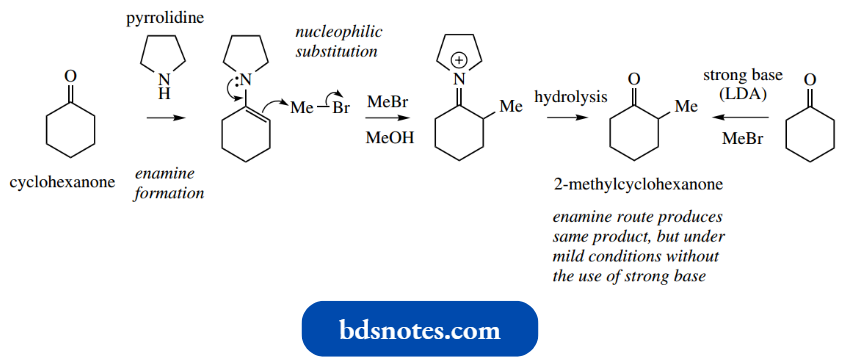
As we have already seen, treating cyclohexanone with LDA gives the enolate anion, which can then be allowed to react with methyl iodide to give 2-methyl cyclohexanone.
“Asymptomatic vs symptomatic effects of outdated enolate practices: Answered”
Alternatively, cyclohexanone may initially be transformed into an enamine with a secondary amine, here pyrrolidine. This intermediate enamine can act as a nucleophile and can be alkylated at the β-position using methyl iodide.
- Finally, 2-methylcyclohexanone may be generated by hydrolysis of the iminium system, effectively a reversal of enamine formation.
- This gives us two routes to 2-methylcyclohexanone, a short process using the very strong base LDA and a longer route that involves no strong base and relatively mild conditions.
- The latter synthesis may well be preferred, depending upon the nature of any other functional groups in the starting substrate.
- The essential feature of enamines is that they are nitrogen analogues of enols and behave as enolate anions.
- They effectively mask a carbonyl function while activating the compound towards nucleophilic substitution.
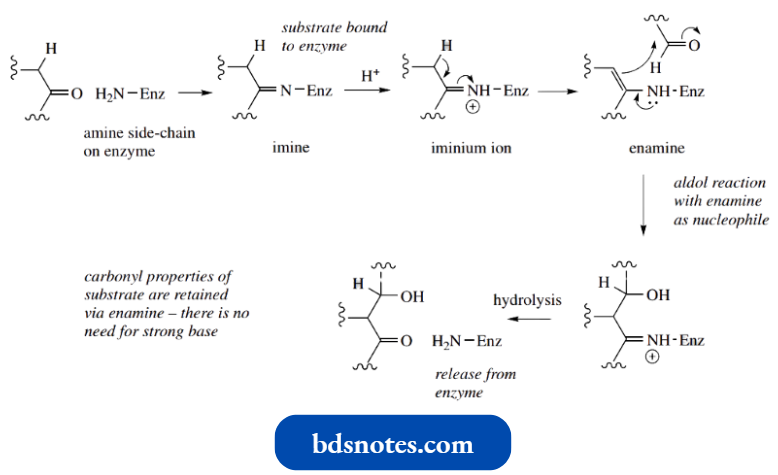
Enamine Reactions In Biochemistry: Aldolase
We saw that an aldol-like reaction could be used to rationalize the biochemical conversion of dihydroxyacetone phosphate (nucleophile) and glyceraldehyde 3-phosphate (electrophile) into fructose 1,6-diphosphate by the enzyme aldolase during carbohydrate biosynthesis.
The reverse reaction, used in the glycolytic pathway for carbohydrate metabolism, was formulated as a reverse aldol reaction.
In a postscript, we noted that nature avoided the use of a strong base to catalyse the reaction by involving an enzyme. Here, we see how this is achieved through an enamine.
- Enzymes are very sophisticated systems that apply sound chemical principles. The side chains of various amino acids are used to supply the necessary bases and acids to help catalyse the reaction.
- Thus, the enzyme aldolase binds the dihydroxyacetone phosphate substrate by reacting the ketone group with an amine, part of a lysine amino acid residue.
This forms an imine that becomes protonated under normal physiological conditions.
“Can preventive measures reduce risks of side reactions in enolate chemistry? FAQs provided”
A basic group removes a proton from the β-carbon of the iminium and forms the enamine.
- This enamine then reacts as a nucleophile towards the aldehyde group of glyceraldehyde 3-phosphate in a simple addition reaction, and the proton necessary for neutralizing the charge is obtained from an appropriately placed amino acid residue.
- Finally, the iminium ion loses a proton and hydrolysis releases the product from the enzyme.
- The reaction is exactly analogous to the chemical aldol reaction (also shown), but it utilizes an enamine as the nucleophile, and it can thus be achieved under typical enzymic conditions, i.e. around neutrality and at room temperature.
- There is one subtle difference though, in that the enzyme produces an enamine from a primary amine.
- We have indicated that enamine formation is a property of secondary amines, whereas primary amines react with aldehydes and ketones to form imine. Thus, a further property of the enzyme is to help stabilize the enamine tautomer relative to the imine.
The Mannich Reaction:
We saw that imines and iminium ions could act as carbonyl analogues and participate in nucleophilic addition reactions.
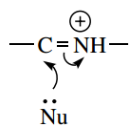
Iminium ion acting as carbonyl analogue for nucleophilic addition reaction
One simple example was the hydrolysis of imines back to carbonyl compounds via nucleophilic attack of water.
- The Mannich reaction is only a special case of nucleophilic addition to iminium ions, where the nucleophile is an enol system, the equivalent of an enolate anion.
- We have to say ‘the equivalent of an enolate anion’ because conditions that favour iminium cations are not going to allow the participation of negatively charged nucleophiles. The Mannich reaction is best discussed via an example.
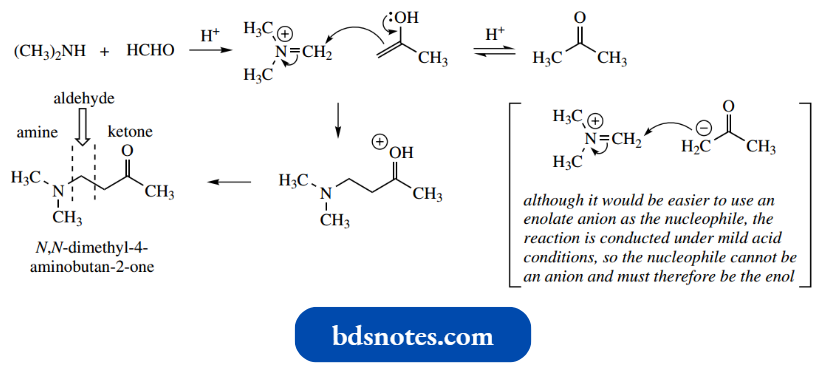
- A mixture of dimethylamine, formaldehyde and acetone under mildly acidic conditions gives N, N dimethyl-4-aminobutan-2-one.
- This is a two-stage iminium ion acting as a carbonyl analogue for the nucleophilic addition reaction process, beginning with the formation of an iminium cation from the amine and the more reactive of the two carbonyl compounds, in this case, the aldehyde.
- This iminium cation then acts as the electrophile for the addition of the nucleophile acetone.
- Now it would be nice if we could use the enolate anion as the nucleophile, as in the other reactions we have looked at, but under the mild acidic conditions we cannot have an anion, and the nucleophile must be portrayed as the enol tautomer of acetone.
The addition is then unspectacular, and, after the loss of a proton from the carbonyl, we are left with the product.
“Differential applications of acid-catalyzed vs base-catalyzed enolate formations: Notes explained”
This is a fairly general reaction and requires an amine plus an aldehyde (usually, but not necessarily, formaldehyde) together with an enolizable ketone, which together a β-aminoketone via an iminium system.
The Mannich reaction is surprisingly important in biochemical processes, especially in the biosynthetic formation of alkaloids. We shall also see several examples in heterocyclic chemistry.
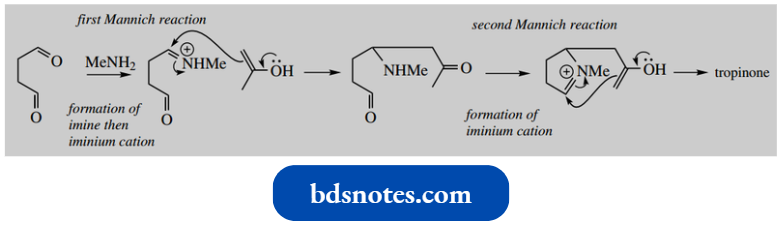
Mannich Reaction: The Synthesis Of Tropine
The Mannich reaction was used for the first synthesis of tropine, the parent alcohol of the tropane alkaloids.
- One of the natural tropane alkaloids used medicinally is hyoscyamine, sometimes in its racemic form atropine.
- Hyoscyamine is an anticholinergic, competing with acetylcholine for the muscarinic site of the parasympathetic nervous system, thus preventing the passage of nerve impulses.
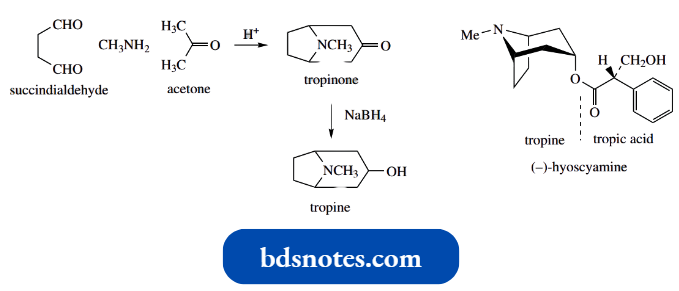
The synthesis involved a reaction of methylamine, succindialdehyde and acetone under mild acid conditions, and although yields were poor, tropinone was formed. This could then be reduced with sodium borohydride to give tropine.
It is instructive to formulate a mechanism for this reaction; note that two Mannich reactions are involved. The scheme below shows the sequence of events, though not all the steps are shown.
Biosynthesis Of Tetrahydroisoquinolines
Mannich and Mannich-like reactions are widely used for the chemical synthesis of heterocycles, and in alkaloid biosynthesis in plants.
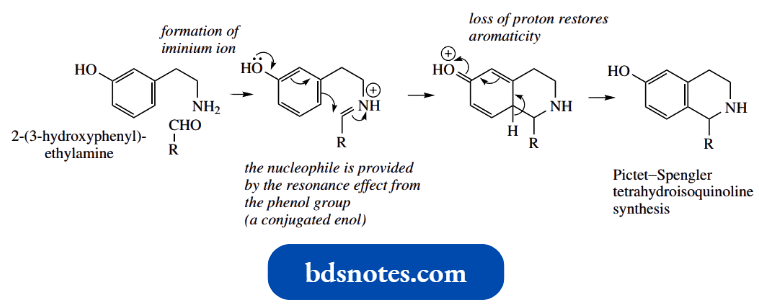
One such reaction important in nature is a biological equivalent of the Pictet–Spengler tetrahydroisoquinoline synthesis, and offers a slight twist, in that the enol nucleophile is a phenol.
“Steps to incorporate AI into analyzing enolate anion reactions: Questions and answers”
Thus, the reaction of 2-(3-hydroxyphenyl)ethylamine with an aldehyde generates initially an imine that will become protonated to an iminium ion.
- The resonance effect from the phenol group will increase electron density at the ortho and para positions in the aromatic ring.
- With the para resonance form, this is equivalent to having a nucleophile located adjacent to the iminium ion and allows the formation of a favourable six-membered ring via the Mannich-like reaction, the nucleophile attacking the C=N.
- Alternatively, we may consider the phenol to be simply a conjugated enol that is participating in a Mannich reaction.
- The final step is the loss of a proton, and this comes from the position para to the oxygen substituent because this allthe ows regeneration of the aromatic ring and phenol group.
- In a chemical reaction, a racemic product will be formed, but enzyme-controlled biochemical reactions normally produce just one enantiomer.
For a simple specific example, the tetrahydroisoquinoline alkaloid salsolinol is found in some plants, and it can also be detected in the urine of humans as a product of dopamine and acetaldehyde.
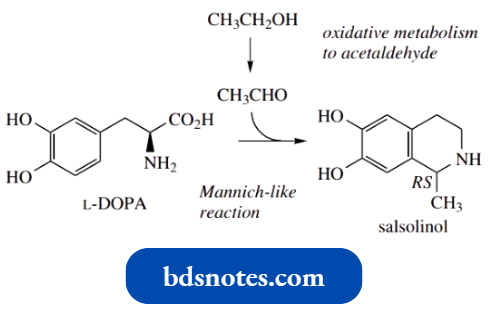
Acetaldehyde is typically formed after ingestion of alcohol. Since the urine product is racemic, it would appear that a chemical Pictet–Spengler synthesis is being observed here rather than an enzymic one.
- We saw that tetrahydroisoquinoline alkaloids with appropriate phenol substituents could be involved in radical coupling processes.
- The complex alkaloids tubocurarine and morphine are derived in nature from simpler tetrahydroisoquinoline alkaloids.
Enolate Anions From Carboxylic Acid Derivatives:
The α-hydrogens of carboxylic acid derivatives show enhanced acidity, as do those of aldehydes and ketones, and for the same reasons, that the carbonyl group stabilizes the conjugate base.
Thus, we can generate enolate anions from carboxylic acid derivatives and use these as nucleophiles in much the same way as we have already seen with enolate anions from aldehydes and ketones.
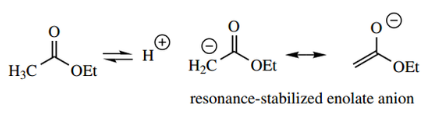
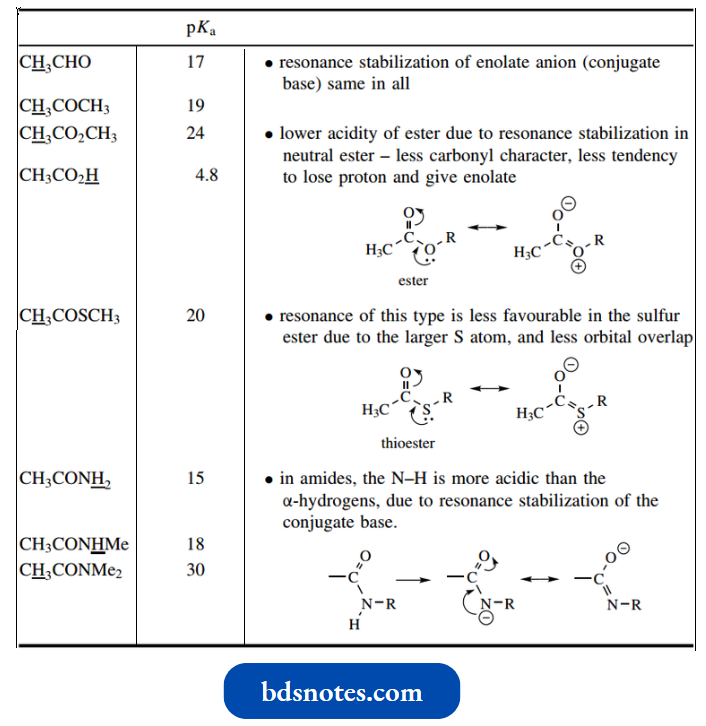
Unfortunately, there are some limitations in the carboxylic acid group of compounds, and the derivatives most often used to form enolate anions are esters. However, esters are less acidic than the corresponding aldehydes or ketones.
“Role of digital tools in improving precision with enolate reaction tracking: FAQs explained”
- Whereas the pKa for the α-protons of aldehydes and ketones is in the region 17–19, for esters such as ethyl acetate it is about 25.
- This difference must relate to the presence of the second oxygen in the ester since resonance stabilization in the enolate anion should be the same.
- To explain this difference, the overlap of the non-carbonyl oxygen lone pair is invoked. Because this introduces charge separation, it is a form of resonance stabilization that can occur only in the neutral ester, not in the enolate anion.
- It thus stabilizes the neutral ester, reduces carbonyl character, and there is less tendency to lose a proton from the α-carbon to produce the enolate. Note that this is not a new concept; we used the same reasoning to explain why amides were not basic like amines.
The α-hydrogens in thioesters are more acidic than in oxygen esters, comparable in fact to those in the equivalent ketone. This can be rationalized by the larger size of sulfur.
- The sulfur lone pair is located in a 3p orbital, whereas oxygen lone pairs are in 2p orbitals; there is consequently less overlap of orbitals. There can be relatively little contribution from this type of resonance stabilization in thioesters.
- Accordingly, normal enolate anion stabilization is not affected.
Note that acids and primary and secondary amides cannot be employed to generate enolate anions. - With acids, the carboxylic acid group has pKa of about 3–5, so the carboxylic proton will be lost much more easily than the α-hydrogens. In primary and secondary amides, the N–H (pKa about 18) will be removed more readily than the α-hydrogens.
- Their acidity may be explained by of resonance stabilization of the anion. Tertiary amides might be used, however, since there are no other protons that are more acidic.
Coenzyme A And Acetyl-CoA
The increased acidity associated with thioesters is one of the reasons that biochemical reactions tend to involve thioesters rather than oxygen esters. The most important thiol encountered in such thioesters is coenzyme A.
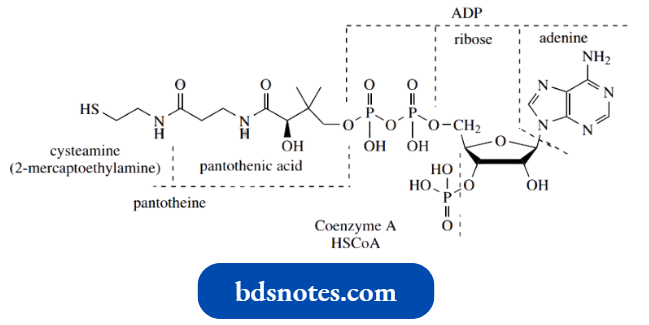
This is a complex molecule, made up of an adenine nucleotide (ADP-3 -phosphate), pantothenic acid (vitamin B5), and cysteamine (2-mercaptoethylamine), but for mechanism purposes can be thought of as a simple thiol, HSCoA. Pre-eminent amongst the biochemical thioesters is the thioester of acetic acid, acetyl-coenzyme A (acetyl-CoA).
- This compound plays a key role in the biosynthesis and metabolism of fatty acids, as well as being a building block for the biosynthesis of a wide range of natural products, such as phenols and macrolide antibiotics.
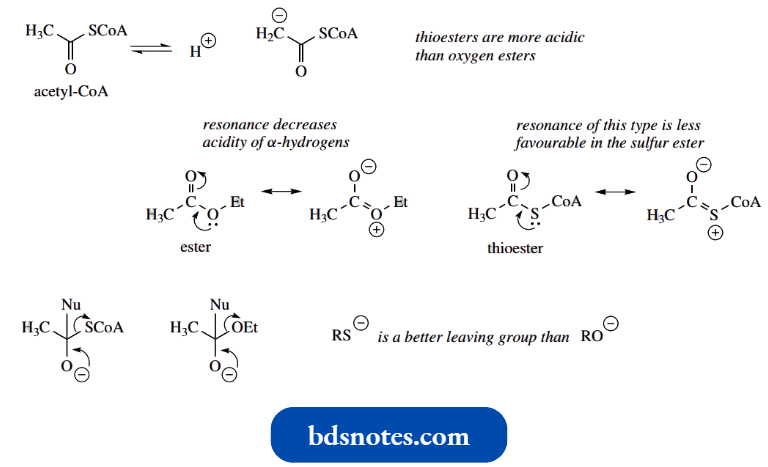
- Acetyl-CoA is a good biochemical reagent for two main reasons. First, the α-protons are more acidic than those in ethyl acetate, comparable in fact to a ketone, and this increases the likelihood of generating an enolate anion.
- As explained above, this derives from sulfur being larger than oxygen, so that electron donation from the lone pair that would stabilize the neutral ester is considerably reduced. This means it is easier for acetyl-CoA to lose a proton and become a nucleophile.
- Second, acetyl-CoA is a better electrophile than ethyl acetate, in that it has a better leaving group; thiols (pKa 10–11) are stronger acids than alcohols (pKa 16). Acetyl-CoA is thus rather well suited to participate in aldol and Claisen reactions.
“How do advancements in technology enhance enolate anion research? Notes guide”
We shall see later that nature can employ yet another stratagem to increase the acidity of the α-protons in thioesters, by converting acetyl-CoA into malonyl-CoA.
- An enolate anion generated from a carboxylic acid derivative may be used in the same sorts of nucleophilic reactions that we have seen with aldehyde and ketone systems.
- It should be noted, however, that the base used to generate the enolate anion must be chosen carefully. If sodium hydroxide were used, then hydrolysis of the carboxylic derivative to the acid would compete with enolate anion formation.
However, the problem is avoided by using the same base, for example., ethoxide, as is present in the ester hydrogen exchange in the α-position function, so that the ester is not hydrolysed.
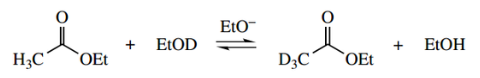
- Larger bases, for example., tert-butoxide, may also be valuable, in that they can remove α-protons but tend to be too large to add to the carbonyl group and form a tetrahedral intermediate.
Using ethoxide as a base, we can get hydrogen exchange by equilibration in a labelled solvent; but, because of the lower acidity of the α-protons compared with aldehydes and ketones, this process is less favourable.
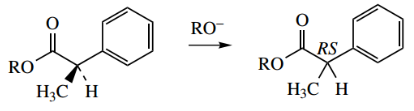
Racemization Of Hyoscyamine To Atropine
The base-catalysed racemization of the alkaloid (−)-hyoscyamine to (±)-hyoscyamine (atropine) is an example of enolate anion participation.
- Alkaloids are normally extracted from plants by using bases, thus liberating the free alkaloid bases from salt combinations.
- (−)-Hyoscyamine is found in belladonna (Atropa belladonna) and stramonium (Datura stramonium) and is used medicinally as an anticholinergic.
- It competes with acetylcholine for the muscarinic site of the parasympathetic nervous system, thus preventing the passage of nerve impulses.
- However, with careless extraction using too much base the product isolated is atropine, which has only half the biological activity of (−)-hyoscyamine, since the enantiomer (+)-hyoscyamine is essentially inactive.
- The racemization process involves the removal of the α-hydrogen to form the enolate anion, which is favoured by both the enolate anion resonance plus additional conjugation with the aromatic ring.
- Since the α protons in esters are not especially acidic, the additional conjugation is an important contributor to enolate anion formation.
The proton may then be restored from either side of the planar system, giving a racemic product.
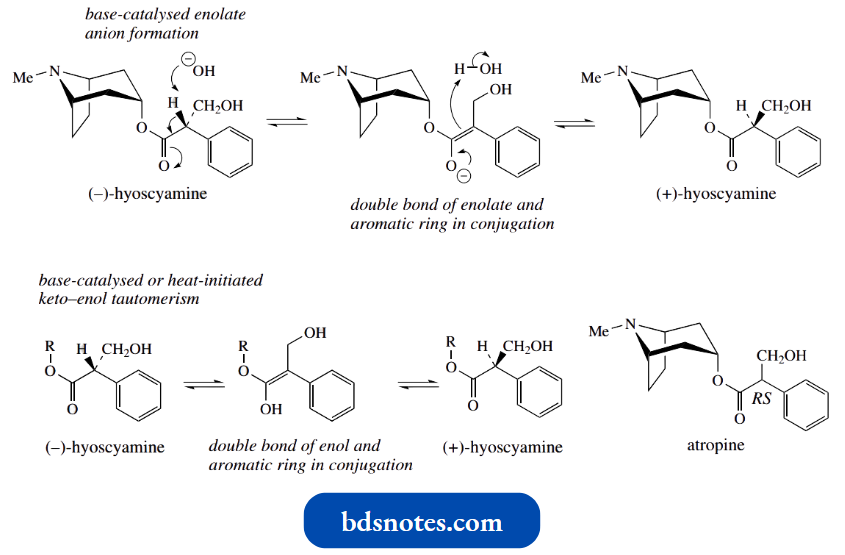
Note that the alcohol portion of hyoscyamine, namely tropine, also contains two chiral centres, but it is a symmetrical molecule and is optically inactive; it can be considered a mesostructure.
- Thus, the optical activity of hyoscyamine stems entirely from the chiral centre in the acid portion, tropic acid.
- Racemization of hyoscyamine may also be brought about by heating, and it is probable that, under these conditions, there is involvement of the enol form, rather than the enolate anion.
- The enol is also stabilized by the additional conjugation that the aromatic ring provides.
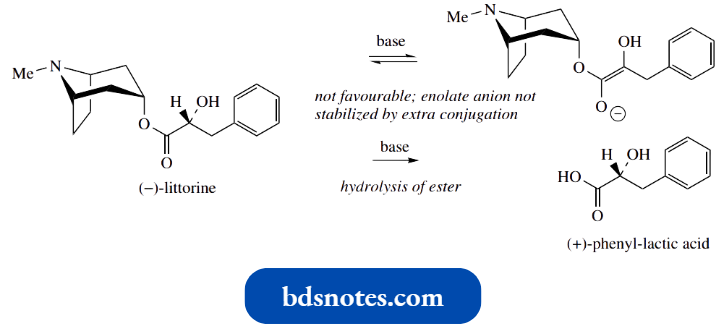
- The importance of this additional conjugation is emphasized by the observation that littorine, an alkaloid from Anthocercis littorea, is not readily racemized by either heat or base.
- The esterifying acid in littorine is phenyl-lactic, and the aromatic ring would not be in conjugation with the double bond of the enol or enolate anion.
Racemization depends entirely on the acidity associated with the isolated ester function.
“Early warning signs of outdated methods in enolate anion studies: Common questions”
Additionally, note that base hydrolysis of hyoscyamine gives (±)-tropic acid and tropine, with racemization preceding hydrolysis.
Base hydrolysis of littorine gives optically pure phenyl-lactic acid, so we deduce that hydrolysis is a more favourable process than racemization.
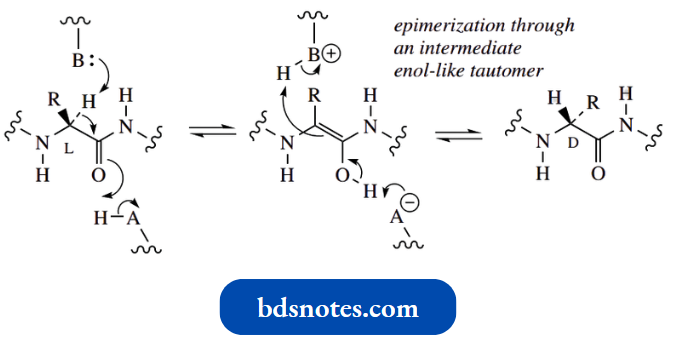
Epimerization Of L-Amino Acids To D-Amino Acids During Peptide Biosynthesis:
Many natural peptide structures, especially peptide antibiotics such as dactinomycin and ciclosporin, contain one or more D-amino acids along with L-amino acids in their structures.
- This contrasts with most proteins, where all the amino acid constituents are of the L-configuration.
- It is now known that the biosynthetic precursors of the D-amino acids are the corresponding L-analogues and that an enzymic epimerization process through an enol-type intermediate is involved.
- However, this does not appear to involve epimerization of the free L-amino acid followed by incorporation of the D-amino acid into the growing peptide chain. There are good reasons for this.
- Enolization in base does not occur, since ionization of the carboxylic acid group predominates. Enolization in acid is also prevented because the basic amino group would be protonated rather than the carbonyl.
- Epimerization appears to take place after the L-amino acid has been incorporated into the peptide and is thus occurring on an amide substrate.
- A simple example is the tripeptide precursor of the penicillin antibiotics, called ACV, an abbreviation for δ-(L-α-aminoacyl)-L-cysteinyl-D-valine.
The amino acid precursors for ACV are L-α-aminoadipic acid (an unusual amino acid derived by modification of L-lysine), L-cysteine, and L-valine (not D-valine).
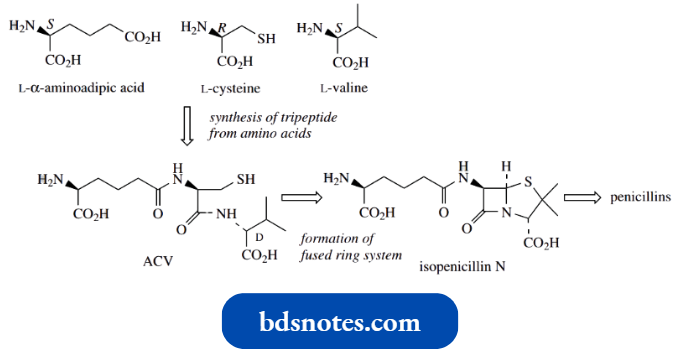
During ACV formation, the stereochemistry of the valine component is changed.
“Differential applications of traditional vs cutting-edge enolate techniques: Notes explained”
- ACV is the linear tripeptide that leads to isopenicillin N, the first intermediate with the fused ring system found in the penicillins.
- Note, that we are using the D and L convention for amino acid stereochemistry rather than the fully systematic R and S.
- This is one occasion where the use of D and L is advantageous, in that the sulfur atom in L-cysteine means this compound has the R configuration, whereas the other L-amino acids have the S configuration.
- Evidence points to the most likely explanation for the epimerization of L- to D-amino acids being the involvement of an enol-like intermediate.
- The carbonyl form is an amide in this example; but, from the comments made earlier, such a transformation could not be achieved chemically in solution, since the N–H proton would be more acidic and would, therefore, be preferentially removed using a base.
- However, this is an enzymic reaction, thus allowing selectivity determined by the functional groups at the enzyme’s binding site.
- A basic residue is responsible for removing the α-hydrogen to generate the enol-like structure, and then a reverse process allows it to be delivered back, though from the opposite side of the planar structure.
- Since this is an enzymic reaction, the product is also produced in just one configuration, rather than as an equimolar mixture of the two configurations typical of a chemical process.
Metabolic Racemization Of Ibuprofen:
The analgesic ibuprofen is supplied for drug use in its racemic form. However, only the (S)-(+)-enantiomer is the biologically active species; the (R)-(−)-form is inactive.
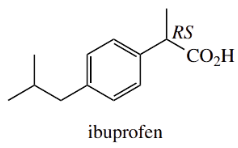
(S)-(+)- isomer active
(R)-(-)- isomer inactive
some metabolic conversion of R → S via racemization
Nevertheless, the racemate provides considerably more analgesic activity than expected, since in the body there is some metabolic conversion of the inactive (R)-isomer into the active (S)-isomer.
- This can be rationalized readily through an enolization mechanism. As we have indicated under D-amino acid formation above, a simple base-catalysed chemical conversion is ruled out by preferential ionization of the carboxylic acid group, though this may have little bearing on a metabolic process.
- An enzyme-mediated process may involve both basic and acidic amino acid side-chains (see D-amino acid formation above), and we could consider the biological transformation as either base-catalysed or acid-catalysed, as shown below.
- Either would generate a planar enediol intermediate, and the reverse process would account for racemization.
The enediol also benefits from favourable conjugation with the aromatic ring.
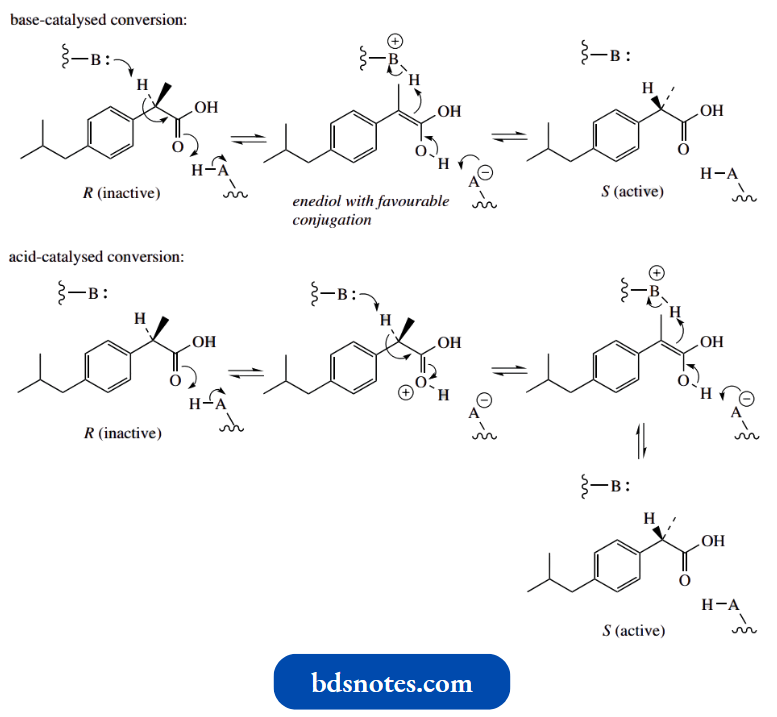
Thus, when (±)-ibuprofen is supplied to the body, the active (+)-isomer can be utilized, with the remaining (−)-isomer then being racemized to provide more of the active isomer.
- Theoretically, almost all of the (−)-isomer could be converted as the (+)-isomer is gradually removed by the body.
- For example, since the racemate contains 50% inactive isomer, racemization of this provides another 25% active isomer, then further racemization of the remaining 25% inactive would leave 12.5%, and so on.
- In practice, transport and excretion differences do not allow total usage of all the material.
- Alkylation of the α-position of suitable carboxylic acid derivatives may be achieved using the enolate anion as a nucleophile in a typical SN2 reaction.
In the example shown, the base used is LDA. This is a strong base that easily removes the weakly acidic α-proton, but because of its size it is a poor nucleophile and so does not affect the ester function.
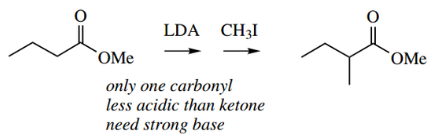
Nucleophilic addition of an enolate anion from a carboxylic acid derivative onto an aldehyde or ketone is simply an aldol-type reaction.
A simple example is shown; again, LDA is used to generate the enolate anion, and addition to the ketone is carried out as a second step.
Addition-To Carbonyl Of Aldehydes/Ketones
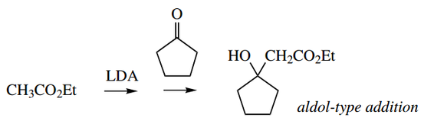
Acylation Of Enolate Anions: The Claisen Reaction
In the aldol reaction, we saw an enolate anion acting as a nucleophile leading to an addition reaction with aldehydes and ketones.
However, if there is a leaving group present, then instead of the intermediate alkoxide anion abstracting a proton from the solvent giving the aldol product, the leaving group may be expelled with the regeneration of the carbonyl group.
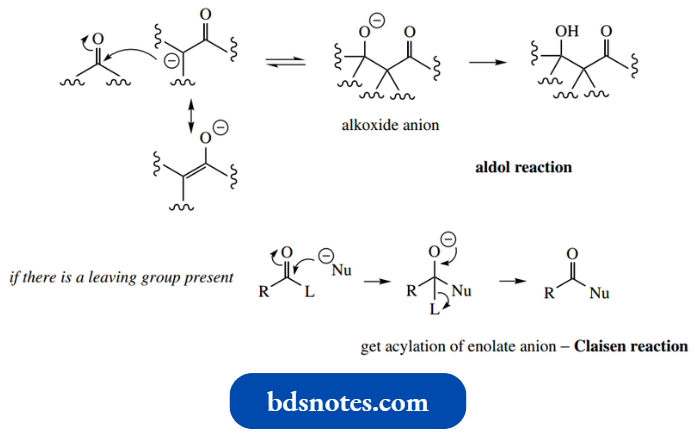
Now this is the same situation we encountered when we compared the reactivity of aldehydes and ketones with that of carboxylic acid derivatives.
- The net result here is acylation of the nucleophile, and in the case of acylation of enolate anions, the reaction is termed a Claisen reaction.
- It is important not to consider aldol and Claisen reactions separately, but to appreciate that the initial addition is the same, and differences in products merely result from the absence or presence get acylation of enolate anion − Claisen reaction of a leaving group.
- This is just how we ratio analyzed the different reactions of aldehydes and ketones compared with carboxylic acid derivatives.
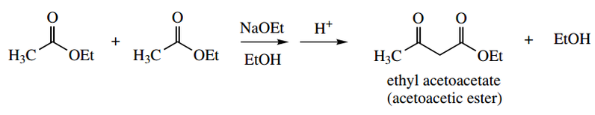
- The Claisen reaction (sometimes Claisen con densation) is formally the base-catalysed reaction between two molecules of ester to give a β-ketoester.
Thus, from two molecules of ethyl acetate, the product is ethyl acetoacetate.
To participate in this sort of reaction, the carboxylic acid derivative acting as a nucleophile must have α-hydrogens to generate an enolate anion.
- In practice, esters are most commonly employed in Claisen-type reactions.
- The Claisen reaction may be visualized as the initial formation of an enolate anion from one molecule of ester, followed by a nucleophilic attack of this species onto the carbonyl group of a second molecule.
The additional anion then loses ethoxide as the leaving group, with the reformation of the carbonyl group.
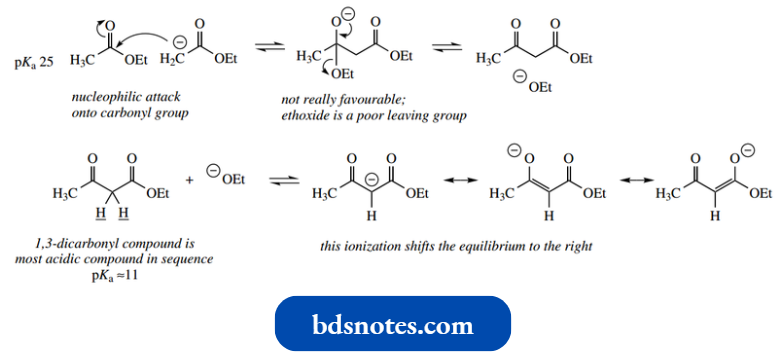
However, the reaction is not quite that simple, and to understand and utilize the Claisen reaction we have to consider pKa values again.
- Loss of ethoxide from the addition anion is not favourable, since ethoxide is not a particularly good leaving group.
- This is because ethoxide is a strong base, the conjugate base of a weak acid. So far then, the reaction will be reversible.
- What makes it proceed further is the fact that ethoxide is a strong base, and able to ionize acids.
- The ethyl acetoacetate product is a 1,3-dicarbonyl compound and has relatively acidic protons on the methylene between the two carbonyls.
- NaOEt a pKa of about 11 makes ethyl acetoacetate the most acidic compound in the sequence.
- Ionization of ethyl acetoacetate, generating a resonance-stabilized enolate anion, removes the product from the reaction mixture and shifts the equilibrium to the right.
- This also explains why, in the simple equation above, two reagents are shown on the arrows, first base and then acid.
- The acid is required in the workup to liberate the β-ketoester from the enolate anion.
The importance of ionization of the β-ketoester product can be illustrated by the attempted Claisen reaction between two molecules of ethyl 2-methyl propionate.
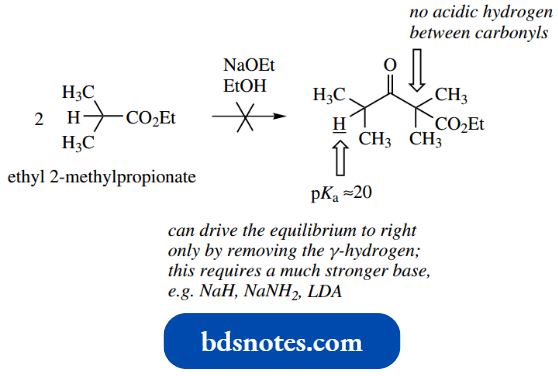
“Can computational chemistry revolutionize enolate anion mechanisms? FAQs provided”
Using sodium ethoxide as a base, the reaction does not proceed. This can be ascribed to the nature of the β-ketoester product, which contains no protons sandwiched between two carbonyls and, therefore, no protons that are sufficiently acidic for the final equilibrium-disturbing step.
- The reaction can be made to proceed, however, and the solution is simple: use a stronger base.
- In this way, the base used is sufficiently powerful to remove a less acidic proton from the product, removing it from the reaction mixture and disturbing the equilibrium.
- Any of the strong bases sodium hydride, sodium amide, or LDA might be employed. Although such bases will produce the enolate anion irreversibly, it is still necessary to ionize the product to overcome the effect of the poor leaving group.
- In the β-ketoester product, the pKa of the only acidic proton is about 20, so this requires a strong base to achieve an equilibrium disturbing ionization.
Claisen And Aldol Reactions In Nature: Hmg-Coa And Mevalonic Acid:
In nature, the biologically active form of acetic acid is acetyl-coenzyme A (acetyl-CoA).
- Two molecules of acetyl-CoA may combine in a Claisen-type reaction to produce acetoacetyl-CoA, the biochemical equivalent of ethyl acetoacetate.
- This reaction features as the start of the sequence to mevalonic acid (MVA), the precursor in animals of the sterol cholesterol.
Later, we shall see another variant of this reaction that employs malonyl-CoA as the nucleophile.
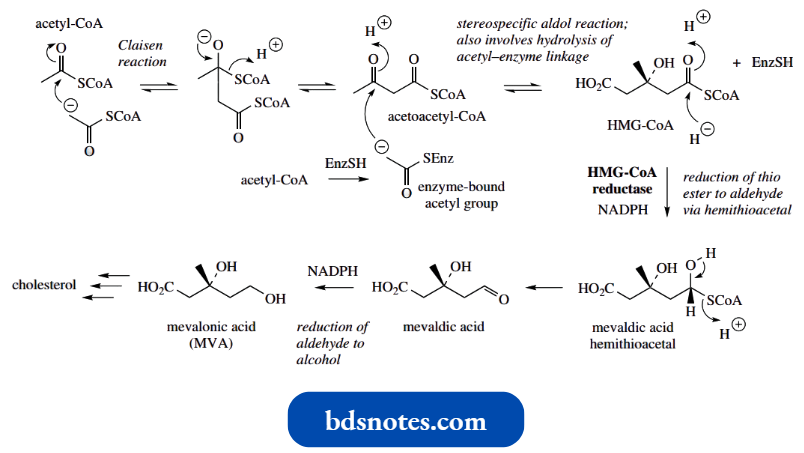
Three molecules of acetyl-CoA are used to form MVA, a third molecule being incorporated via a stereospecific aldol addition to give the branched-chain ester β-hydroxy-β-methylglutaryl-CoA (HMG-CoA).
- This third acetyl CoA molecule appears to be bound to the enzyme via a thiol group, and this linkage is subsequently hydrolysed to form the free acid group of HMG-CoA.
- It should be noted that, on purely chemical grounds, acetoacetyl-CoA is the more acidic substrate in this reaction, and might be expected to act as the nucleophile rather than the third acetyl-CoA molecule.
- The enzyme thus achieves what is a less favourable reaction. There is a rather similar reaction in the Krebs cycle, where acetyl-CoA adds on to oxaloacetate via an aldol reaction, again with the enzymic reaction employing the less acidic substrate as the nucleophile.
- The subsequent conversion of HMG-CoA into MVA involves a two-step reduction of the thioester group a primary alcohol and provides an essentially irreversible and rate-limiting transformation.
- Drug-mediated inhibition of this enzyme, HMG-CoA reductase (HMGR), can be used to regulate the biosynthesis of the steroid cholesterol.
High levels of blood cholesterol are known to contribute to the incidence of coronary heart disease and heart attacks.
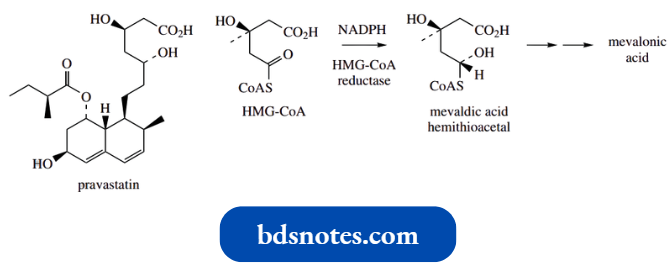
The statins, for example., pravastatin, are a group of HMGR inhibitors that possess functionalities that mimic the half-reduced substrate evaluate hemithioacetal.
- The affinity of these agents towards HMG-CoA reductase is some 104-fold more than the natural substrate, making them extremely effective inhibitors of the enzyme, and powerful drugs in coronary care.
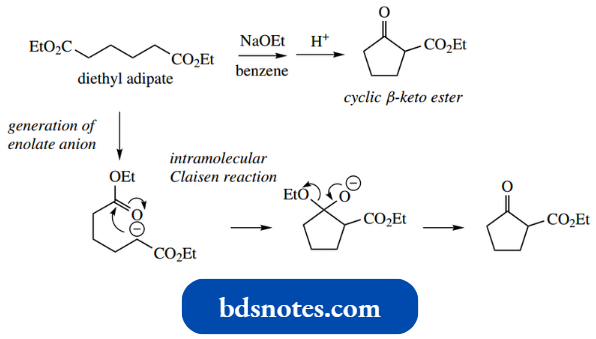
- Should there be two ester functions in the same molecule, then it is possible to achieve an intramolecular Claisen reaction, particularly if this results in a favourable five- or six-membered ring.
- This reaction is usually given a separate name, a Dieckmann reaction, but should be thought of as merely an intramolecular extension of the Claisen reaction.
- As we have seen previously, intramolecular reactions are favoured over intermolecular reactions when the reaction is carried out at high dilution, conditions that minimize the interaction of two separate molecules.
A simple example involving the transformation of diethyl adi pate into a cyclic β-ketoester is shown.
We saw the possibilities for a mixed aldol reaction above, in which the reaction could become useful if we restricted the number of couplings possible.
- The same considerations can be mixed with the Claisen reaction applied to the Claisen reaction.
Thus, it is possible to have four products from two esters, depending on which ester became the nucleophile and which was acting as the electrophile.
Mixed Claisen Reaction
⇒ \(\mathrm{RCH}_2 \mathrm{CO}_2 \mathrm{Et}+\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{Et} \longrightarrow 4 \text { products }\)
Nucleophile And Electrophile
⇒ \(\begin{aligned}\mathrm{RCH}_2 \mathrm{CO}_2 \mathrm{Et}+\mathrm{RCH}_2 \mathrm{CO}_2 \mathrm{Et}\)
⇒ \(\mathrm{RCH}_2 \mathrm{CO}_2 \mathrm{Et}+\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{Et}\)
⇒ \(\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{Et}+\mathrm{RCH}_2 \mathrm{CO}_2 \mathrm{Et}\)
⇒ \(\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{Et}+\mathrm{R}^{\prime} \mathrm{CH}_2 \mathrm{CO}_2 \mathrm{Et}\)
To be synthetically useful, a mixed Claisen reaction (crossed Claisen reaction) needs one ester with no α-hydrogens so that it cannot become the nucleophile. Such reactants include oxalate, formate and benzoate esters. An example is shown below.
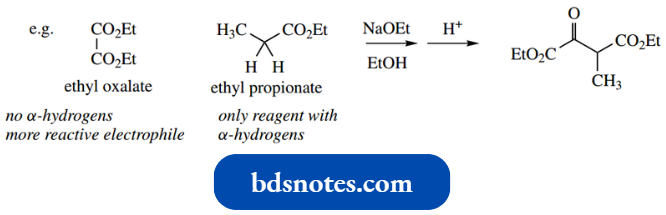
However, one might expect that the product from two molecules of ethyl propionate could also be formed.
- In practice, ethyl oxalate, because of its second electron-withdrawing carboxylate group, is a more reactive electrophile, so the major product is as shown.
- Formates are also more susceptible to nucleophilic attack; they lack the electron-donating inductive effect of an alkyl group and provide no steric hindrance.
- Benzoates are not as reactive as formates and oxalates, but the phenyl ring is electron-withdrawing and they also lack α-hydrogens.
- To minimize self-condensation of the nucleophilic reagent, it helps to add this gradually to the electrophilic species, so that the latter is always present in excess.
- Alternatively, and much more satisfactory from a synthetic point of view, it is possible to carry out a two-stage process, forming the enolate anion first.
We also saw this approach with a mixed aldol reaction. Thus, ethyl acetate could be converted into its enolate anion by reaction with the strong base LDA in a reaction that is essentially irreversible.
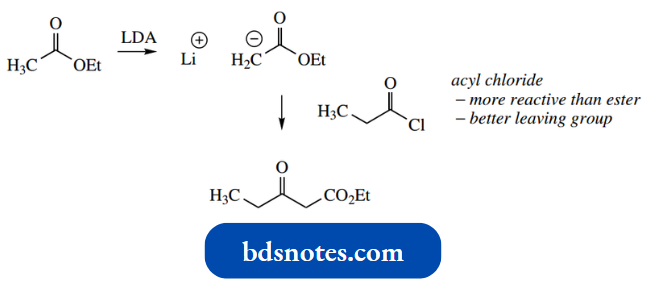
This nucleophile can then be treated with the electrophile. This could be a second ester, but there is an even better idea.
- If one is going to use a two-stage process, one can now employ an electrophile with a better-leaving group than ethoxide, and also get over the final ionization problem.
It would not be possible to use an acyl halide in a one-pot reaction because it would be quickly attacked by a base. An acyl halide could be used in a two-stage reaction, as shown here.
Ester–Ketone Condensations: Predicting The Product:
Let us use a systematic approach to consider what product is most likely to result when a mixture of an ester and a ketone, both capable of forming enolate anions, is treated with base.
For example, consider an ethyl acetate–acetone mixture treated with sodium hydride in ether solution.
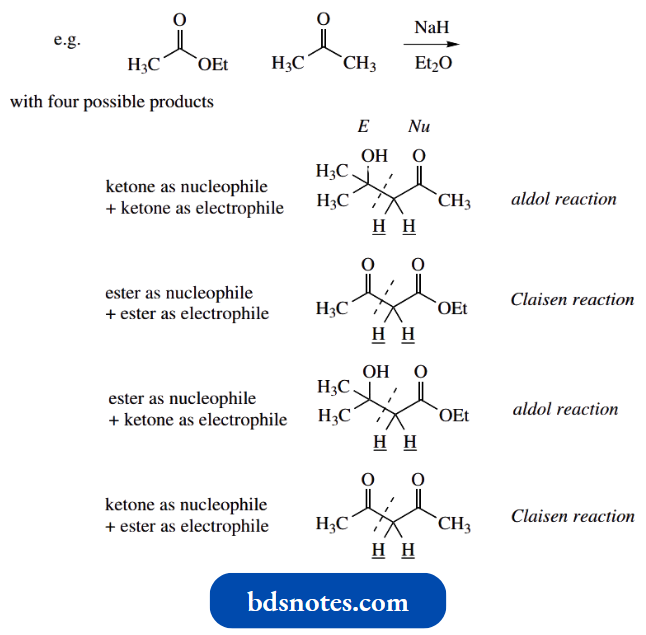
Four reactions and products can be considered, involving either ketone or ester as the nucleophile, with ketone as the electrophile (aldol reactions) or ester as the electrophile (Claisen reactions).
- Both aldol and Claisen reactions are equilibria, and product formation is a result of disturbing these equilibria. This would be dehydration in Aldol reactions and ionization in Claisen reactions.
- Ionization would be the more immediate determinant. On that basis, it is obvious that the 1,3-dicarbonyl products from Claisen reactions are going to be more acidic than the aldol products, which possess just one carbonyl group.
- Now let us look at the ease of forming the enolate anion nucleophiles. Ketones are more acidic than esters.
Taken together, these factors mean the more favoured product is going to be the β-diketone (acetylacetone), formed from a ketone nucleophile by a Claisen reaction with an ester. This is the reaction observed.
The product is β-diketone acetylacetone
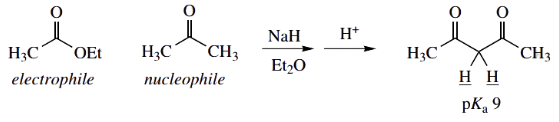
- ketone is more acidic than ester − ketone enolate favoured
- β-diketone is the most acidic of the four possible products
Aldol And Claisen Reactions In The Biosynthesis Of Phenols:
Many natural aromatic compounds are produced from the cyclization of poly-β-keto chains by enzymic aldol and Claisen reactions.
Examples include simple structures like orsellinic acid and phloracetophenone, and more complex highly modified structures of medicinal interest, such as mycophenolic acid, used as an immunosuppressant drug, the antifungal agent griseofulvin, and antibiotics of the tetracycline group, for example., tetracycline itself.
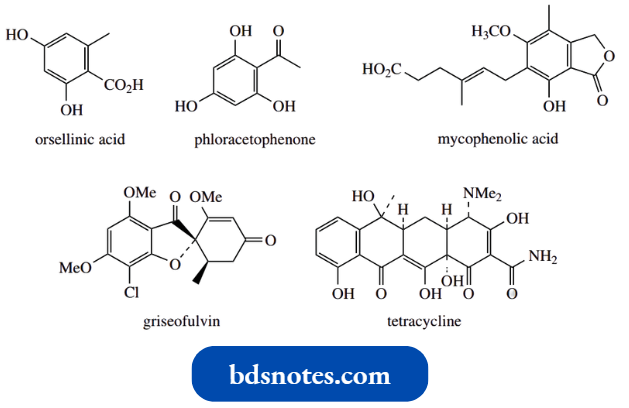
The more complex structures are inappropriate for consideration here, but the two compounds orsellinic acid and phloracetophenone exemplify nicely the enolate anion mechanisms we have been considering, as well as the concept of keto-enol tautomerism.
- A multifunctional enzyme complex is responsible for producing a poly-β-keto chain via a sequence of several Claisen reactions, together with subsequent reactions that achieve cyclization and aromatization.
- The C8 poly-β-keto chain shown is bonded to the enzyme through a thioester linkage.
- Because of the number of functional groups in this molecule, it is very reactive, and the enzyme plays a significant role in stabilizing it and preventing any unwanted chemical reactions.
- In addition, the enzyme binds the substrate in a folded conformation, allowing the atoms to be held in positions approximating to those occupied in the desired product.
- There are various possibilities for undergoing intramolecular aldol or Claisen reactions, dictated by the nature of the enzyme and how the substrate is folded on the enzyme surface.
Methylenes flanked by two carbonyl groups are the more acidic, allowing the formation of enolate anions.
- These may then participate in intramolecular reactions with ketone or ester carbonyl groups, with a natural tendency to form strain-free six-membered rings.
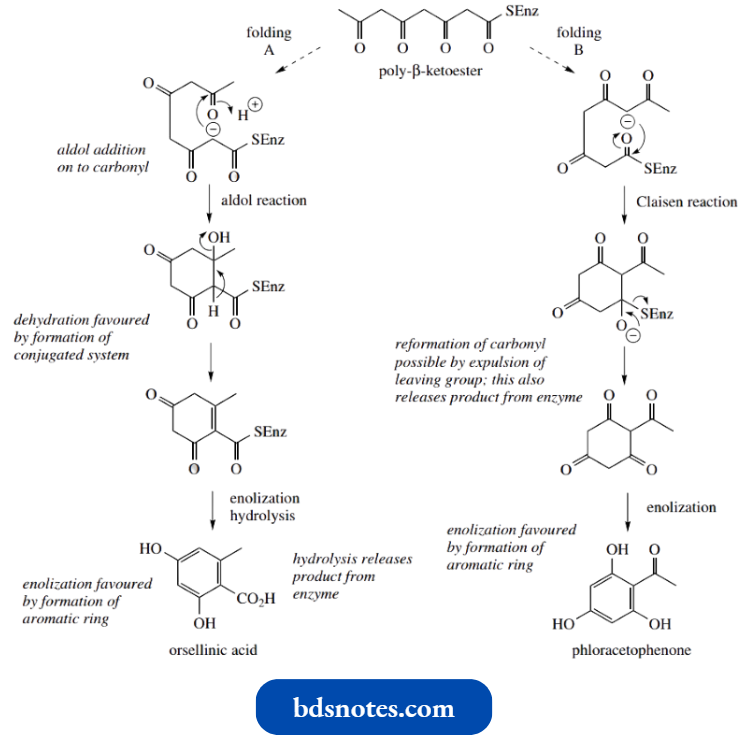
- To produce the compounds orsellinic acid and phloracetophenone, we can envisage the same substrate being folded in two different ways. Which folding occurs will be dependent on the organism and the enzyme it contains.
- With folding A, ionization of the α-methylene allows aldol addition onto the carbonyl six carbons distant along the chain, giving the tertiary alcohol.
- Dehydration occurs as in most chemical aldol reactions, giving the conjugated system, and enolization follows to attain the stability conferred by the aromatic ring.
- The thioester bond is then hydrolysed to produce orsellinic acid, at the same time releasing the product from the enzyme.
Alternatively, folding B allows a Claisen reaction to occur, which, although mechanistically analogous to the aldol reaction, is terminated by expulsion of the leaving group and direct release from the enzyme. Enolization of the cyclohexanone produces phloracetophenone.
“Asymptomatic vs symptomatic effects of ignoring new trends in enolate anion research: Answered”
Essentially the same sort of enolate anion aldol and Claisen reactions occur in the production of the more complex structures mycophenolic acid, griseofulvin, and tetracycline. However, the final structure is only obtained after a series of further modifications.
Reverse Claisen Reactions:
The driving force for the Claisen reaction is the formation of the enolate anion of the β-ketoester product. If this cannot form, the reverse reaction controls the equilibrium.
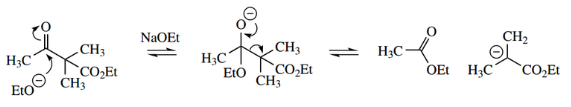
This means that a reverse Claisen reaction can occur if a β-ketoester is treated with base. This is most likely to occur if we attempt to hydrolyse the β-ketoester to give a β-ketoacid using aqueous base.
- Note that the alcoholic base used for the Claisen reaction does not affect the ester group, since the nucleophile is the same as the leaving group.
Aqueous base treatment of a β-ketoester will, however, result in both ester hydrolysis and a reverse Claisen reaction, and poses a problem if one only wants to hydrolyse the ester.
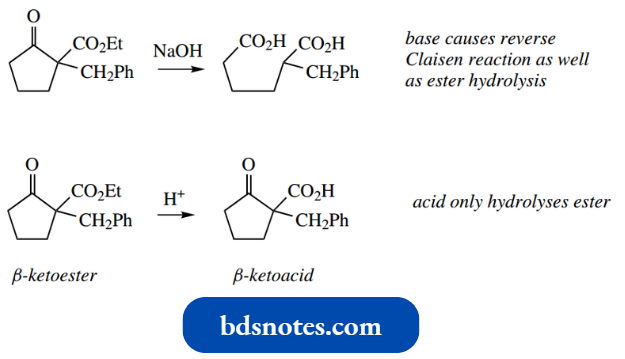
The reverse Claisen reaction is common, especially with cyclic β-ketoesters, such as one gets from the Dieckmann reaction.
- If one only wants to hydrolyse the ester, it thus becomes necessary to use the rather less effective acid-catalysed hydrolysis method.
- Cleavage of β-diketones, the products of a mixed Claisen reaction between an ester electrophile and a ketone nucleophile, behave similarly towards the base, and a reverse Claisen reaction ensues.
Again, this is prevalent with cyclic systems.
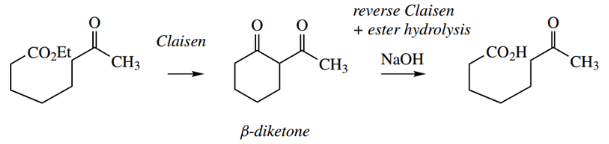
b-diketone
Nevertheless, as we shall, it is also possible to exploit the reverse Claisen reaction to achieve useful transformations.
Decarboxylation Reactions:
- Hydrolysis of the ester function of the β-ketoester Claisen product under acidic conditions yields a β-ketoacid, but these compounds are especially susceptible to loss of carbon dioxide, i.e. decar boxylation.
- Although β-ketoacids may be quite stable, decarboxylation occurs readily on mild heating and is ascribed to the formation of a six-membered hydrogen-bonded transition state.
- Decarboxylation is represented as a cyclic flow of electrons, leading to an enol product that rapidly reverts to the more favourable keto tautomer.
Reverse Claisen reaction in biochemistry: β-oxidation of fatty acids:
Perhaps the most important example of the reverse Claisen reaction in biochemistry is that involved in the β-oxidation of fatty acids, used to optimize energy release from storage fats, or fats ingested as food.
In common with most biochemical sequences, thioesters rather than oxygen esters are utilized.
β-oxidation Of Fatty Acids
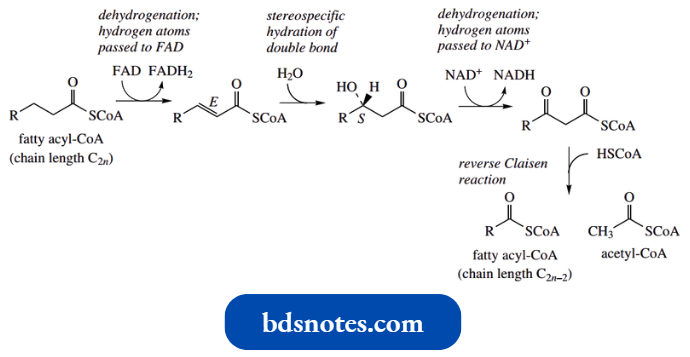
The β-oxidation sequence involves three reactions, dehydrogenation, hydration, and then oxidation of a secondary alcohol to a ketone, thus generating a β-keto thioester from a thioester.
We shall study these reactions in more detail later. The β-keto thioester then suffers a reverse Claisen reaction, initiated by nucleophilic attack of the thiol coenzyme A.
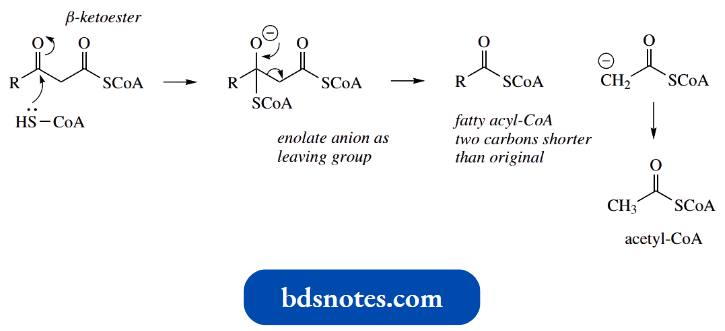
The leaving group is the enolate anion of acetyl-CoA, and the reaction thus cleaves off a two-carbon fragment from the original fatty acyl-CoA.
- Since the nucleophile is coenzyme A, the other product is also a coenzyme A ester.
- The reaction generates a new fatty acyl-CoA, shorter by two carbons, which can re-enter the β-oxidation cycle.
Most natural fatty acids have an even number of carbons, so the process continues until the original fatty acid chain is cleaved completely into acetyl-CoA fragments.
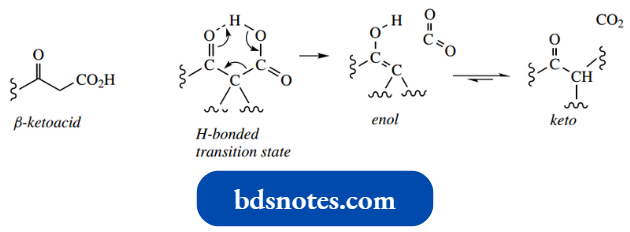
We can now see how a number of the reactions recently studied fit together.
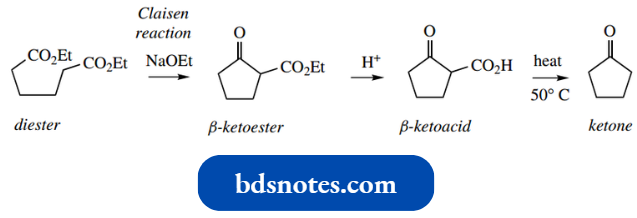
Decarboxylation Of Β-Ketoacids In Biochemistry: Isocitrate Dehydrogenase:
The enzyme isocitrate dehydrogenase is one of the enzymes of the Krebs or citric acid cycle, a major feature in carbohydrate metabolism.
- This enzyme has two functions, the major one being the dehydrogenation (oxidation) of the secondary alcohol group in isocitric acid to a ketone, forming oxalosuccinic acid.
This requires the cofactor NAD+. For convenience, we are showing non-ionized acids here, for example., isocitric acid, rather than anions, for example., isocitrate.
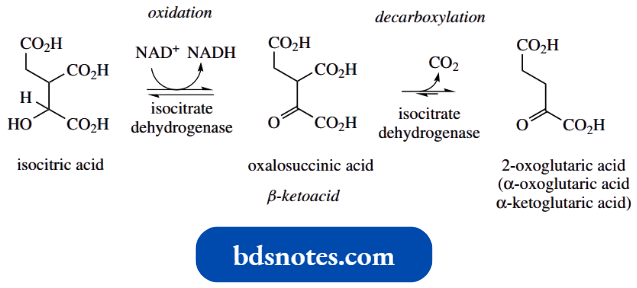
The second function, and the one pertinent to this section, is the decarboxylation of oxalosuccinic acid to 2-oxoglutaric acid.
- This is simply a biochemical example of the ready decarboxylation of a β-ketoacid, involving an intramolecular hydrogen-bonded system.
This reaction could occur chemically without an enzyme, but it is known that isocitric acid, the product of the dehydrogenation, is still bound to the enzyme isocitrate dehydrogenase when decarboxylation occurs.
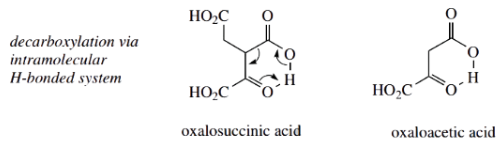
It is appropriate here to look at the structure of oxaloacetic acid, a critical intermediate in the Krebs cycle, and to discover that it too is a β-ketoacid.
- In contrast to oxalosuccinic acid, it does not suffer decarboxylation in this enzyme-mediated cycle but is used as the electrophile for an aldol reaction with acetyl-CoA.
- Decarboxylation of 1,1-diacids (gem-diacids) is a similar reaction involving a hydrogen-bonded transition state.
1,1-Diacids may be stable entities, for example., malonic acid, but they are susceptible to decarboxy location upon heating; malonic acid decarboxylates at 150 ◦C.
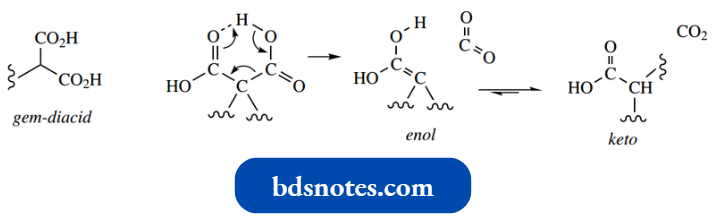
gem-Diacids are typical products that might be obtained from synthetic sequences using esters of malonic acid, for example., diethyl malonate, a 1,3-dicarbonyl compound.
- Since the methylene group in diethyl malonate is sandwiched between two carbonyls, the enol keto protons are considerably more acidic than those in ethyl acetate.
The pKa is of the order of 13, compared with about 24 for ethyl acetate, so it becomes much easier to form the enolate anion.
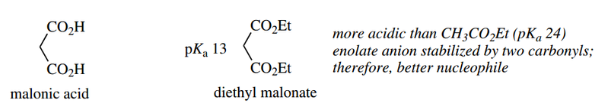
These decarboxylation reactions must not be viewed as unwanted processes that complicate reactions, but reactions that can be put to very good use. There were hints in the last paragraph.
- Two carbonyl groups in a 1,3 relationship increase the acidity of the α-protons between the two groups compared with protons adjacent to just one carbonyl group. It is easier to form enolate anions and then carry out nucleophilic reactions.
Therefore, since we may subsequently remove an ester function by hydrolysis and decarboxylation, we can view an ester group as a useful and temporary activating group. This is exemplified by the two sequences below.
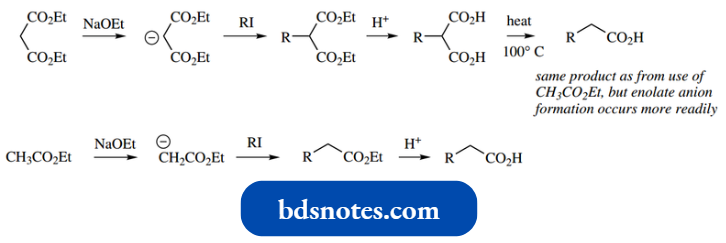
Diethyl malonate can be converted into its enolate anion, which may then be used to participate in an SN2 reaction with an alkyl halide.
- Ester hydrolysis and mild heating leads to production of an alkylated acetic acid.
- The same product might be obtained by starting with ethyl acetate, but this would be less efficient and possibly require a stronger base because the lower acidity of the α-protons makes the generation of the enolate anion less effective.
One of the ester groups in diethyl malonate can thus be regarded as a temporary activating group to increase the acidity of the α-protons.

The same viewpoint can taken for the ester function in a β-ketoester such as ethyl acetoacetate.
- Again, the acidity of the α-protons is increased because there are two carbonyl groups, and the generation of an enolate anion is facilitated.
Although mono- or di-alkylation of a ketone might be achieved through enolate anions, it would be easier to use the more acidic β-ketoester and follow this with hydrolysis and decarboxylation.
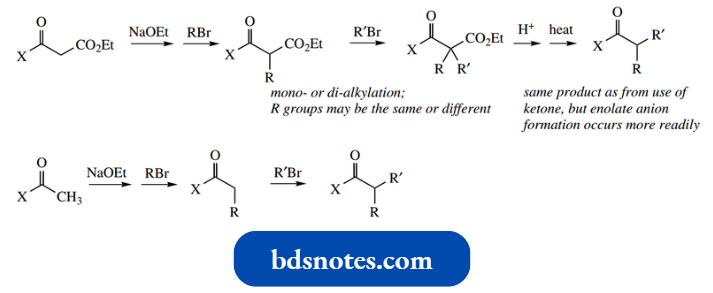
In general terms, a β-ketoester like ethyl acetoacetate can be considered as a pathway to substituted ketones, and diethyl malonate is a source of substituted acids.
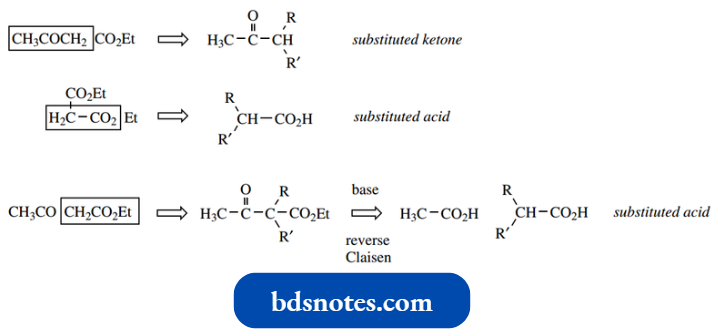
“Success rate of interventions using modern enolate anion techniques: FAQ”
Note also that we can even make good use of the reverse Claisen reaction. Thus, alkylation of ethyl acetoacetate followed by suitable base treatment to effect a reverse Claisen reaction would also generate a substituted acid.
- An alcoholic base would be used for the enolate anion chemistry, whereas an aqueous base would initiate the reverse Claisen reaction and ester hydrolysis.
- In this sequence, we are using the acyl group as a temporary activating group.
- Back we saw two methods of synthesizing 2-methylcyclohexanone, i.e. by direct alkylation of the enolate anion derived from cyclohexanone and by using an enamine derivative as the nucleophilic species.
- The latter route had the advantage of not using a strong base to generate the nucleophile.
- We can now add a further approach for synthesis of the same compound, via a β-ketoester.
This also has the advantage of proceeding smoothly and, although it does use base to generate the enolate anion, the base required would be considerably less strong than for the ketone route.
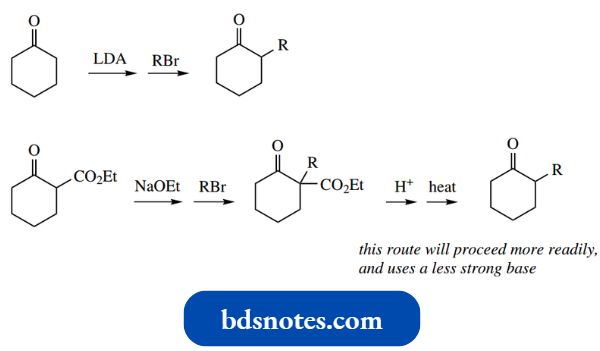
On several occasions, we have noted that reactions involving enolate anions could be improved significantly by utilizing strongly basic reagents, such as sodium hydride, sodium amide, or LDA, and carrying out the reaction in two stages.
This stratagem removed the constrictions imposed by unfavourable equilibria, by preparing the enolate anion in an essentially irreversible reaction, then adding the electrophile that could have a more reactive leav ing group.
This is further exemplified by the synthesis of a β-diketone from a β-ketoester, as shown below, again exploiting a decarboxylation reaction.
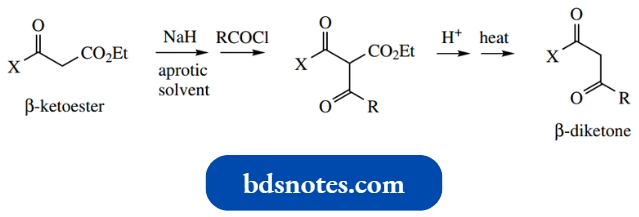
Claisen Reactions In Nature Involving Malonyl-CoA:
We saw that nature employs a Claisen reaction between two molecules of acetyl-CoA to form acetoacetyl-CoA as the first step in the biosynthesis of mevalonic acid and subsequently cholesterol.
- This was a direct analogy for the Claisen reaction between two molecules of ethyl acetate.
- In fact, in nature, the formation of acetoacetyl-CoA by this particular reaction using the enolate anion from acetyl-CoA is pretty rare.
- We have just seen that diethyl malonate can be used instead of ethyl acetate as a nucleophile.
- The second ester group is effectively used to activate the system for producing a nucleophile and then is removed when the required reaction has been achieved.
Would it surprise you to know that, concerning this strategy, nature got there first?
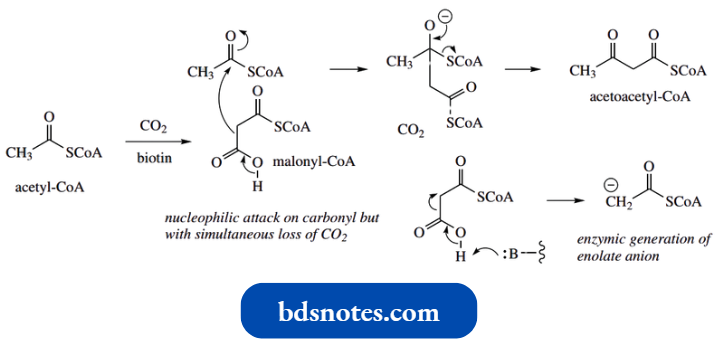
“Is enolate-related risk reversible if addressed promptly? Answer provided”
The nucleophile in biological Claisen reactions that effectively adds on acetyl-CoA is almost always malonyl CoA.
- This is synthesized from acetyl-CoA by a reaction that utilizes a biotin-cyt–enzyme complex to incorporate carbon dioxide into the molecule.
- This has now flanked the α-protons with two carbonyl groups and increases their acidity. The enzymic Claisen reaction now proceeds, but, during the reaction, the added carboxyl is lost as carbon dioxide.
- Having done its job, it is immediately removed. In contrast to the chemical analogy, a carboxylated intermediate is not formed.
- Mechanistically, one could perhaps write a concerted decarboxylase tion–nucleophilic attack, as shown.
- An alternative rationalization is that decarboxylation of the malonyl ester is used by the enzyme to effectively generate the acetyl enolate anion without the requirement for a strong base.
- Malonyl-CoA is used as the nucleophilic species in the biosynthesis of fatty acids and a whole host of other natural products, including aromatic compounds.
Nucleophilic Addition To Conjugated Systems: Conjugate Addition And Michael Reactions:
We are familiar with the concept that the reactivity of a carbonyl group can be ascribed to the difference in electronegativity between carbon and oxygen, and the resultant unequal sharing of electrons.
The polarization δ+/δ− can be considered as a contribution from the resonance form having full charge separation.

Now let us go a step further, and conjugate the carbonyl group with a double bond.
If we polarize the carbonyl as before, then conjugation allows another resonance form to be written, in which the β-carbon now carries a positive charge.
Thus, as well as the carbonyl carbon being electrophilic, the β-carbon is also an electrophilic centre.
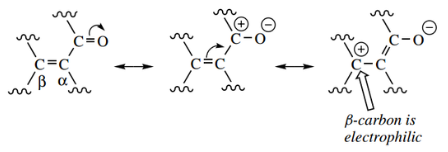
Conjugation of the carbonyl with a double bond transfers the electronic characteristics δ+/δ− of the carbonyl group along the carbon chain.
- The alkene would normally be nucleophilic and react with electrophiles. When conjugated with a carbonyl, it now becomes electrophilic and reacts with nucleophiles.
- A typical nucleophilic attack on the β-position is now shown, resulting in a transfer of negative charge onto the carbonyl. The product is a resonance form of an enolate anion with charge on the oxygen.
Abstraction of a proton from solvent will thus ultimately result in the production of the more favourable keto tautomer, and restoration of the carbonyl group.
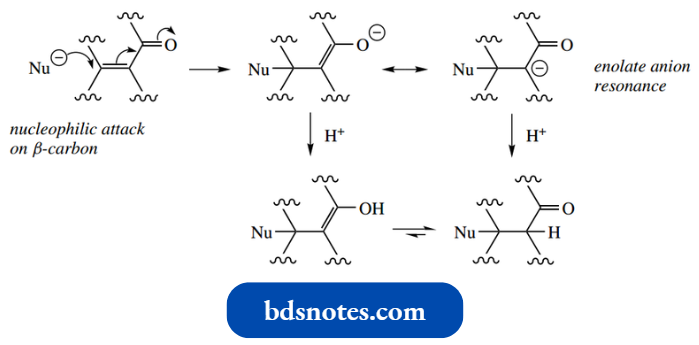
It is possible to get either the typical addition reaction onto the carbonyl group, termed a 1,2- addition, or this form of conjugate addition, termed
1,4-addition, terminology that is understandable if the enol tautomer is considered as the product formed first.
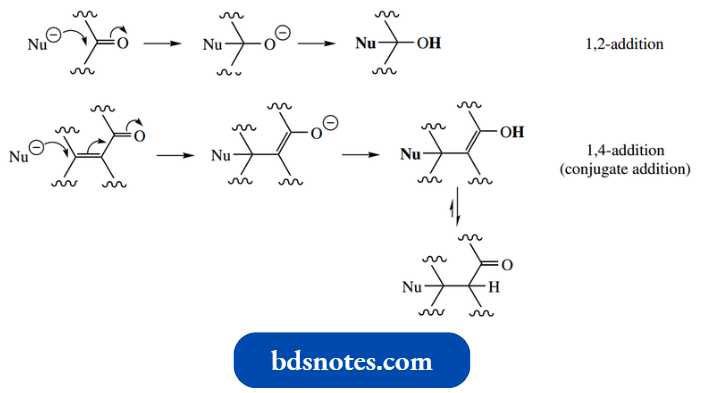
“Treatment scope in academia vs industry for enolate anion applications: FAQs”
The less-reactive sodium borohydride may reduce unsaturated aldehydes by 1,2-addition, whereas unsaturated ketones tend to undergo conjugate addition. This allows selective reduction processes to be exploited.
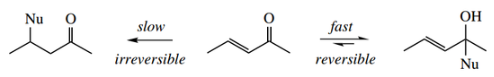
For example, in the unsaturated ketone shown, we may achieve a reduction of the carbonyl using LAH, a reduction of the double bond via catalytic hydrogenation, or conjugate reduction using sodium borohydride.
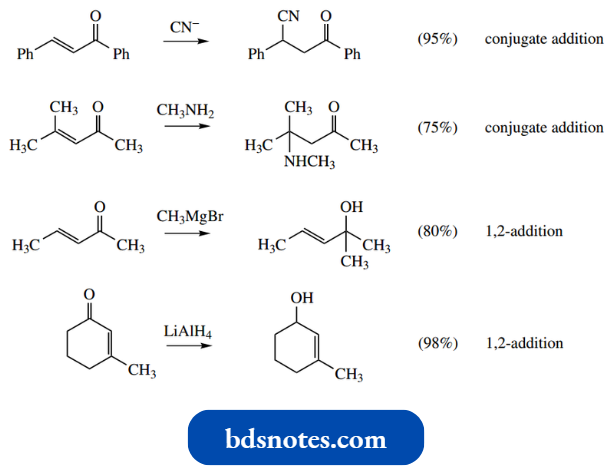
The conjugate addition of a thiol, methanethiol, to the α, β-unsaturated aldehyde acrolein may be used in the synthesis of the amino acid methionine.
- Under basic conditions, the nucleophile will be the thiolate anion, and 1,4-addition leads to this aldehyde.
- Methionine may then be obtained via the Strecker synthesis, a sequence that involves imine formation, then nucleophilic attack of cyanide on this imine/carbonyl analogue.
The reaction is completed by acidic hydrolysis of the nitrile function to a carboxylic acid.
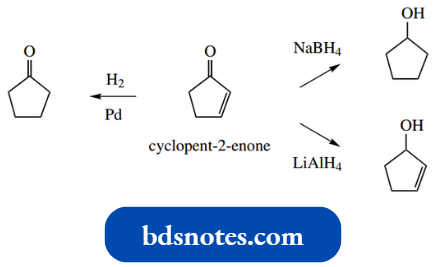
It should also be noted, as we have seen earlier, that other electron-withdrawing groups, for example., esters and nitriles, can achieve the same end as aldehydes or ketones.
“Cost of ignoring enolate anion principles vs benefits of systematic approaches: Q&A”
Conjugate addition can be observed when such groups are conjugated with a double bond.
Flavonoids: Conjugate addition And Heterocyclic Ring Formation:
Flavonoids are natural plant phenols containing a six-membered oxygen heterocyclic ring.
- Considerable quantities of flavonoids are consumed daily in our vegetable diet, and there is growing belief that they have beneficial properties, acting as antioxidants and giving protection against cardiovascular disease, and perhaps cancer.
- Their polyphenolic nature enables them to scavenge injurious free radicals, such as superoxide and hydroxyl radicals, which can cause serious cell damage.
- In particular, flavonoids in red wine and tea have been demonstrated to be effective antioxidants.
One of the simplest natural flavonoids is flavanone liquiritigenin, a material that contributes to the bright yellow colour of liquorice root.
Liquiritigenin may be synthesized readily, as shown, by a two-stage process starting from the phenolic ketone and aldehyde.
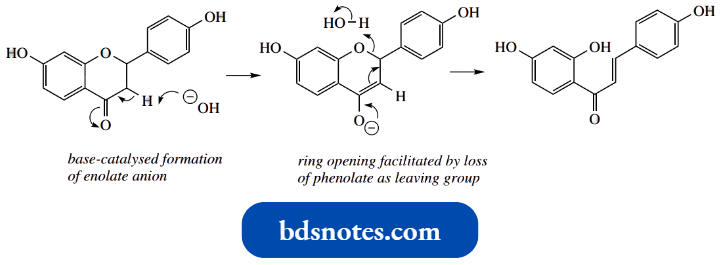
The conjugate addition of enolate anions onto α, β-unsaturated systems is an important synthetic reaction and is termed the Michael reaction, though this terminology may often be used in the broader context for the other conjugate additions considered above.
A typical example of the Michael reaction is the base-catalysed reaction of ethyl acetoacetate with the α, β-unsaturated ester ethyl acrylate.
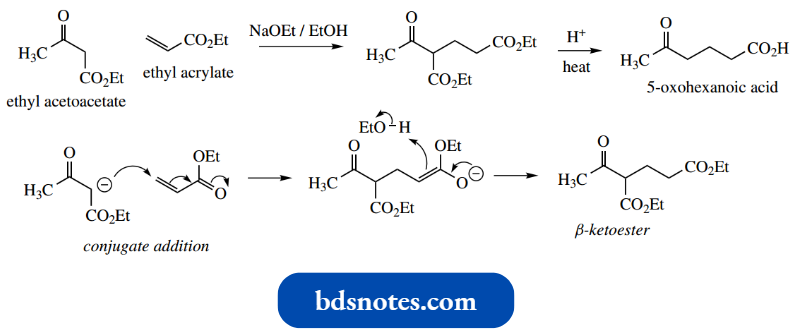
“Why are enolate anion mechanisms often misunderstood in practice? Questions answered”
The nucleophile will be the enolate anion from ethyl acetoacetate, which attacks the β-carbon of the electrophile, generating an addition complex that then acquires a proton at the α-position with restoration of the carbonyl group.
- The product is an δ-ketoester with an ester side-chain that has a β-relationship to the keto group.
- This group may thus be removed by a sequence of acid-catalysed hydrolysis, followed by thermal decarboxylation. The final product in this sequence is therefore a δ-ketoacid, i.e. a 1,5-dicarbonyl compound.
Other examples of the Michael reaction are shown below. Note the relatively mild bases that are employed in these reactions where the nucleophiles are 1,3-dicarbonyl compounds.
Michael’s Reaction: The Robinson Annulation
A rather nice example of enolate anion chemistry involving the Michael reaction and the aldol reaction is provided by the Robinson annulation, a ring-forming sequence used in the synthesis of steroidal systems (Latin: annulus, ring).
In the partial synthesis shown, there are two reagents, the α, β-unsaturated ketone methyl vinyl ketone and the 1,3-diketone 2-methylcyclohexa-1,3-dione.
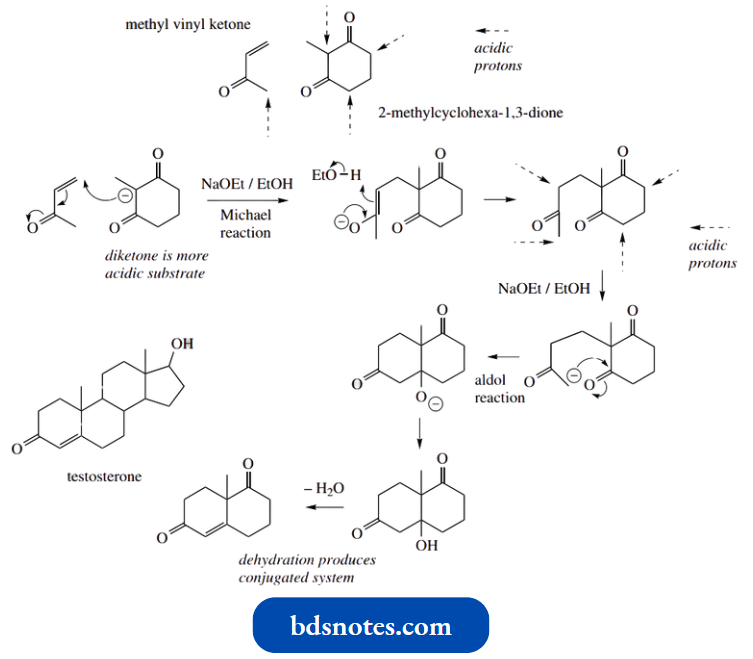
“Most common complications of poorly understood enolate anion concepts: FAQs”
These are reacted together in a basic solution. It can be deduced that the 1,3-diketone is more acidic than the monotone substrate, so will be ionized by the removal of a proton from the carbon between the two carbonyls to give the enolate anion as a nucleophile.
- This attacks the α, β-unsaturated ketone in a Michael reaction. Understandably, this large nucleophile prefers to attack the unhindered β-position rather than the more congested ketone carbonyl.
- The product from the Michael reaction will be a triketone. Now this substrate has four potential sites for proton removal, all flanked by a single ketone group, and thus all hydrogens are of similar acidity.
- The reaction that occurs is the intramolecular reaction that generates a strain-free six-membered ring system.
- This involves generating an enolate anion through the loss of a proton from the terminal methyl of the sidechain, followed by an aldol reaction involving the appropriate ring carbonyl as electrophile.
- Dehydration follows to generate the conjugated system, and it is this dehydration that disturbs the equilibrium. This annulation process was of considerable value in early approaches to steroid synthesis.
- The structural relationship of the bicyclic product obtained here to the male sex hormone testosterone is immediately apparent.
- Further, the non-conjugated carbonyl is now activating the adjacent carbon that subsequently features in building up the third ring system.
Michael Acceptors Can Be Carcinogens:
The Michael reaction involves conjugate addition of a nucleophile onto an α, β-unsaturated carbonyl compound, or similar system.
- Such reactions take place in nature as well, and some can be potentially dangerous to us.
- For example, the α, β-unsaturated ester ethyl acrylate is a cancer suspect agent.
- This electrophile can react with biological nucleophiles and, in so doing, bind irreversibly to the nucleophile, rendering it unable to carry out its normal functions.
- A particularly important enzyme that can act as a nucleophile is DNA polymerase, which is responsible for the synthesis of strands of DNA, especially as part of a DNA repair mechanism.
The nucleophilic centre is a thiol grouping, and this may react with ethyl acrylate as shown.
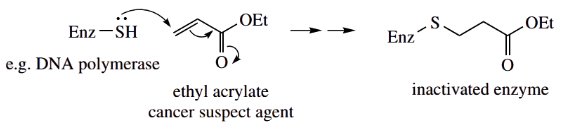
All is not doom and gloom, however, in that nature has provided in our bodies an alternative nucleophile to react with stray electrophiles like Michael acceptors.
This rather important compound is the tripeptide glutathione, a combination of glutamic acid, cysteine, and glycine.
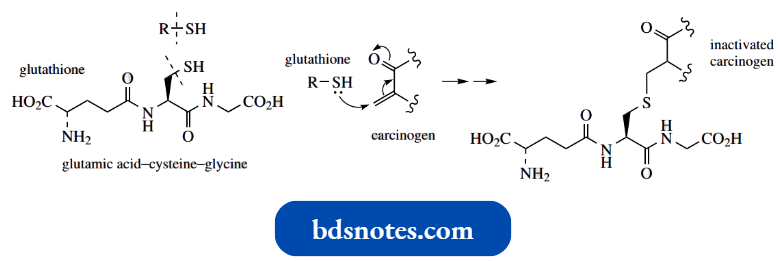
It is the thiol group in glutathione that reacts with a carcinogenic α, β-unsaturated carbonyl compound in the same way as did the thiol group of DNA polymerase.
- As a result, the carcinogen becomes irreversibly bound to glutathione, and can no longer interact with other biochemicals.
- Furthermore, as a result of the amino acid functionalities, the inactivated carcinogen now has increased polarity compared with the original compound.
- This compound is likely to be water-soluble, and can thus be excreted from the body. We have also seen glutathione inactivating other electrophiles, such as toxic epoxides.
- Glutathione is also implicated in the removal of toxic metabolites from the analgesic paracetamol (USA: acetaminophen).
The oxidative metabolism of paracetamol produces an N-hydroxy derivative, and this readily loses water to generate a reactive and toxic quinone imine, which interacts with proteins to cause cell damage.
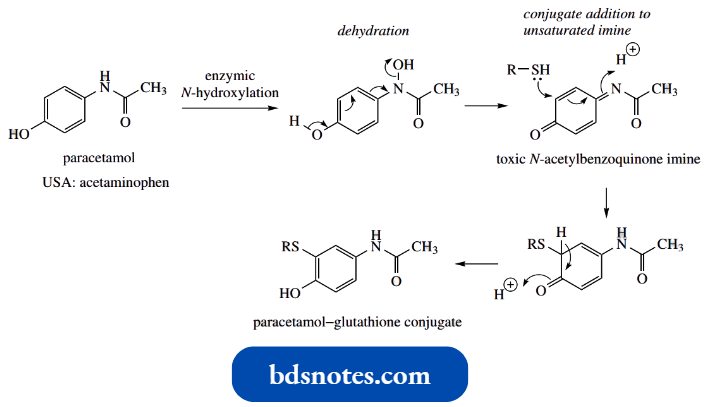
“Difference between aldol condensation and Michael addition: Notes explained”
Glutathione normally deactivates this reactive electrophile through a conjugate addition reaction.
- This time, we see conjugate addition onto an unsaturated imine rather than an unsaturated ketone. Rearomatization produces a non-toxic paracetamol–glutathione adduct.
- Unfortunately, if someone takes a large overdose of paracetamol, there may be insufficient glutathione available to detoxify all the metabolites.
- This can precipitate cell damage, particularly to the liver. Paracetamol is a safe analgesic unless taken in overdose.
- Multiple conjugate additions: anionic polymerization and superglue We have seen several reactions in which alkene derivatives can be polymerized.
Radical polymerization is the usual process of producing industrial polymers, but we also saw the implications of cationic polymerization.
- Here we see how an anionic process can lead to polymerization, and that this is an example of multiple conjugate additions.
- Alkene polymers such as poly(methyl methacrylate) and polyacrylonitrile are easily formed via anionic polymerization because the intermediate anions are resonance stabilized by the additional functional group, the ester or the nitrile.
- The process is initiated by a suitable anionic species, a nucleophile that can add to the monomer through conjugate addition in Michael fashion.
- The intermediate resonance-stabilized addition anion can then act as a nucleophile in further conjugate addition processes, eventually giving a polymer.
The process will terminate by proton abstraction, probably from solvent.
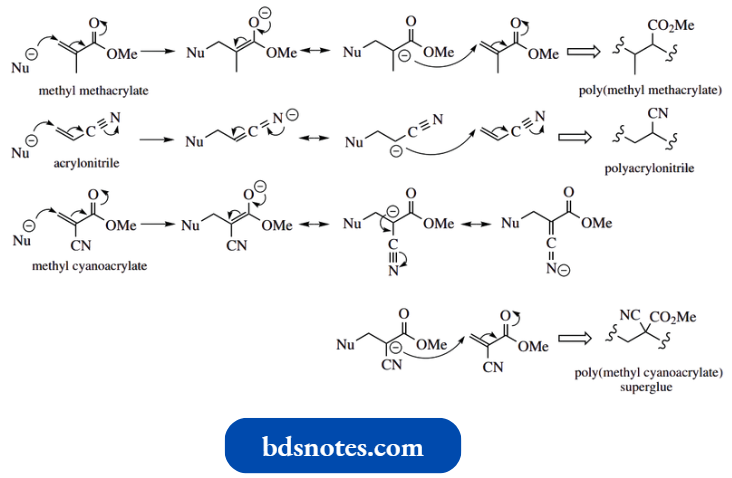
Methyl cyanoacrylate combines the anion-stabilizing features of both an ester group and a nitrile.
- Addition anions form very easily because of their enhanced resonance stability, and this polymerization process forms the basis of superglue.
- Traces of moisture on surfaces (including fingers) initiate anionic polymerization and the bonding together of almost any material.
- Superglue now has some value in surgery, bonding tissue without the need for stitches.
Leave a Reply